

Computational Analysis of Molnupiravir

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
3. Methods
3.1. DFT Calculations
3.2. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- First Oral Antiviral for COVID-19, Lagevrio (Molnupiravir), Approved by MHRA. Available online: https://www.gov.uk/government/news/first-oral-antiviral-for-covid-19-lagevrio-molnupiravir-approved-by-mhra (accessed on 29 December 2021).
- Available online: https://www.indiatoday.in/science/story/merck-covid-19-pill-molnupiravir-efficacy-omicron-vaccine-1889024-2021-12-17 (accessed on 29 December 2021).
- Toots, M.; Plemper, R.K. Next-Generation Direct-Acting Influenza Therapeutics. Transl. Res. 2020, 220, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Toots, M.; Yoon, J.-J.; Cox, R.M.; Hart, M.; Sricher, Z.M.; Makhsous, N.; Plesker, R.; Barrena, A.H.; Reddy, P.G.; Mitchell, D.G.; et al. Characterization of orally efficacious influenza drug with high resistance barrier in ferrets and human airway epithelia. Sci. Transl. Med. 2019, 11, eaax5866. [Google Scholar] [CrossRef] [PubMed]
- Toots, M.; Yoon, J.-J.; Hart, M.; Natchus, M.G.; Painter, G.R.; Plemper, R.K. Quantitative efficacy paradigms of the influenza clinical drug candidate EIDD-2801 in the ferret model. Transl. Res. 2020, 218, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.who.int/health-topics/coronavirus#tab=tab_1 (accessed on 29 December 2021).
- Available online: https://www.who.int/dg/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---11-march-2020 (accessed on 29 December 2021).
- Available online: https://covid19.who.int/ (accessed on 15 January 2022).
- Abdelnabi, R.; Foo, C.S.; Kaptein, S.J.F.; Zhang, X.; Do, T.N.D.; Langendries, L.; Vangeel, L.; Breuer, J.; Pang, J.; Williams, R.; et al. The combined treatment of Molnupiravir and Favipiravir results in a potentiation of antiviral efficacy in a SARS-CoV-2 hamster infection model. EBioMedicine 2021, 72, 103595. [Google Scholar] [CrossRef]
- Wahl, A.; Gralinski, L.E.; Johnson, C.E.; Yao, W.; Kovarova, M.; Dinnon, K.H., III; Liu, H.; Madden, V.J.; Krzystek, H.M.; De, C.; et al. SARS-CoV-2 infection is effectively treated and prevented by EIDD-2801. Nature 2021, 591, 451–457. [Google Scholar] [CrossRef]
- Hashemian, S.M.R.; Pourhanifeh, M.H.; Hamblin, M.R.; Shahrzad, M.K.; Mirzaei, H. RdRp inhibitors and COVID-19: Is molnupiravir a good option? Biomed. Pharmacother. 2022, 146, 112517. [Google Scholar] [CrossRef]
- Cox, R.M.; Wolf, J.D.; Plemper, R.K. Therapeutically administered ribonucleoside analogue MK-4482/EIDD-2801 blocks SARS-CoV-2 transmission in ferrets. Nat. Microbiol. 2021, 6, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Rosenke, K.; Hansen, F.; Schwarz, B.; Feldmann, F.; Haddock, E.; Rosenke, R.; Meade-White, K.; Okumura, A.; Leventhal, S.; Hawman, D.W.; et al. Orally delivered MK-4482 inhibits SARS-CoV-2 replication in the Syrian hamster model. Nat. Commun. 2021, 12, 2295. [Google Scholar] [CrossRef]
- Kabinger, F.; Stiller, C.; Schmitzová, J.; Dienemann, C.; Kokic, G.; Hillen, H.S.; Höbartner, C.; Cramer, P. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat. Struct. Mol. Biol. 2021, 28, 740–746. [Google Scholar] [CrossRef]
- Fischer, W.A.; Eron, J.J.; Holman, W.; Cohen, M.S.; Fang, L.; Szewczyk, L.G.; Sheahan, T.P.; Baric, R.; Mollan, K.R.; Wolfe, C.R.; et al. A Phase 2a clinical trial of Molnupiravir in patients with COVID-19 shows accelerated SARS-CoV-2 RNA clearance and elimination of infectious virus. Sci. Transl. Med. 2022, 14, eabl7430. [Google Scholar] [CrossRef]
- Malone, B.; Campbell, E.A. Molnupiravir: Coding for catastrophe. Nat. Struct. Mol. Biol. 2021, 28, 706–708. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.merck.com/news/merck-and-ridgebacks-investigational-oral-antiviral-molnupiravir-reduced-the-risk-of-hospitalization-or-death-by-approximately-50-percent-compared-to-placebo-for-patients-with-mild-or-moderat (accessed on 29 December 2021).
- The Emergence of Powerful Oral Anti-COVID-19 Drugs in the Post-Vaccine Era. Available online: https://www.thelancet.com/journals/lanmic/article/PIIS2666-5247(21)00278-0/fulltext (accessed on 29 December 2021).
- Available online: https://www.indiatoday.in/science/story/covid-19-pills-drug-omicron-pfizer-molnupiravir-paxlovid-ronapreve-1889039-2021-12-17 (accessed on 29 December 2021).
- Available online: https://www.reuters.com/world/us/merck-pfizer-covid-19-pills-effective-against-omicron-us-fda-official-2021-12-23/ (accessed on 29 December 2021).
