
Combined Transcriptomics and Metabolomics Analysis Reveals the Molecular Mechanism of Salt Tolerance of Huayouza 62, an Elite Cultivar in Rapeseed (Brassica napus L.)

 , ,
, ,
Abstract
:1. Introduction
2. Results
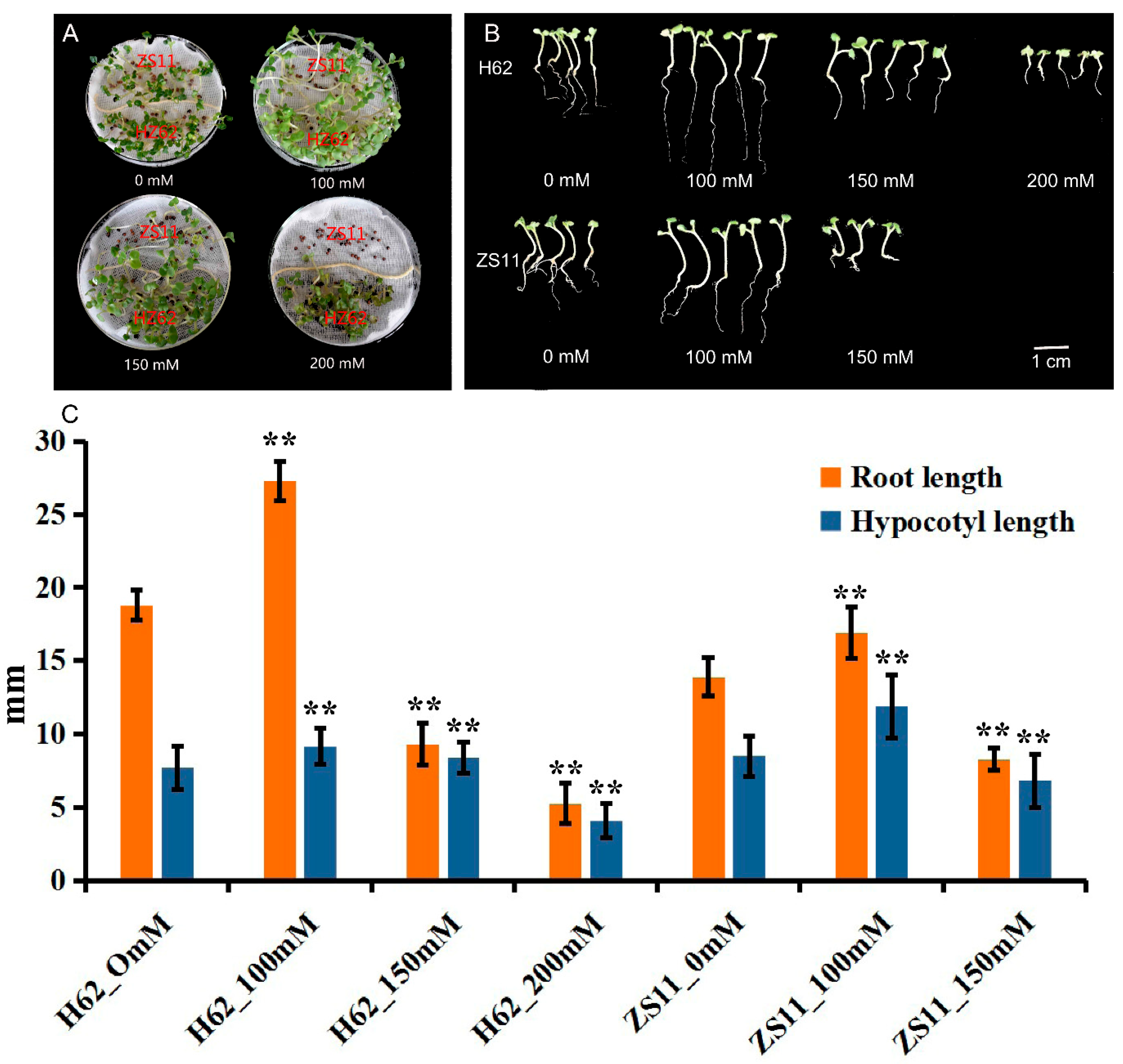
2.1. Effects of Different NaCl Concentrations on the Germination of Rapeseed Seeds
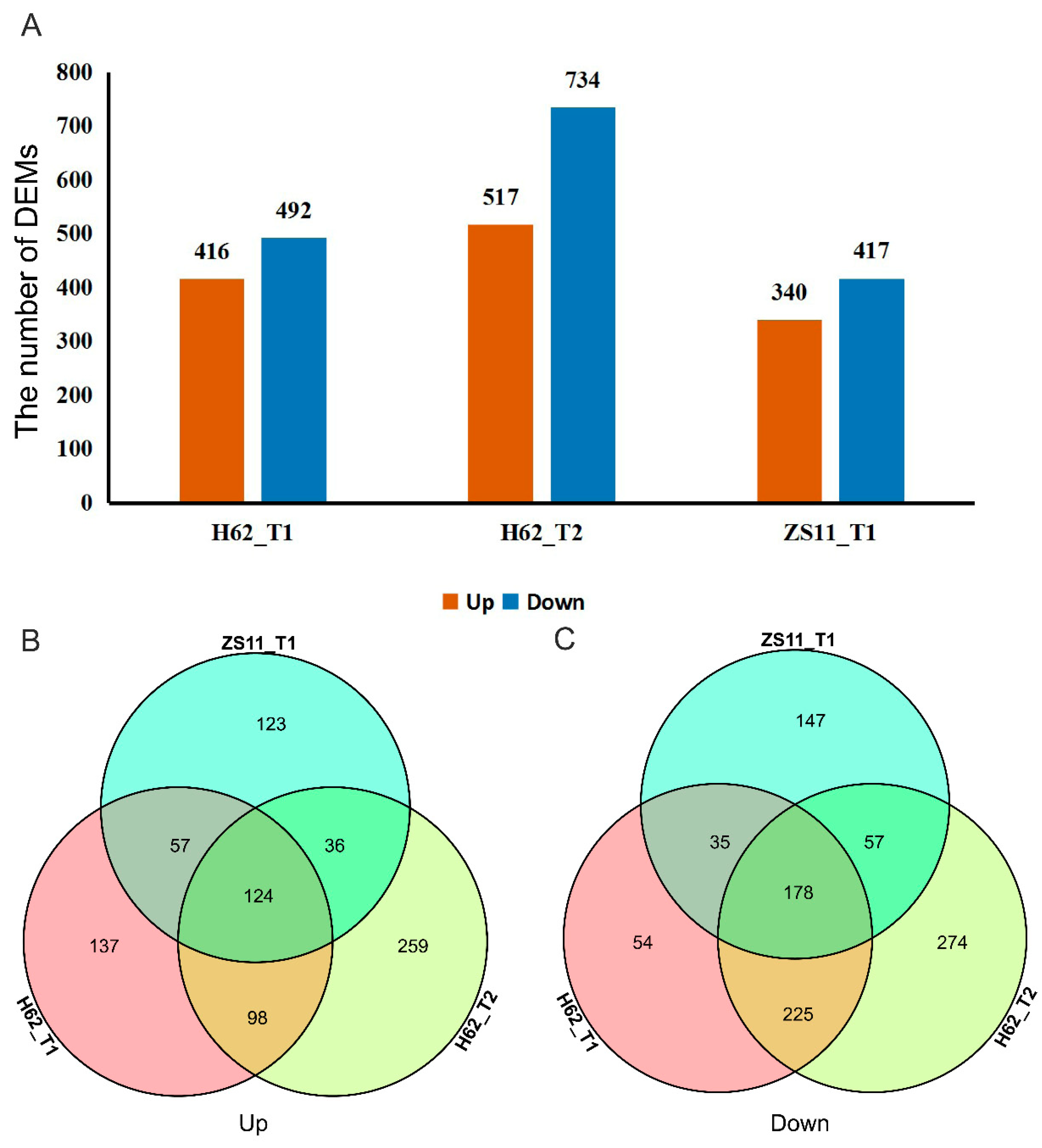
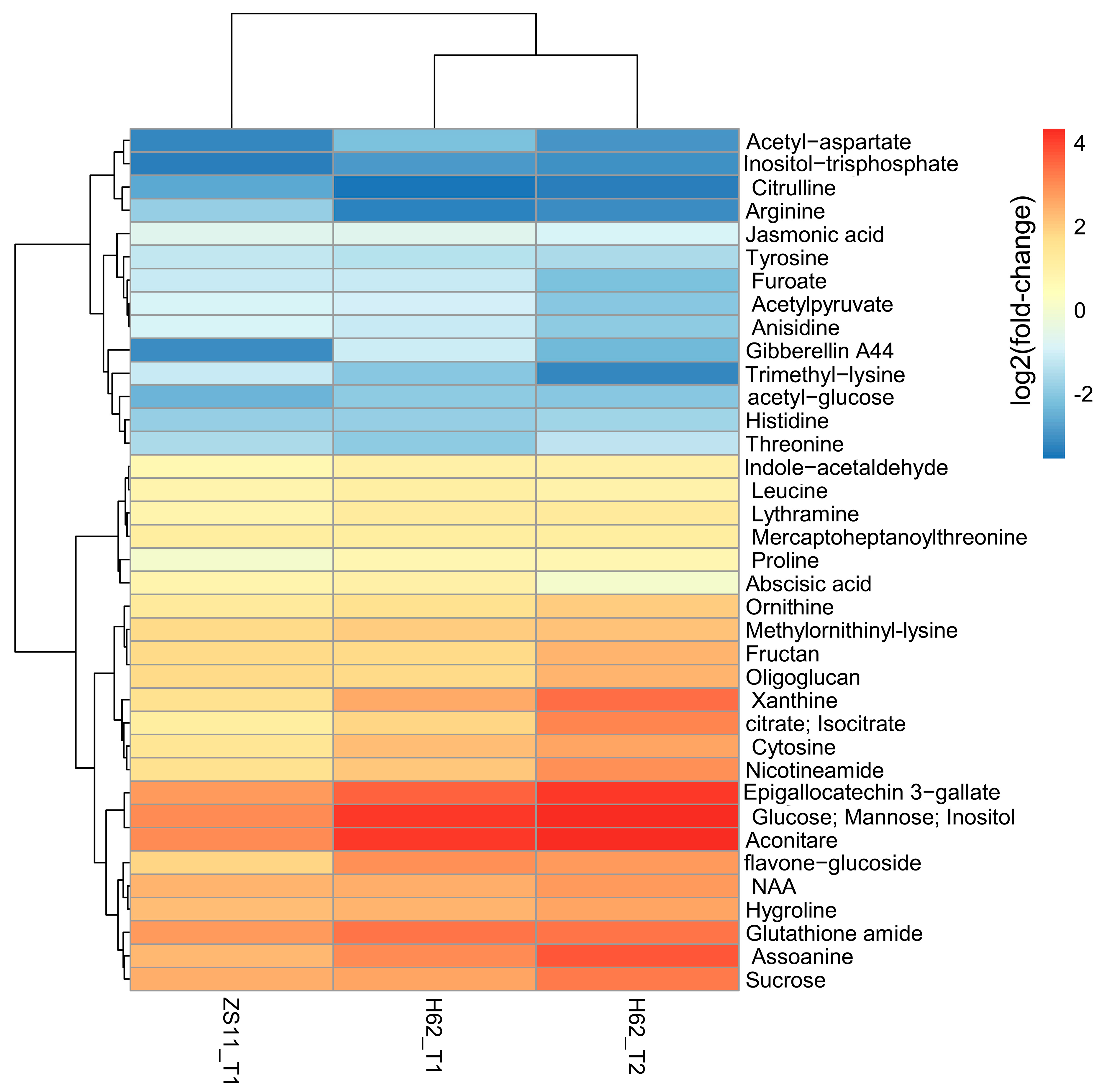
2.2. DEMs Description of Two Rapeseed Varieties under Salt Stress
2.3. Sequencing Output and Assembly
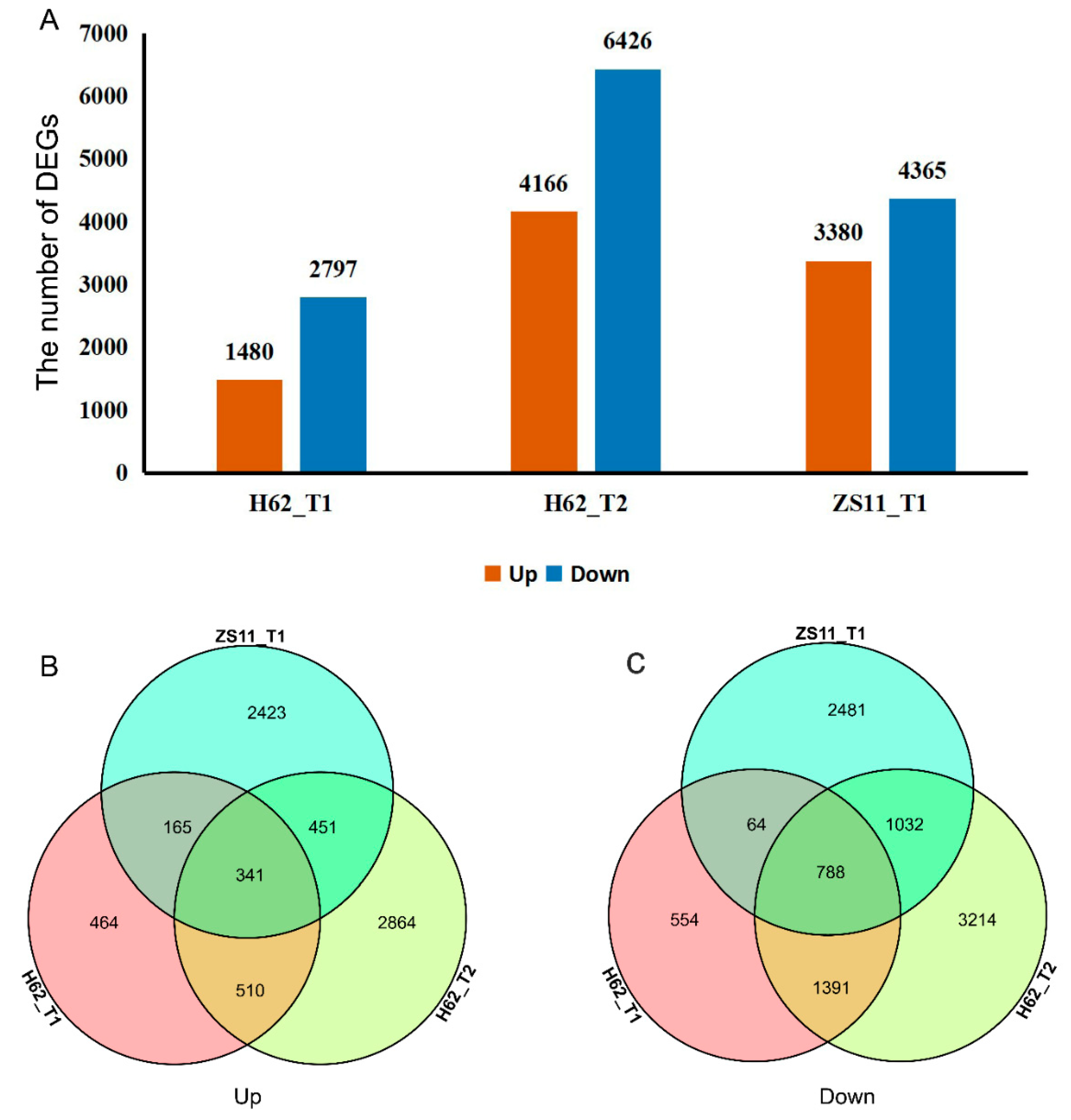
2.4. General Gene-Expression Description under Salt Stress
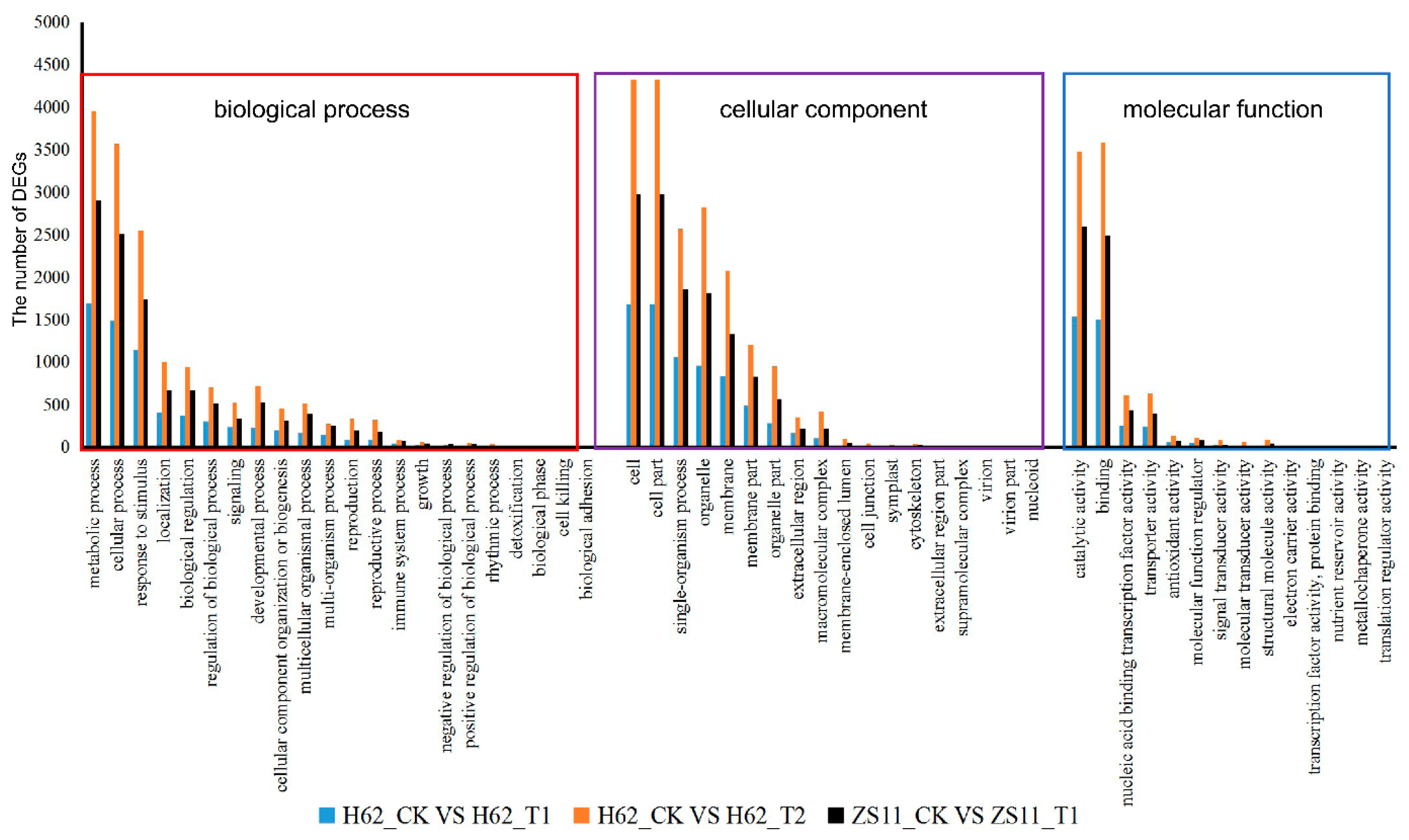
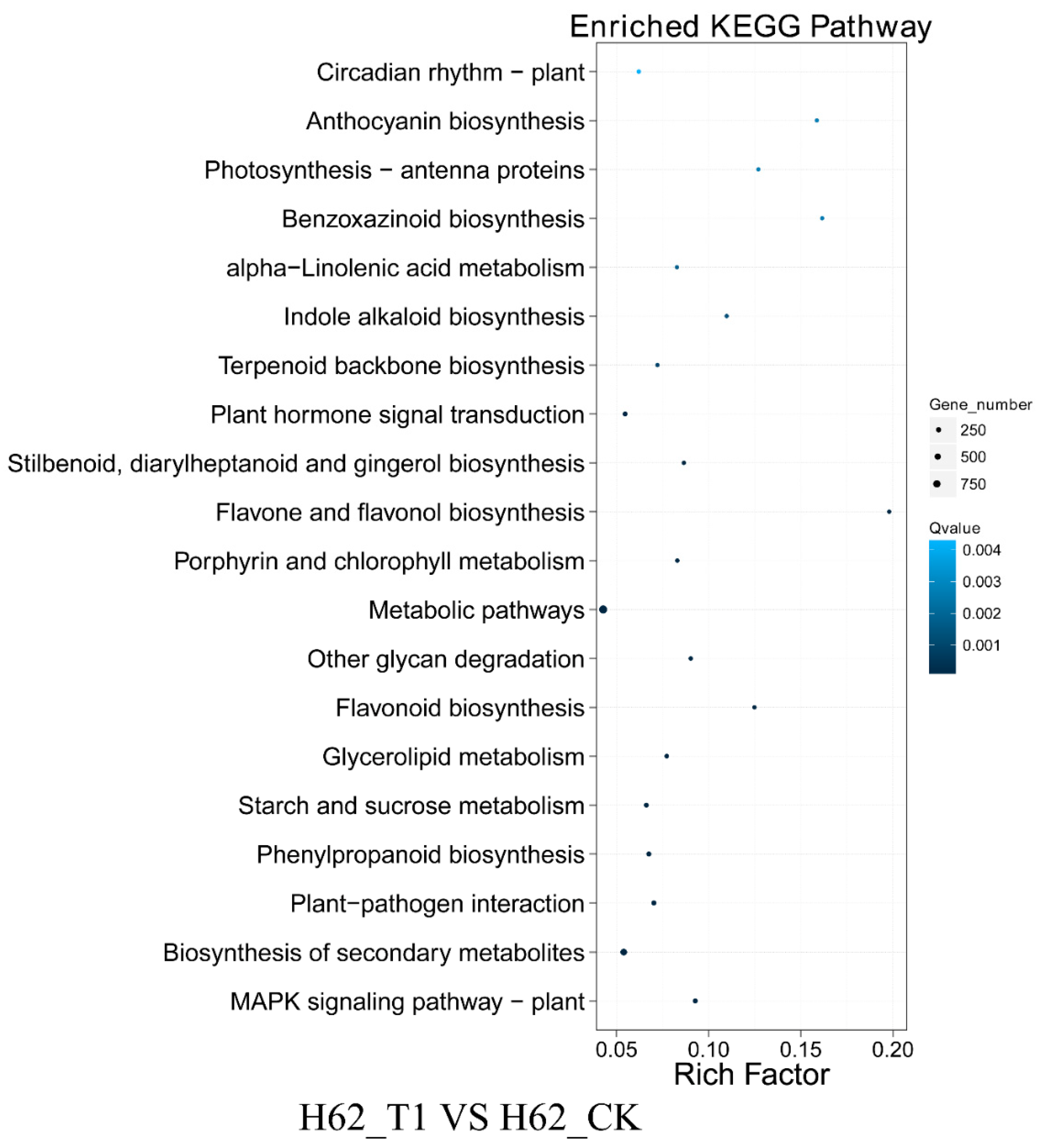
2.5. GO and KEGG Analysis of DEGs Two Rapeseed Varieties under Different NaCl Condition
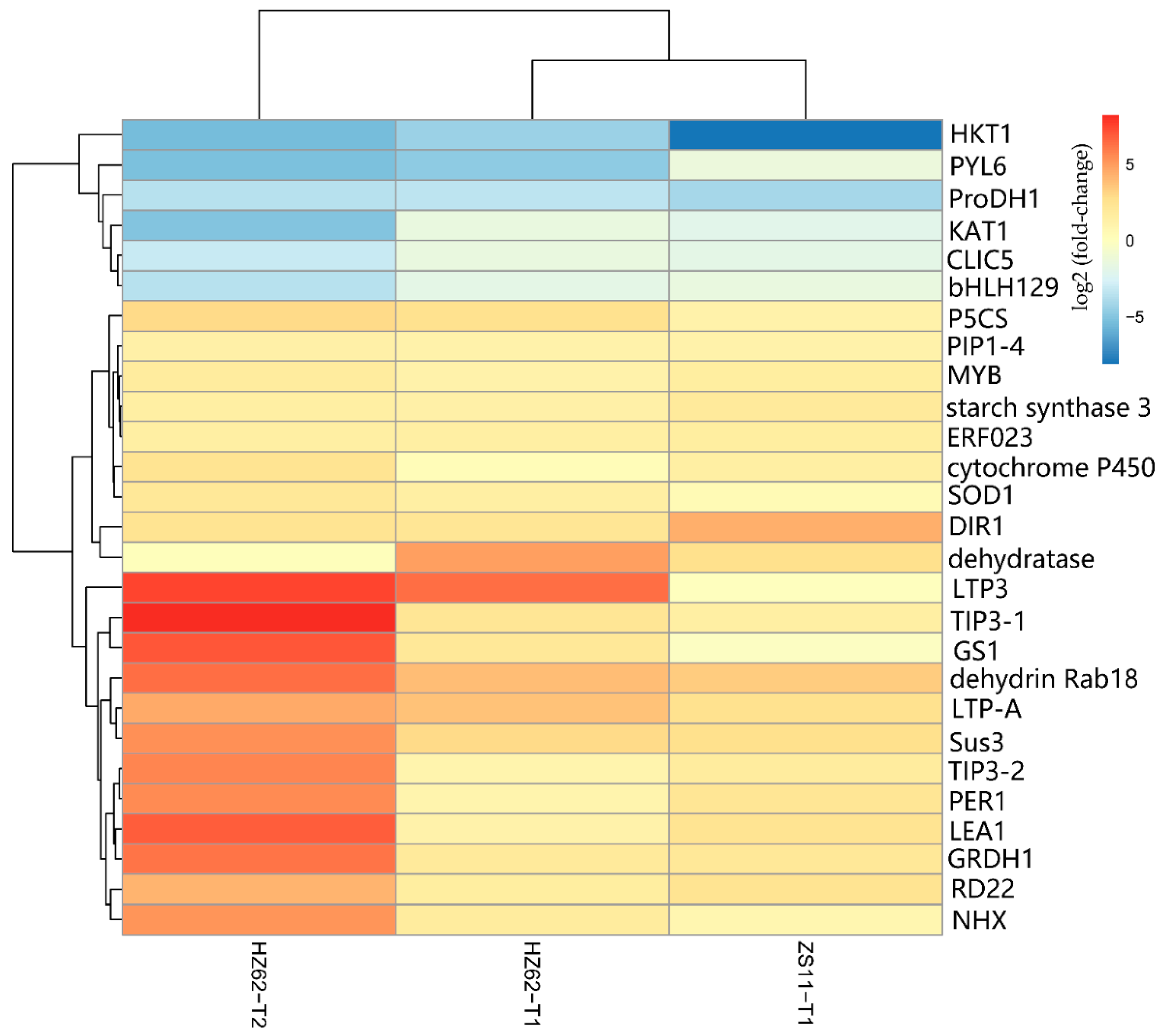
2.6. Identification of Genes Responding to Salt Stress
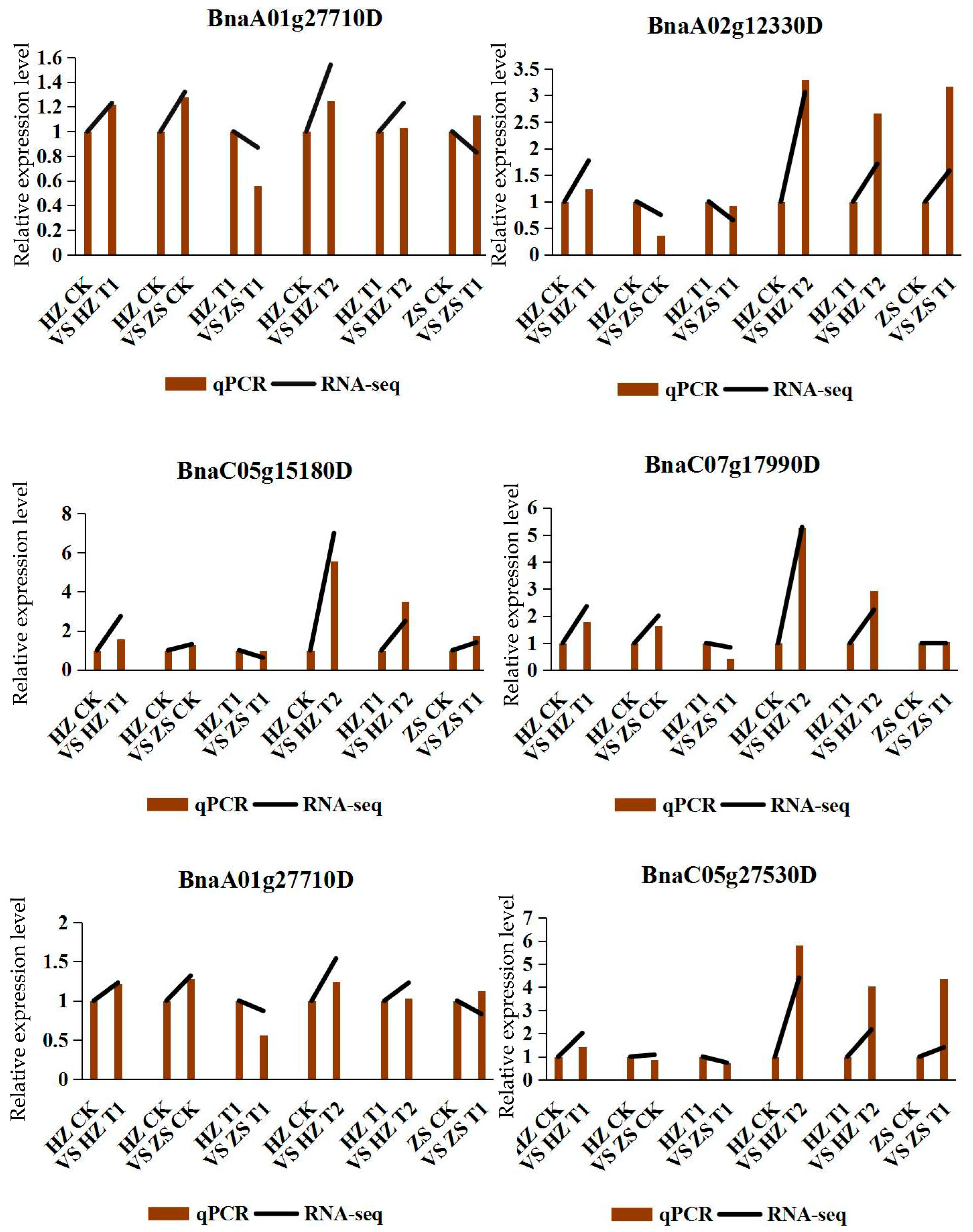
2.7. Confirmation of DEG Profiles by qPCR Analysis
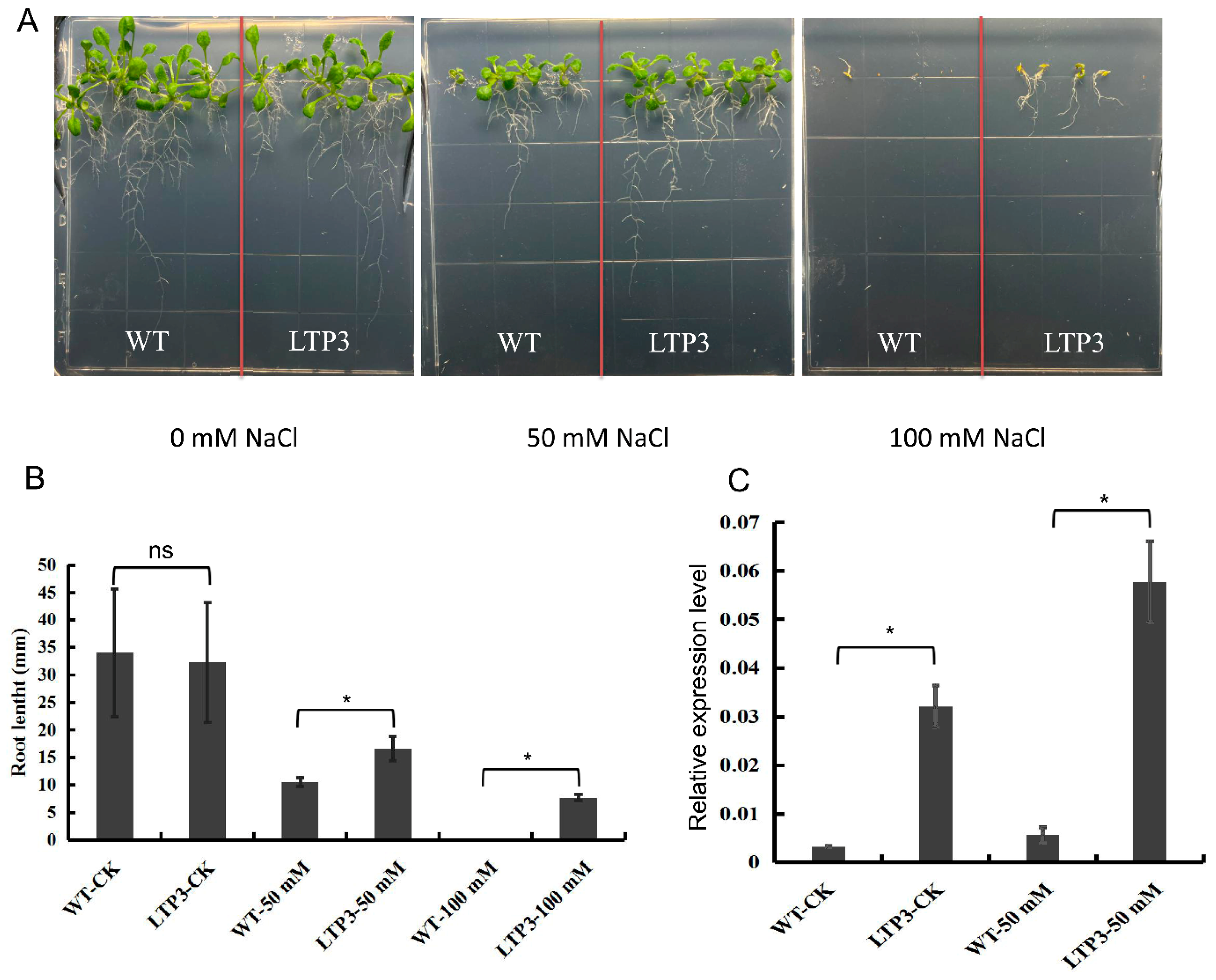
2.8. Over-Expression of BnLTP3 Increased Salt Tolerance in Arabidopsis thaliana
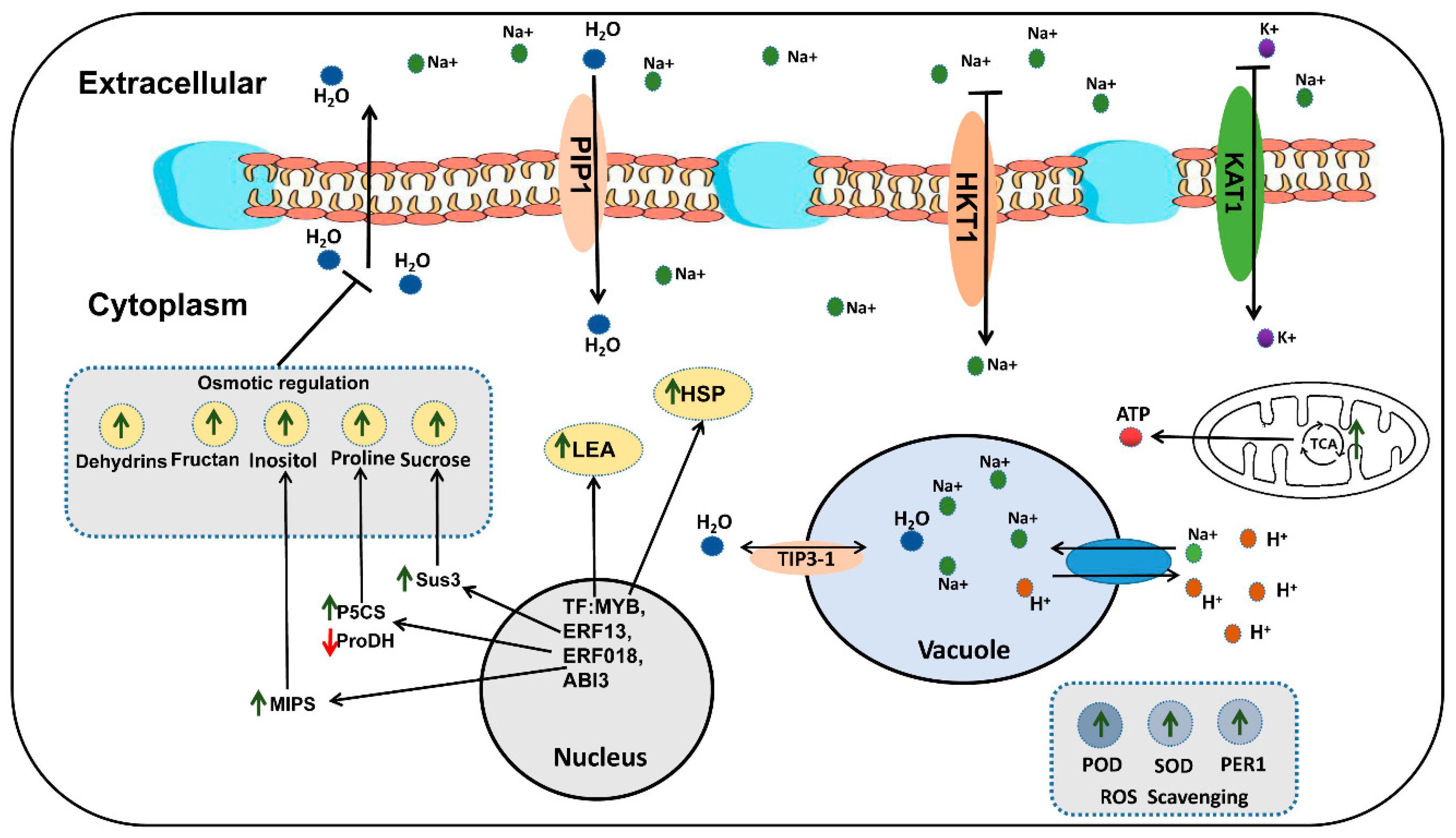
3. Discussion
4. Materials and Methods
4.1. Plant Material, Salt Stress Treatment, and Sample Collection
4.2. LC-MS for Metabolite Determination
4.3. LC-MS (Gas Chromatography-Mass Spectrometry) Analysis
4.4. Metabolite Data Analysis to Obtain Differential Metabolites
4.5. Transcriptome Sequencing Analysis
4.6. Sequencing Analysis
4.7. RT-PCR and Real-Time Quantitative PCR
4.8. BnLTP3 Gene Function Verification
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhu, J.K. Abiotic stress signaling and responses in plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef] [Green Version]
- Ruan, C.J.; da Silva, J.A.T.; Mopper, S.; Qin, P.; Lutts, S. Halophyte improvement for a salinized world. Crit. Rev. Plant Sci. 2010, 6, 329–359. [Google Scholar] [CrossRef]
- Flowers, T.J. Improving crop salt tolerance. J. Exp. Bot. 2004, 396, 307–319. [Google Scholar] [CrossRef]
- Munns, R.; Tester, M. Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Zhang, H.; Song, C.; Zhu, J.K.; Shabala, S. Mechanisms of plant responses and adaptation to soil salinity. Innovation 2020, 1, 100017. [Google Scholar] [CrossRef]
- Ren, Z.H.; Gao, J.P.; Li, G.; Cai, X.L.; Huang, W.; Chao, D.Y.; Zhu, M.; Wang, Z.; Luan, S.; Lin, H. A rice quantitative trait locus for salt tolerance encodes a sodium transporter. Nat. Genet. 2005, 37, 1141–1146. [Google Scholar] [CrossRef]