- Shiryaev, A.A.; Goncharenko, A.N.; Burkhanova, T.M.; Alkhimova, L.A.; Babashkina, M.G.; Chandrasekaran, R.; Safin, D.A. A chiral (1R,2R)-N,N′-bis-(salicylidene)-1,2-diphenyl-1,2-ethanediamine Schiff base dye: Synthesis, crystal structure, Hirshfeld surface analysis, computational study, photophysical properties and in silico antifungal activity. J. Iran. Chem. Soc. 2021, 18, 2897–2911. [Google Scholar] [CrossRef]
- Babashkina, M.G.; Frontera, A.; Kertman, A.V.; Saygideger, Y.; Murugavel, S.; Safin, D.A. Favipiravir: Insight into the crystal structure, Hirshfeld surface analysis and computational study. J. Iran. Chem. Soc. 2022, 19, 85–94. [Google Scholar] [CrossRef]
- Alkhimova, L.E.; Babashkina, M.G.; Safin, D.A. Computational analysis of aspirin. J. Mol. Struct. 2022, 1251, 131975. [Google Scholar] [CrossRef]
- Burkhanova, T.M.; Babashkina, M.G.; Taskin-Tok, T.; Sharov, A.V.; Safin, D.A. Naphthalene-based bis-N-salicylidene aniline dyes: Crystal structures, Hirshfeld surface analysis, computational study and molecular docking with the SARS-CoV-2. J. Iran. Chem. Soc. 2022. [Google Scholar] [CrossRef]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef]
- Paymode, D.J.; Vasudevan, N.; Ahmad, S.; Kadam, A.L.; Cardoso, F.S.P.; Burns, J.M.; Cook, D.W.; Stringham, R.W.; Snead, D.R. Toward a Practical, Two-Step Process for Molnupiravir: Direct Hydroxyamination of Cytidine Followed by Selective Esterification. Org. Process Res. Dev. 2021, 25, 1822–1830. [Google Scholar] [CrossRef]
- Abraham, J.P.; Sajan, D.; Joe, I.H.; Jayakumar, V.S. Molecular structure, spectroscopic studies and first-order molecular hyperpolarizabilities of p-amino acetanilide. Spectrochim. Acta Part A 2008, 71, 355–367. [Google Scholar] [CrossRef]
- Karamanis, P.; Pouchan, C.; Maroulis, G. Structure, stability, dipole polarizability and differential polarizability in small gallium arsenide clusters from all-electron ab initio and density-functional-theory calculations. Phys. Rev. A 2008, 77, 013201–013203. [Google Scholar] [CrossRef]
- Eme, A.; Sağdınç, S.M. Spectroscopic (FT–IR, FT–Raman, UV–Vis) analysis, conformational, HOMO-LUMO, NBO and NLO calculations on monomeric and dimeric structures of 4–pyridazinecarboxylic acid by HF and DFT methods. J. Mol. Struct. 2017, 1147, 322–334. [Google Scholar] [CrossRef]
- Cully, M. A tale of two antiviral targets—And the COVID-19 drugs that bind them. Nat. Rev. Drug Discov. 2022, 21, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.organic-chemistry.org/prog/peo/ (accessed on 29 December 2021).
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery. Curr. Comp.-Aid. Drug 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L. The many roles of computation in drug discovery. Science 2004, 303, 1813–1818. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, H.-Y.; Kang, S.; Silverman, R.B.; Poulos, T.L. Electrostatic Control of Isoform Selective Inhibitor Binding in Nitric Oxide Synthase. Biochemistry 2016, 55, 3702–3707. [Google Scholar] [CrossRef]
- Tok, T.T.; Tatar, G. Structures and functions of coronavirus proteins: Molecular modeling of viral nucleoprotein. Int. J. Virol. Infect. Dis. 2017, 2, 001–007. [Google Scholar]
- Tok, T.T.; Gowder, S.J.T. An Updated Review on COVID-19 with Special Reference to Structural Elucidation and Functional Properties. Biomed. J. Sci. Tech. Res. 2020, 31, 24345–24351. [Google Scholar] [CrossRef]
- Shamsi, A.; Mohammad, T.; Anwar, S.; Amani, S.; Khan, M.S.; Husain, F.M.; Rehman, M.T.; Islam, A.; Hassan, M.I. Potential drug targets of SARS-CoV-2: From genomics to therapeutics. Int. J. Biol. Macromol. 2021, 177, 1–9. [Google Scholar] [CrossRef]
- Ullrich, S.; Nitsche, C. The SARS-CoV-2 main protease as drug target. Bioorg. Med. Chem. Let. 2020, 30, 127377. [Google Scholar] [CrossRef]
- Ding, X.-C.; He, J.; Zhang, X.; Jiang, C.; Sun, Y.; Zhang, Y.; Chen, Q.; He, H.; Li, W.; Xie, J.; et al. Crucial Mutations of Spike Protein on SARS-CoV-2 Evolved to Variant Strains Escaping Neutralization of Convalescent Plasmas and RBD-Specific Monoclonal Antibodies. Front. Immunol. 2021, 12, 693775. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView; Version 6.0; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 8, 3265–3269. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Modeling 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Rose, Y.; Duarte, J.M.; Lowe, R.; Segura, J.; Bi, C.; Bhikadiya, C.; Chen, L.; Rose, A.S.; Bittrich, S.; Burley, S.K.; et al. RCSB protein data bank: Architectural advances towards integrated searching and efficient access to macromolecular structure data from the PDB archive. J. Mol. Biol. 2021, 433, 166704. [Google Scholar] [CrossRef] [PubMed]
- Accelrys Software Inc. Discovery Studio Modeling Environment; Release 3.5; Accelrys Software Inc.: San Diego, CA, USA, 2013. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Thermodynamic Parameter | Keto-oxime | Keto-hydroxylamine | Hydroxyl-oxime |
|---|---|---|---|
| Self-consistent field energy (a.u.) | −1197.962 | −1197.951 | −1197.922 |
| Total energy (thermal) (kcal mol−1) | 225.112 | 224.768 | 224.572 |
| Electronic energy (thermal) (kcal mol−1) | 0.000 | 0.000 | 0.000 |
| Translational energy (thermal) (kcal mol−1) | 0.889 | 0.889 | 0.889 |
| Rotational energy (thermal) (kcal mol−1) | 0.889 | 0.889 | 0.889 |
| Vibrational energy (thermal) (kcal mol−1) | 223.335 | 222.990 | 222.794 |
| Total heat capacity (thermal) (cal mol−1 K−1) | 85.340 | 84.962 | 86.088 |
| Electronic heat capacity (thermal) (cal mol−1 K−1) | 0.000 | 0.000 | 0.000 |