- Moez, H.; Chantal, E.; Mariama, N.; Laurent, L.; Khaled, M. New insights on plant salt tolerance mechanisms and their potential use for breeding. Front. Plant Sci. 2016, 7, 1787. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.Q.; Deyholos, M.K. Comprehensive transcriptional profiling of NaCl-stressed Arabidopsis roots reveals novel classes of responsive genes. BMC Plant Biol. 2006, 6, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahajan, S.; Pandey, G.K.; Tuteja, N. Calcium- and salt-stress signaling in plants: Shedding light on SOS pathway. Arch. Biochem. Biophys. 2008, 471, 146–158. [Google Scholar] [CrossRef]
- Sánchez-Barrena, M.J.; Martínez-Ripoll, M.; Zhu, J.K.; Albert, A. The structure of the Arabidopsis thaliana SOS3: Molecular mechanism of sensing calcium for salt stress response. J. Mol. Biol. 2015, 345, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Vivek, P.J.; Tuteja, N.; Soniya, E.V. CDPK1 from ginger promotes salinity and drought stress tolerance without yield penalty by improving growth and photosynthesis in Nicotiana tabacum. PLoS ONE 2013, 8, e76392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Wang, X.; Yang, L.; Nan, W.; Bi, Y. Involvement of active MKK9-MAPK3/MAPK6 in increasing respiration in salt-treated Arabidopsis callus. Protoplasma 2020, 257, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Purty, R.S.; Kumar, G.; Singla-Pareek, S.L.; Pareek, A. Towards salinity tolerance in Brassica: An overview. Physiol. Mol. Biol. Plants 2008, 14, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.F.; Guo, S.X.; Li, X.X.; Ren, X.Q. Comparative analysis of salt-responsive phosphoproteins in maize leaves using Ti4+-IMAC enrichment and ESI-Q-TOF MS. Electrophoresis 2013, 34, 485–492. [Google Scholar] [CrossRef]
- Yang, H.; Deng, L.; Liu, H.; Fan, S.; Hua, W.; Liu, J. Overexpression of BnaAOX1b confers tolerance to osmotic and salt stress in rapeseed. G3-Genes Genom. Genet. 2019, 9, 3501–3511. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Yang, P.; Cui, F.; Zhang, F.; Luo, X.; Xie, J. Transcriptome analysis of salt stress responsiveness in the seedlings of Dongxiang wild rice (Oryza rufipogon Griff.). PLoS ONE 2016, 11, e0146242. [Google Scholar] [CrossRef] [Green Version]
- Chandran, A.N.; Kim, J.W.; Yoo, Y.H.; Park, H.L.; Kim, Y.J.; Cho, M.H.; Jung, K.H. Transcriptome analysis of rice-seedling roots under soil-salt stress using RNA-Seq method. Plant Biotechnol. Rep. 2019, 13, 567–578. [Google Scholar] [CrossRef]