| Translational heat capacity (thermal) (cal mol−1 K−1) | 2.981 | 2.981 | 2.981 |
| Rotational heat capacity (thermal) (cal mol−1 K−1) | 2.981 | 2.981 | 2.981 |
| Vibrational heat capacity (thermal) (cal mol−1 K−1) | 79.379 | 79.001 | 80.127 |
| Total entropy (thermal) (cal mol−1 K−1) | 164.370 | 163.370 | 165.868 |
| Electronic entropy (thermal) (cal mol−1 K−1) | 0.000 | 0.000 | 0.000 |
| Translational entropy (thermal) (cal mol−1 K−1) | 43.269 | 43.269 | 43.269 |
| Rotational entropy (thermal) (cal mol−1 K−1) | 35.425 | 35.425 | 35.428 |
| Vibrational entropy (thermal) (cal mol−1 K−1) | 85.676 | 84.677 | 87.170 |
| Zero-point vibrational energy (thermal) (kcal mol−1) | 210.837 | 210.592 | 210.174 |
| Rotational constants (GHz) | |||
| A | 0.38535 | 0.39715 | 0.41586 |
| B | 0.12511 | 0.12266 | 0.11980 |
| C | 0.10302 | 0.10192 | 0.09932 |
| Bond | Keto-oxime | Keto-hydroxylamine | Hydroxyl-oxime |
|---|---|---|---|
| C1–C4 | 1.525 | 1.526 | 1.529 |
| C1–C9 | 1.528 | 1.527 | 1.527 |
| C2–C3 | 1.551 | 1.551 | 1.546 |
| C3–C4 | 1.539 | 1.540 | 1.538 |
| C6–C7 | 1.344 | 1.358 | 1.341 |
| C7–C8 | 1.443 | 1.426 | 1.451 |
| C10–C11 | 1.528 | 1.528 | 1.527 |
| C11–C12 | 1.544 | 1.544 | 1.544 |
| C11–C13 | 1.534 | 1.534 | 1.534 |
| C1–O1 | 1.451 | 1.452 | 1.452 |
| C2–O1 | 1.409 | 1.411 | 1.409 |
| C3–O3 | 1.405 | 1.403 | 1.413 |
| C4–O2 | 1.416 | 1.415 | 1.416 |
| C5–O4 | 1.228 | 1.229 | 1.359 |
| C9–O6 | 1.432 | 1.431 | 1.430 |
| C10–O6 | 1.369 | 1.369 | 1.371 |
| C10–O7 | 1.200 | 1.200 | 1.200 |
| C5–N1 | 1.384 | 1.422 | 1.365 |
| C5–N2 | 1.373 | 1.358 | 1.281 |
| C6–N1 | 1.395 | 1.363 | 1.409 |
| C8–N2 | 1.396 | 1.323 | 1.407 |
| C8–N3 | 1.289 | 1.359 | 1.291 |
| N3–O5 | 1.421 | 1.398 | 1.389 |
| Tautomer | D–X∙∙∙A | d(D–X) | d(X∙∙∙A) | d(D∙∙∙A) | ∠(DXA) |
|---|---|---|---|---|---|
| keto-oxime | O2–H1∙∙∙O3 | 0.968 | 2.095 | 2.648 | 114.52 |
| O3–H2∙∙∙O4 | 0.974 | 1.843 | 2.718 | 147.92 | |
| N2–H3∙∙∙O5 | 1.012 | 2.163 | 2.537 | 99.62 | |
| keto-hydroxylamine | O2–H1∙∙∙O3 | 0.967 | 2.165 | 2.676 | 111.59 |
| O3–H2∙∙∙O4 | 0.966 | 2.064 | 2.917 | 146.30 | |
| O5–H4∙∙∙N2 | 0.975 | 2.032 | 2.622 | 116.97 | |
| hydroxyl-oxime | O2–H1∙∙∙O3 | 0.967 | 2.165 | 2.676 | 111.59 |
| O3–H2∙∙∙O4 | 0.966 | 2.064 | 2.917 | 146.30 | |
| O5–H4∙∙∙N2 | 0.975 | 2.032 | 2.622 | 116.97 |
| Molecular Vibration 1 | Frequency (cm−1) | IR Intensity (KM∙mol−1) | Raman Activity (Å4∙amu−1) | Force Constant, k (mDyne A−1) |
|---|---|---|---|---|
| keto-oxime | ||||
| νO5–H4 | 3856 | 189.04 | 220.68 | 9.3559 |
| νO2–H3 | 3754 | 73.43 | 60.22 | 8.8462 |
| νO3–H2 | 3629 | 417.06 | 96.23 | 8.2735 |
| νNH | 3605 | 109.93 | 48.16 | 8.2416 |
| νs(C6–H9 + C7–H10) | 3261 | 12.72 | 74.49 | 6.8628 |
| νas(C6–H9 + C7–H10) | 3237 | 6.62 | 66.59 | 6.7326 |
| νC13–H14 | 3120 | 20.70 | 48.34 | 6.3159 |
| νasH14–C12–H15 | 3106 | 15.07 | 37.91 | 6.2675 |
| νC1–H5 | 3102 | 21.15 | 84.32 | 6.1803 |
| νC13–H18 | 3099 | 25.87 | 71.32 | 6.2376 |
| νasH15–C12–H16 | 3095 | 29.03 | 43.98 | 6.2194 |
| νasH11–C9–H12 + νC1–H5 | 3077 | 13.81 | 48.81 | 6.1665 |
| νC11–H13 | 3058 | 14.12 | 130.10 | 5.9645 |
| νC2–H6 | 3046 | 37.37 | 58.52 | 5.9362 |
| νC4–H8 + νC2–H6 | 3043 | 22.81 | 157.47 | 5.9235 |
| νs(C13–H17 + C13–H18 + C13–H19) | 3037 | 22.14 | 204.18 | 5.6303 |
| νs(C12–H14 + C12–H15 + C12–H16) | 3031 | 25.20 | 121.28 | 5.6016 |
| νs(C9–H11 + C9–H12) | 3027 | 19.80 | 43.60 | 5.7009 |
| νC3–H7 | 2994 | 23.23 | 49.13 | 5.7226 |
| νC10=O7 | 1814 | 367.75 | 20.51 | 22.8748 |
| νC5=O4 + βN2–H3 + βO3–H2 | 1733 | 836.72 | 22.71 | 11.9387 |
| νC8=N3 + νC5=O4 + νC6=C7 +βO5–H4 + βN2–H3 + βC6–H9 + βC7–H10 | 1714 | 140.42 | 371.53 | 13.9766 |
| keto-hydroxylamine | ||||
| νO2–H1 | 3745 | 76.60 | 59.88 | 8.8028 |
| νN3–H3 | 3619 | 108.95 | 137.56 | 8.3276 |
| νO3–H2 | 3569 | 526.21 | 126.65 | 8.0039 |
| νO5–H4 | 3512 | 85.58 | 129.66 | 7.7383 |
| νs(C6–H9 + C7–H10) | 3244 | 9.42 | 77.27 | 6.7877 |
| νas(C6–H9 + C7–H10) | 3216 | 0.59 | 82.40 | 6.6483 |
| νC13–H17 + νC13–H18 + νC13–H19 | 3120 | 19.99 | 48.02 | 6.3164 |
| νC12–H14 + νC12–H15 + νC12–H16 | 3106 | 15.40 | 37.56 | 6.2662 |
| νC1–H5 | 3101 | 20.69 | 86.39 | 6.1751 |
| νasH18–C13–H19) | 3099 | 26.15 | 71.95 | 6.2365 |
| νasH15–C12–H16) | 3095 | 28.63 | 44.13 | 6.2181 |
| νasH11–C9–H12 + νC1–H5 | 3075 | 14.97 | 46.70 | 6.1604 |
| νC11–H13 | 3056 | 13.35 | 110.25 | 5.9559 |
| νC2–H6 | 3055 | 21.17 | 105.53 | 5.9716 |
| νC4–H8 | 3041 | 33.92 | 129.47 | 5.9150 |
| νs(C13–H17 + C13–H18 + C13–H19) | 3037 | 22.96 | 204.79 | 5.6307 |
| νs(C12–H14 + C12–H15 + C12–H16) | 3031 | 23.47 | 110.87 | 5.5975 |
| νsH11–C9–H12 | 3025 | 21.21 | 50.87 | 5.6943 |
| νC3–H7 | 2987 | 27.06 | 45.99 | 5.6959 |
| νC10=O7 | 1815 | 365.00 | 19.83 | 22.8983 |
| νC5=O4 + βO5–H4 + βO3–H2 + βC6–H9 | 1715 | 785.23 | 33.24 | 17.0337 |
| νC8=N3 + νC5=O4 + νC6=C7 +βC6–H9 + βC7–H10 + βC2–H6 | 1663 | 291.45 | 25.55 | 10.4411 |
| βN3–H3 + βO5–H4 | 1591 | 20.24 | 20.25 | 2.7871 |
| hydroxyl-oxime | ||||
| νO2–H1 + νO3–H2 + νO4–H3 | 3782 | 94.84 | 36.15 | 8.9787 |
| νO2–H1 + νO4–H3 | 3774 | 27.16 | 36.51 | 8.9377 |
| νO5–H4 | 3615 | 16.28 | 90.73 | 8.2026 |
| νC6–H9 | 3273 | 9.92 | 75.23 | 6.9078 |
| νC7–H10 | 3222 | 2.37 | 85.81 | 6.6768 |
| νC13–H17 + νC13–H18 + νC13–H19 | 3121 | 19.94 | 49.24 | 6.3195 |
| νC12–H14 + νC12–H15 + νC12–H16 | 3106 | 14.34 | 42.24 | 6.2714 |
| νasH18–C13–H19 | 3100 | 23.66 | 67.74 | 6.2426 |
| νC1–H5 + νasH11–C9–H12 | 3099 | 22.56 | 82.80 | 6.1714 |
| νC12–H14 + νC12–H15 + νC12–H16 | 3095 | 30.72 | 47.70 | 6.2155 |
| νC1–H5 + νasH11–C9–H12 | 3077 | 12.97 | 50.18 | 6.1565 |
| νC2–H6 | 3061 | 20.82 | 56.13 | 5.9950 |