- Amirbakhtiar, N.; Ismaili, A.; Ghaffari, M.R.; Firouzabadi, F.N.; Shobbar, Z.S. Transcriptome response of roots to salt stress in a salinity-tolerant bread wheat cultivar. PLoS ONE 2019, 14, e0213305. [Google Scholar] [CrossRef]
- Long, W.; Zou, X.; Zhang, X. Transcriptome analysis of canola (Brassica napus) under salt stress at the germination stage. PLoS ONE 2015, 10, e0116217. [Google Scholar] [CrossRef]
- Yong, H.Y.; Zou, Z.; Kok, E.P.; Kwan, B.H.; Chow, K.; Nasu, S.; Nanzyo, M.; Kitashiba, H.; Nishio, T. Comparative transcriptome analysis of leaves and roots in response to sudden increase in salinity in Brassica napus by RNA-seq. J. Biomed. Biotechnol. 2014, 2014, 467395. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, J.K.; Lindon, J.C.; Holmes, E. Metabonomics: Understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological nmr spectroscopic data. Xenobiotica 1999, 29, 1181–1189. [Google Scholar] [CrossRef]
- Fiehn, O. Metabolomics—The link between genotypes and phenotypes. Plant Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef]
- Wishart, D.S. Current progress in computational metabolomics. Brief. Bioinform. 2007, 5, 279–293. [Google Scholar] [CrossRef] [Green Version]
- Fernie, A.R.; Schauer, N. Metabolomics-assisted breeding: A viable option for crop improvement? Trends Genet. 2009, 25, 39–48. [Google Scholar] [CrossRef]
- Gavaghan, C.L.; Jia, V.L.; Hadfield, S.T.; Hole, S.; Nicholson, J.K.; Wilson, I.D.; Howe, P.W.; Stanley, P.D.; Holmes, E. Application of NMR-based metabolomics to the investigation of salt stress in maize (Zea mays). Phytochem. Anal. 2011, 22, 214–224. [Google Scholar] [CrossRef]
- Richter, J.A.; Erban, A.; Kopka, J.; Zörb, C. Metabolic contribution to salt stress in two maize hybrids with contrasting resistance. Plant Sci. 2015, 233, 107–115. [Google Scholar] [CrossRef]
- Widodo; Patterson, J.H.; Newbigin, E.; Tester, M.; Bacic, A.; Roessner, U. Metabolic responses to salt stress of barley (Hordeum vulgare L.) cultivars, sahara and clipper, which differ in salinity tolerance. J. Exp. Bot. 2009, 60, 4089–4103. [Google Scholar] [CrossRef] [Green Version]
- Cui, F.; Sui, N.; Duan, G.; Liu, Y.; Han, Y.; Liu, S.; Wan, S.; Li, G. Identification of Metabolites and Transcripts Involved in Salt Stress and Recovery in Peanut. Front. Plant Sci. 2018, 9, 217. [Google Scholar] [CrossRef]
- Mohamed, I.A.A.; Shalby, N.; El-Badri, A.M.; Batool, M.; Wang, C.; Wang, Z.; Salah, A.; Rady, M.; Jie, K.; Wang, B.; et al. NA-seq analysis revealed key genes associated with salt tolerance in rapeseed germination through carbohydrate metabolism, hormone, and MAPK signaling pathways. Ind. Crop. Prod. 2022, 176, 114262. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y.; Du, S.; Chen, S.; Tang, H. Dynamic metabonomic responses of tobacco (Nicotiana tabacum) plants to salt stress. J. Proteome Res. 2011, 10, 1904–1914. [Google Scholar] [CrossRef]
- Urano, K.; Kurihara, Y.; Seki, M.; Shinozaki, K. ‘Omics’ analyses of regulatory networks in plant abiotic stress responses. Curr. Opin. Plant Biol. 2010, 13, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ding, Q.; Shao, H.; Wang, H. Overexpression of KvP5CS1 increases salt tolerance in transgenic tobacco. Pak. J. Bot. 2019, 51, 831–836. [Google Scholar] [CrossRef]
- Xue, X.; Liu, A.; Hua, X. Proline accumulation and transcriptional regulation of proline biosynthesis and degradation in Brassica napus. BMB Rep. 2009, 42, 28–34. [Google Scholar] [CrossRef] [Green Version]
- Saadia, M.; Jamil, A.; Akram, N.A.; Ashraf, M.A. Study of proline metabolism in canola (Brassica napus L.) seedlings under salt stress. Molecules 2012, 17, 5803–5815. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, F.M.; Arrabaça, M.C.; Carvalho, L.M.M. Sucrose Metabolism in Lupinus albus L. under salt stress. Biol. Plant. 2004, 48, 317–319. [Google Scholar] [CrossRef]
- Kusuda, H.; Koga, W.; Kusano, M.; Oikawa, A.; Saito, K.; Hirai, M.Y.; Yoshida, K. Ectopic expression of myo-inositol 3-phosphate synthase induces a wide range of metabolic changes and confers salt tolerance in rice. Plant Sci. 2015, 232, 49–56. [Google Scholar] [CrossRef]
- Zhang, Y.; Fang, J.; Wu, X.; Dong, L. Na+/K+ balance and transport regulatory mechanisms in weedy and cultivated rice (Oryza sativa L.) under salt stress. BMC Plant Biol. 2018, 18, 375. [Google Scholar] [CrossRef]
- Jarvis, D.E.; Ryu, C.H.; Beilstein, M.A.; Schumaker, K.S. Distinct roles for SOS1 in the convergent evolution of salt tolerance in Eutrema salsugineum and Schrenkiella parvula. Mol. Biol. Evol. 2014, 31, 2094–2107. [Google Scholar] [CrossRef] [Green Version]
- Abdelaziz, M.E.; Kim, D.; Ali, S.; Fedoroff, N.V.; Al-Babili, S. The endophytic fungus Piriformospora indica enhances Arabidopsis thaliana growth and modulates Na+/K+ homeostasis under salt stress conditions. Plant Sci. 2017, 263, 107–115. [Google Scholar] [CrossRef] [Green Version]