| νC11–H13 | 3056 | 14.50 | 140.64 | 5.9601 |
| νC4–H8 | 3047 | 29.86 | 139.46 | 5.9408 |
| ν(C13–H17 + C13–H18 + C13–H19) | 3038 | 21.16 | 199.10 | 5.6333 |
| ν(C12–H14 + C12–H15 + C12–H16) | 3031 | 24.44 | 122.92 | 5.5991 |
| νsH11–C9–H12 | 3025 | 23.18 | 61.34 | 5.7005 |
| νC3–H7 | 3006 | 17.27 | 35.96 | 5.7667 |
| νC10=O7 | 1817 | 363.72 | 20.82 | 22.9946 |
| νC5=N2 + νC6=C7 + βO4–H3 + βC6–H9 + βC7–H10 | 1718 | 556.37 | 146.50 | 12.9824 |
| νC8=N3 + νC5=N2 + νC6=C7 +βO4–H3 + βC7–H10 | 1658 | 71.05 | 288.48 | 13.6289 |
| νC8=N3 + νC5=N2 + νC6=C7 +βO4–H3 + βO5–H4 + βC6–H9 + βC7–H10 | 1609 | 85.65 | 63.89 | 13.3853 |
| Parameter | Keto-oxime | Keto-hydroxylamine | Hydroxyl-oxime |
|---|---|---|---|
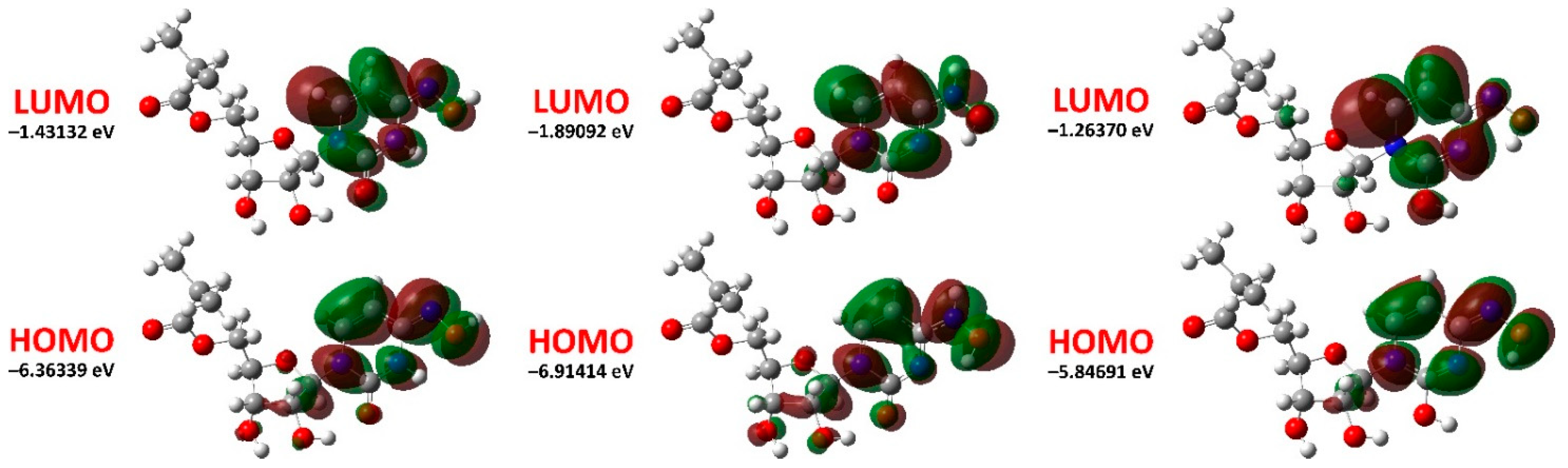
| EHOMO (eV) | −6.36339 | −6.91414 | −5.84691 |
| ELUMO (eV) | −1.43132 | −1.89092 | −1.26370 |
| ΔELUMO−HOMO = ELUMO − EHOMO (eV) | 4.93207 | 5.02322 | 4.58321 |
| Ionization energy, I = −EHOMO (eV) | 6.36339 | 6.91414 | 5.84691 |
| Electron affinity, A = −ELUMO (eV) | 1.43132 | 1.89092 | 1.26370 |
| Electronegativity, χ = (I + A)/2 (eV) | 3.89736 | 4.40253 | 3.55531 |
| Chemical potential, μ = −χ (eV) | −3.89736 | −4.40253 | −3.55531 |
| Global chemical hardness, η = (I − A)/2 (eV) | 2.46604 | 2.51161 | 2.29161 |
| Global chemical softness, S = 1/(2η) (eV−1) | 0.20275 | 0.19908 | 0.21819 |
| Global electrophilicity index, ω = μ2/(2η) (eV) | 3.07972 | 3.85854 | 2.75794 |
| Maximum additional electric charge, ΔNmax = −μ/ƞ | 1.58041 | 1.75287 | 1.55145 |
| λmax (nm) | Osc. Strength | Transition | λmax (nm) | Osc. Strength | Transition |
|---|---|---|---|---|---|
| keto-oxime | |||||
| 164.8 | 0.0514 | HOMO−7 → LUMO+3 (32.5%) | 186.4 | 0.0353 | HOMO−8 → LUMO (16.5%) |
| HOMO−6 → LUMO+5 (5.8%) | HOMO−2 → LUMO+2 (11.6%) | ||||
| HOMO−5 → LUMO+5 (7.2%) | HOMO−1 → LUMO+2 (47.8%) | ||||
| 178.3 | 0.0351 | HOMO−3 → LUMO+2 (21.2%) | 214.5 | 0.0445 | HOMO−5 → LUMO (8.9%) |
| HOMO → LUMO+15 (11.6%) | HOMO−2 → LUMO (13.4%) | ||||
| HOMO → LUMO+16 (27.3%) | HOMO−1 → LUMO (36.5%) | ||||
| 180.6 | 0.0655 | HOMO−4 → LUMO+1 (8.0%) | HOMO → LUMO+6 (18.4%) | ||
| HOMO−3 → LUMO+1 (14.4%) | 231.0 | 0.4040 | HOMO → LUMO (15.2%) | ||
| HOMO−2 → LUMO+2 (43.7%) | HOMO → LUMO+2 (64.7%) | ||||
| HOMO−1 → LUMO+2 (9.5%) | 281.3 | 0.0987 | HOMO → LUMO (79.3%) | ||
| HOMO → LUMO+2 (17.5%) | |||||
| keto-hydroxylamine | |||||
| 159.1 | 0.0406 | HOMO−15 → LUMO (7.8%) | 199.6 | 0.0340 | HOMO−1 → LUMO+3 (29.8%) |
| HOMO−13 → LUMO (9.0%) | HOMO−1 → LUMO+4 (28.8%) | ||||
| HOMO−9 → LUMO+1 (16.9%) | HOMO → LUMO+6 (13.5%) | ||||
| HOMO−9 → LUMO+2 (34.7%) | 0.1085 | HOMO−1 → LUMO+2 (23.1%) | |||
| 169.3 | 0.0516 | HOMO−10 → LUMO (19.8%) | HOMO → LUMO+5 (25.1%) | ||
| HOMO−3 → LUMO+7 (9.2%) | HOMO → LUMO+6 (14.2%) | ||||