- Obata, T.; Kitamoto, H.K.; Nakamura, A.; Fukuda, A.; Tanaka, Y. Rice Shaker potassium channel OsKAT1 confers tolerance to salinity stress on yeast and rice cells. Plant Physiol. 2007, 144, 1978–1985. [Google Scholar] [CrossRef] [Green Version]
- Tyerman, S.D.; Niemietz, C.M.; Bramley, H.H. Plant aquaporins: Multifunctional water and solute channels with expanding roles. Plant Cell Environ. 2010, 25, 173–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fotiadis, D.; Jeno, P.; Mini, T.; Wirtz, S.; Muller, S.A.; Fraysse, L.; Kjellbom, P.; Engel, A. Structural characterization of two aquaporins isolated from native spinach leaf plasma membranes. J. Biol. Chem. 2001, 276, 1707–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoni, V.; Gerbeau, P.; Javot, H.; Maurel, C. The high diversity of aquaporins reveals novel facets of plant membrane functions. Curr. Opin. Plant Biol. 2000, 3, 476–481. [Google Scholar] [CrossRef]
- Chaumont, F.; Barrieu, F.; Wojcik, E.; Chrispeels, M.; Jung, R. Aquaporins constitute a large and highly fivergent protein family in maize. Plant Physiol. 2001, 125, 1206–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.; Yuan, Q.; Wang, Y.; Cai, R.; Deng, X.; Wang, J.S.; Zhou, S.; Chen, M.; Chen, L.; Huang, C.; et al. Overexpression of a wheat aquaporin gene, TaAQP8, enhances salt stress tolerance in transgenic tobacco. Plant Cell Physiol. 2012, 53, 2127–2141. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Wang, J.; Li, S.; Zhang, L.; Qi, C.; Weeda, S.L.; Guo, Y.D. Conferring enhanced drought stress tolerance in tomato. Sci. Rep. 2016, 6, 31814. [Google Scholar] [CrossRef] [Green Version]
- Ayadi, M.; Brini, F.; Masmoudi, K. Overexpression of a wheat aquaporin gene, TdPIP2;1, enhances salt and drought tolerance in transgenic durum wheat cv. Maali. Int. J. Mol. Sci. 2019, 20, 2389. [Google Scholar] [CrossRef] [Green Version]
- Dure, L.; Crouch, M.; Harada, J.; Ho, T.H.D.; Mundy, J.; Quatrano, R.; Thomas, T.; Sung, Z.R. Common amino acid sequence domains among the LEA proteins of higher plants. Plant Mol. Biol. 1989, 12, 475–486. [Google Scholar] [CrossRef]
- Hu, T.Z. Oslea3, a late embryogenesis abundant protein gene from rice, confers tolerance to water deficit and salt stress to transgenic rice. Russ. J. Plant Physiol. 2008, 55, 530–537. [Google Scholar] [CrossRef]