| HOMO−2 → LUMO+7 (10.6%) | HOMO → LUMO+7 (9.8%) | ||||
| HOMO−1 → LUMO+10 (7.3%) | 201.1 | 0.0902 | HOMO−1 → LUMO+2 (50.2%) | ||
| 170.7 | 0.0589 | HOMO−10 → LUMO (23.3%) | HOMO → LUMO+5 (20.6%) | ||
| HOMO−2 → LUMO+7 (7.7%) | 231.0 | 0.0581 | HOMO → LUMO+2 (70.8%) | ||
| HOMO−1 → LUMO+10 (12.0%) | HOMO → LUMO+4 (9.9%) | ||||
| 190.6 | 0.0396 | HOMO−5 → LUMO+2 (8.0%) | 238.0 | 0.0473 | HOMO → LUMO+1 (73.5%) |
| HOMO−3 → LUMO+2 (8.5%) | HOMO → LUMO+2 (9.9%) | ||||
| HOMO−2 → LUMO+1 (22.9%) | 267.0 | 0.2593 | HOMO → LUMO (85.7%) | ||
| HOMO−2 → LUMO+2 (35.1%) | |||||
| hydroxyl-oxime | |||||
| 158.7 | 0.0413 | HOMO−9 → LUMO+2 (8.7%) | 182.4 | 0.0330 | HOMO−5 → LUMO (8.7%) |
| HOMO → LUMO+32 (10.7%) | HOMO−4 → LUMO+1 (50.6%) | ||||
| HOMO → LUMO+34 (10.2%) | HOMO−3 → LUMO+2 (11.7%) | ||||
| 168.1 | 0.0366 | HOMO−9 → LUMO (22.2%) | 182.6 | 0.0374 | HOMO−5 → LUMO (38.7%) |
| HOMO−8 → LUMO+1 (7.5%) | HOMO−3 → LUMO+2 (15.6%) | ||||
| HOMO−1 → LUMO+7 (10.3%) | HOMO−2 → LUMO+3 (16.5%) | ||||
| HOMO → LUMO+30 (8.7%) | 234.6 | 0.0434 | HOMO → LUMO+6 (7.1%) | ||
| 171.3 | 0.0855 | HOMO−9 → LUMO (15.4%) | HOMO → LUMO+7 (77.8%) | ||
| HOMO−8 → LUMO+1 (16.6%) | 248.0 | 0.0521 | HOMO → LUMO+3 (9.0%) | ||
| HOMO−5 → LUMO+2 (10.9%) | HOMO → LUMO+4 (8.4%) | ||||
| HOMO−4 → LUMO+3 (7.7%) | HOMO → LUMO+5 (76.9%) | ||||
| 173.4 | 0.0315 | HOMO−7 → LUMO+1 (12.8%) | 249.6 | 0.3958 | HOMO → LUMO+1 (10.6%) |
| HOMO−6 → LUMO+1 (16.9%) | HOMO → LUMO+2 (8.3%) | ||||
| HOMO−5 → LUMO+1 (8.3%) | HOMO → LUMO+3 (55.2%) | ||||
| HOMO−4 → LUMO+3 (30.5%) | HOMO → LUMO+5 (10.4%) | ||||
| 275.5 | 0.0308 | HOMO → LUMO+2 (61.6%) | |||
| HOMO → LUMO+3 (23.7%) | |||||
| Hydrogen | Keto-oxime | Keto-hydroxylamine | Hydroxyl-oxime |
|---|---|---|---|
| H1 | 2.74 | 2.90 | 2.33 |
| H2 | 5.47 | 6.24 | 2.87 |
| H3 | 7.54 | 6.35 | 4.89 |
| H4 | 5.29 | 7.88 | 6.82 |
| H5 | 4.59 | 4.61 | 4.54 |
| H6 | 5.39 | 5.55 | 5.59 |
| H7 | 3.82 | 3.77 | 3.82 |
| H8 | 4.40 | 4.38 | 4.35 |
| H9 | 7.07 | 7.80 | 6.51 |
| H10 | 5.39 | 5.24 | 5.84 |
| H11 | 3.54 | 3.53 | 3.59 |
| H12 | 3.96 | 3.95 | 4.05 |
| H13 | 2.41 | 2.38 | 2.46 |
| H14 | 0.96–1.06 | 1.02–1.06 | 1.03–1.06 |
| H15 | 0.96–1.06 | 0.94–0.95 | 1.03–1.06 |
| H16 | 0.96–1.06 | 0.94–0.95 | 0.96 |
| H17 | 1.59 | 1.60 | 1.62 |
| H18 | 0.73 | 0.71 | 0.76 |
| H19 | 0.96–1.06 | 1.02–1.06 | 1.03–1.06 |
| Parameter | Keto-oxime | Keto-hydroxylamine | Hydroxyl-oxime | Urea [29] |
|---|---|---|---|---|
| μx (Debye) | −2.7182 | −0.6997 | 1.0585 | |
| μy (Debye) | −4.9139 | −7.5858 | 1.0487 | |
| μz (Debye) | −0.0544 | −1.3843 | 0.7004 | |
| μD (Debye) | 5.6159 | 7.7428 | 1.6465 | |
| αxx (a.u.) | 256.320 | 254.188 | 272.451 | |
| αyy (a.u.) | 213.808 | 216.526 | 212.955 | |
| αzz (a.u.) | 153.182 | 152.609 | 144.264 | |
| αxy (a.u.) | 26.209 | 22.665 | 22.940 | |
| αxz (a.u.) | −14.113 | −12.219 | −12.450 | |
| αyz (a.u.) | 6.759 | 9.801 | 1.162 | |
| α (a.u.) | 207.770 | 207.774 | 208.89 | |
| α (esu) | 30.792 × 10−24 | 30.792 × 10−24 | 31.106 × 10−24 | 3.8312 × 10−24 |
| αtautomer/αurea | 8.0 | 8.0 | 8.1 | |
| Δα (a.u.) | 104.189 | 100.937 | 119.970 | |
| Δα (esu) | 15.441 × 10−24 | 14.959 × 10−24 | 17.780 × 10−24 | |
| βxxx (a.u.) | −263.723 | −47.212 | 16.843 | |
| βyyy (a.u.) | −5.671 | −49.597 | 46.796 | |
| βzzz (a.u.) | 2.192 | 0.908 | 0.007 | |
| βxyy (a.u.) | −0.326 | −25.778 | 53.700 | |
| βxxy (a.u.) | −86.296 | −66.014 | 130.374 | |
| βxxz (a.u.) | 23.622 | −18.584 | 6.807 | |
| βxzz (a.u.) | 9.757 | 13.287 | 11.574 | |
| βyzz (a.u.) | −4.432 | −2.785 | 5.898 | |
| βyyz (a.u.) | 5.210 | −7.786 | 4.231 | |
| βxyz (a.u.) | 18.340 | −1.679 | −11.407 | |
| β (a.u.) | 273.715 | 135.020 | 200.946 | |
| β (esu) | 2.365 × 10−30 | 1.166 × 10−30 | 1.736 × 10−30 | 0.1947 × 10−30 |
| βtautomer/βurea | 12.1 | 6.0 | 8.9 |
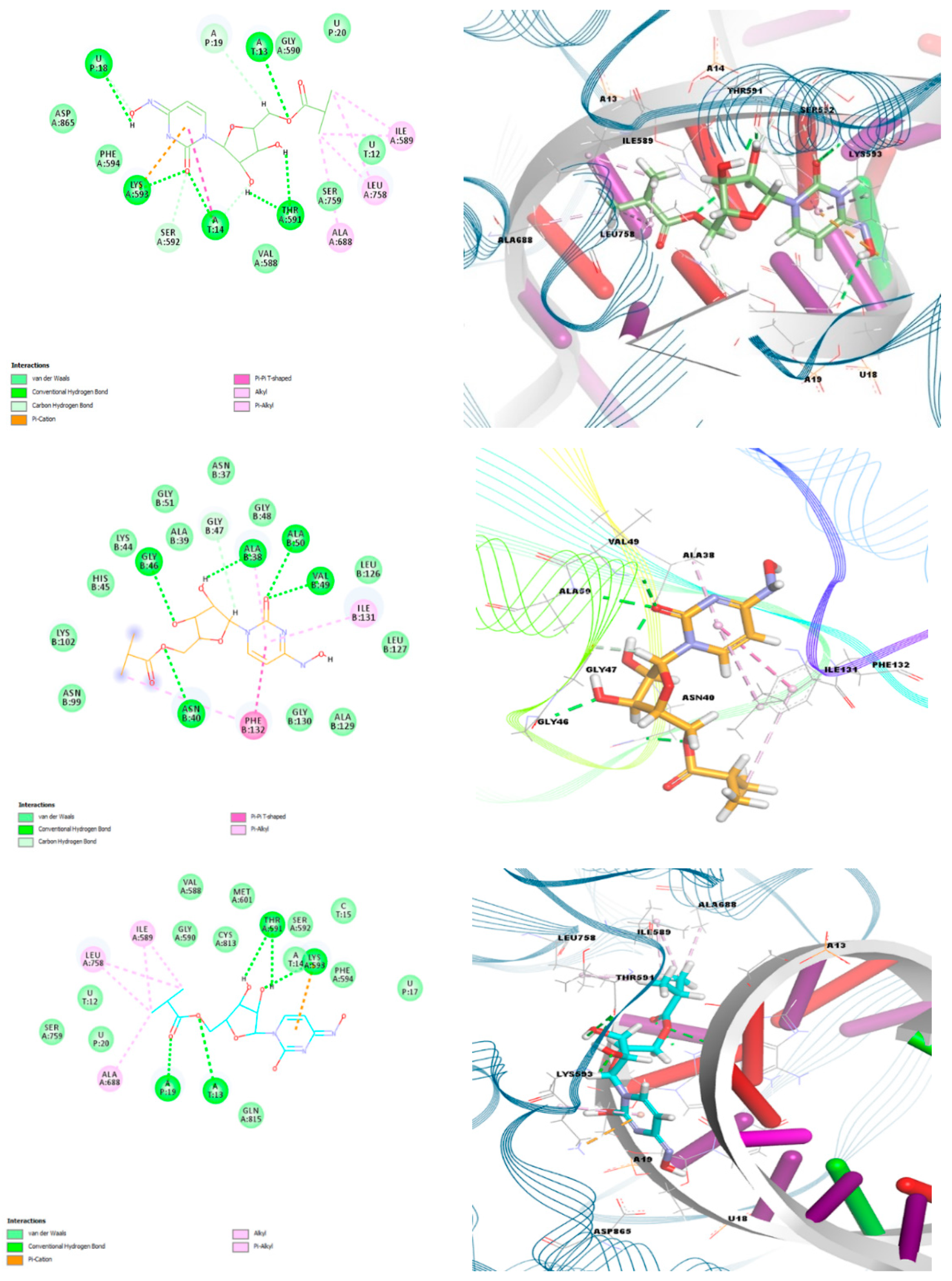
| Protein | PDB Code | Keto-oxime | Keto-hydroxylamine | Hydroxyl-oxime |
|---|---|---|---|---|
| Main protease (Mpro) | 6LU7 | −6.60 | −7.00 | −7.30 |
| Papain-like protease (PLpro) | 6WUU | −7.50 | −7.40 | −7.40 |
| Nonstructural protein 3 (Nsp3_range 207–379-AMP) | 6W6Y | −6.90 | −7.20 | −6.90 |
| Nonstructural protein 3 (Nsp3_range 207–379-MES) | 6W6Y | −8.10 | −7.90 | −7.80 |
| Helicase (Nsp13)-adp | 6JYT | −6.30 | −6.50 | −6.20 |
| Helicase (Nsp13)-ncb | 6JYT | −6.80 | −6.90 | −6.60 |
| RdRp-RTP | 7BV2 | −9.90 | −7.50 | −9.30 |
| RdRp-RNA | 7BV2 | −7.00 | −6.60 | −6.80 |
| Nsp14 (ExoN) | 5C8S | −6.80 | −7.00 | −6.60 |
| Nsp14 (N7-MTase) | 5C8S | −7.50 | −7.80 | −7.80 |
| Nsp15 (endoribonuclease) | 6WLC | −6.30 | −6.40 | −6.50 |
| Nsp16 (GTA site) | 6WVN | −7.70 | −7.70 | −7.60 |
| Nsp16 (MGP site) | 6WVN | −6.30 | −6.10 | −6.10 |
| Nsp16 (SAM site) | 6WVN | −7.60 | −7.30 | −7.40 |
| N protein (NCB site) | 6WXD | −6.90 | −7.20 | −6.80 |
| Spike protein, RBD (Native) | 6M0J | −5.75 | −5.91 | −5.18 |
| Spike protein, RBD (Mutated) | 6M0J | −5.88 | −5.85 | −5.74 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharov, A.V.; Burkhanova, T.M.; Taskın Tok, T.; Babashkina, M.G.; Safin, D.A. Computational Analysis of Molnupiravir. Int. J. Mol. Sci. 2022, 23, 1508. https://doi.org/10.3390/ijms23031508
Sharov AV, Burkhanova TM, Taskın Tok T, Babashkina MG, Safin DA. Computational Analysis of Molnupiravir. International Journal of Molecular Sciences. 2022; 23(3):1508. https://doi.org/10.3390/ijms23031508
Chicago/Turabian StyleSharov, Artem V., Tatyana M. Burkhanova, Tugba Taskın Tok, Maria G. Babashkina, and Damir A. Safin. 2022. "Computational Analysis of Molnupiravir" International Journal of Molecular Sciences 23, no. 3: 1508. https://doi.org/10.3390/ijms23031508