- Bies-Ethève, N.; Gaubier-Comella, P.; Debures, A.; Lasserre, E.; Jobet, E.; Raynal, M.; Cooke, R.; Delseny, M. Inventory, evolution and expression profiling diversity of the LEA (late embryogenesis abundant) protein gene family in Arabidopsis thaliana. Plant Mol. Biol. 2008, 67, 107–124. [Google Scholar] [CrossRef]
- Yan, L.; Pei, L.; Zhao, S.X.; Zhang, R.Y.; Liu, L.; Li, L.C.; Chen, M.; Ye, X.G.; Chen, Y.F.; Ma, Y.Z. Overexpression of W6 gene tncreases salt tolerance in transgenic tobacco plants. Acta Agron. Sinica 2008, 34, 984–990. [Google Scholar] [CrossRef]
- Liu, Y.; Song, Q.P.; Li, D.X.; Yang, X.H.; Li, D.Q. Multifunctional roles of plant dehydrins in response to environmental stresses. Front. Plant Sci. 2017, 8, 1018. [Google Scholar] [CrossRef] [Green Version]
- Lindorff-Larsen, K.; Winther, J.R. Surprisingly high stability of barley lipid transfer protein, LTP1, towards denaturant, heat and proteases. FEBS Lett. 2001, 488, 145–148. [Google Scholar] [CrossRef] [Green Version]
- Haslekås, C.; Stacy, R.A.P.; Nygaard, V.; Culiáñez-Macià, F.A.; Aalen, R.B. The expression of a peroxiredoxin antioxidant gene, atper1, in Arabidopsis thaliana is seed-specific and related to dormancy. Plant Mol. Biol. 1998, 36, 833–845. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.H.; Chu, P.; Zhou, Y.L.; Ding, Y.; Li, Y.; Liu, J.; Jiang, L.; Huang, S.Z. Ectopic expression of NnPER1, a Nelumbo nucifera 1-cysteine peroxiredoxin antioxidant, enhances seed longevity and stress tolerance in Arabidopsis. Plant J. 2016, 88, 608–619. [Google Scholar] [CrossRef]
- Sun, X.; Li, Y.; Cai, H.; Bai, X.; Ji, W.; Ding, X.; Zhu, Y. The Arabidopsis AtbZIP1 transcription factor is a positive regulator of plant tolerance to salt, osmotic and drought stresses. J. Plant Res. 2012, 125, 429–438. [Google Scholar] [CrossRef]
- Zheng, X.; Bo, C.; Lu, G.; Han, B. Overexpression of a NAC transcription factor enhances rice drought and salt tolerance. Biochem. Bioph. Res. Commun. 2009, 379, 985–989. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, N.N.; Gong, S.Y.; Lu, R.; Li, Y.; Li, X.B. Overexpression of a cotton (Gossypium hirsutum) WRKY gene, GhWRKY34, in Arabidopsis enhances salt-tolerance of the transgenic plants. Plant Physiol. Biochem. 2015, 96, 311–320. [Google Scholar] [CrossRef]
- Tominaga-Wada, R.; Nukumizu, Y.; Wada, T. Tomato (Solanum lycopersicum) Homologs of TRIPTYCHON(SlTRY) and GLABRA3 (SlGL3) are involved in anthocyanin accumulation. Plant Signal. Behav. 2013, 8, e24575. [Google Scholar] [CrossRef] [Green Version]
- Long, L.; Yang, W.W.; Liao, P.; Guo, Y.W.; Kumar, A.; Gao, W. Transcriptome analysis reveals differentially expressed erf transcription factors associated with salt response in cotton. Plant Sci. 2019, 281, 72–81. [Google Scholar] [CrossRef]
- Seo, Y.J.; Park, J.B.; Cho, Y.J.; Jung, C.; Seo, H.S.; Park, S.K.; Nahm, B.; Song, J. Overexpression of the ethylene-responsive factor gene BrERF4 from Brassica rapa increases tolerance to salt and drought in Arabidopsis plants. Mol. Cells 2010, 30, 271–277. [Google Scholar] [CrossRef]
- Lu, C.W.; Shao, Y.; Li, L.; Chen, A.J.; Xu, W.Q.; Wu, K.J.; Luo, Y.; Zhu, B. Overexpression of SlERF1 tomato gene encoding an ERF-type transcription activator enhances salt tolerance. Russ. J. Plant Physiol. 2011, 58, 118–125. [Google Scholar] [CrossRef]
- Drincovich, M.A.F.; Casati, P.; Andreo, C.S. NADP-malic enzyme from plants: A ubiquitous enzyme involved in different metabolic pathways. FEBS Lett. 2001, 490, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Long, M.A. cytosolic nadp-malic enzyme gene from rice (Oryza sativa L.) confers salt tolerance in transgenic arabidopsis. Biotechnol. Lett. 2007, 29, 1129–1134. [Google Scholar] [CrossRef]
- Li, X.; Li, Y.; Ahammed, G.J.; Zhang, X.L.; Ying, L.; Zhang, L.; Li, Q.; Han, W. RBOH1-dependent apoplastic H2O2 mediates epigallocatechin-3-gallate-induced abiotic stress tolerance in Solanum lycopersicum L. Environ. Exp. Bot. 2019, 161, 357–366. [Google Scholar] [CrossRef]
- Jalal, A.G.; Yang, L.; Li, X.; Han, W.Y.; Chen, S. Epigallocatechin-3-Gallate alleviates salinity-retarded seed germination and oxidative stress in tomato. J. Plant Growth Regul. 2018, 37, 1349–1356. [Google Scholar] [CrossRef]
- Wan, H.; Wei, Y.; Qian, J.; Gao, Y.; Wen, J.; Yi, B.; Ma, C.; Tu, J.; Fu, T.; Shen, J. Association mapping of salt tolerance traits at germination stage of rapeseed (Brassica napus L.). Euphytica 2018, 214, 190. [Google Scholar] [CrossRef]
- Dunn, W.B.; Broadhurst, D.; Begley, P.; Zelena, E.; Francis-Mcintyre, S.; Anderson, N.; Brown, M.; Knowles, J.D.; Halsall, A.; Haselden, J.N.; et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2011, 6, 1060–1083. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNAseq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thornton, B.; Basu, C. Real-Time PCR (qPCR) primer design using free online software. Biochem. Mol. Biol. Educ. 2011, 39, 145–154. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2011, 25, 402–408. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | TRR | TCR | CRR | TMR | UMR | TGN | KGN |
|---|---|---|---|---|---|---|---|
| H62_CK_1 | 54.04 | 45.39 | 84.01 | 73.44 | 21.63 | 77,996 | 72,412 |
| H62_CK_2 | 54.04 | 44.55 | 82.44 | 71.43 | 21.90 | 79,639 | 73,968 |
| H62_CK_3 | 54.03 | 44.55 | 82.44 | 71.71 | 21.35 | 77,173 | 71,656 |
| H62_T1_1 | 54.03 | 44.18 | 81.76 | 71.6 | 21.01 | 77,378 | 71,886 |
| H62_T1_2 | 54.04 | 44.35 | 82.08 | 73.06 | 20.64 | 77,245 | 71,684 |
| H62_T1_3 | 55.67 | 44.71 | 80.30 | 74.1 | 20.98 | 75,820 | 70,371 |
| H62_T2_1 | 54.03 | 44.84 | 82.98 | 76.88 | 19.52 | 72,893 | 67,757 |
| H62_T2_2 | 54.03 | 44.76 | 82.84 | 75.4 | 21.7 | 75,294 | 69,933 |
| H62_T2_3 | 54.03 | 44.22 | 81.83 | 75.19 | 20.19 | 75,088 | 69,839 |
| ZS11_CK_1 | 54.03 | 44.56 | 82.46 | 74.48 | 21.96 | 76,629 | 71,160 |
| ZS11_CK_2 | 54.04 | 45.22 | 83.69 | 74.13 | 22.2 | 77,841 | 72,342 |
| ZS11_CK_3 | 54.03 | 44.28 | 81.94 | 73.32 | 21.53 | 76,138 | 70,811 |
| ZS11_T1_1 | 54.03 | 44.13 | 81.68 | 75.23 | 22.35 | 75,167 | 69,754 |
| ZS11_T1_2 | 57.14 | 45.15 | 79.00 | 76.26 | 22.34 | 72,940 | 67,636 |
| ZS11_T1_3 | 57.14 | 45.30 | 79.28 | 75.21 | 22.64 | 73,875 | 68,597 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wan, H.; Qian, J.; Zhang, H.; Lu, H.; Li, O.; Li, R.; Yu, Y.; Wen, J.; Zhao, L.; Yi, B.; et al. Combined Transcriptomics and Metabolomics Analysis Reveals the Molecular Mechanism of Salt Tolerance of Huayouza 62, an Elite Cultivar in Rapeseed (Brassica napus L.). Int. J. Mol. Sci. 2022, 23, 1279. https://doi.org/10.3390/ijms23031279
Wan H, Qian J, Zhang H, Lu H, Li O, Li R, Yu Y, Wen J, Zhao L, Yi B, et al. Combined Transcriptomics and Metabolomics Analysis Reveals the Molecular Mechanism of Salt Tolerance of Huayouza 62, an Elite Cultivar in Rapeseed (Brassica napus L.). International Journal of Molecular Sciences. 2022; 23(3):1279. https://doi.org/10.3390/ijms23031279
Chicago/Turabian StyleWan, Heping, Jiali Qian, Hao Zhang, Hongchen Lu, Ouqi Li, Rihui Li, Yi Yu, Jing Wen, Lun Zhao, Bin Yi, and et al. 2022. "Combined Transcriptomics and Metabolomics Analysis Reveals the Molecular Mechanism of Salt Tolerance of Huayouza 62, an Elite Cultivar in Rapeseed (Brassica napus L.)" International Journal of Molecular Sciences 23, no. 3: 1279. https://doi.org/10.3390/ijms23031279