
Adenosine Receptors in Neuropsychiatric Disorders: Fine Regulators of Neurotransmission and Potential Therapeutic Targets

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Adenosine
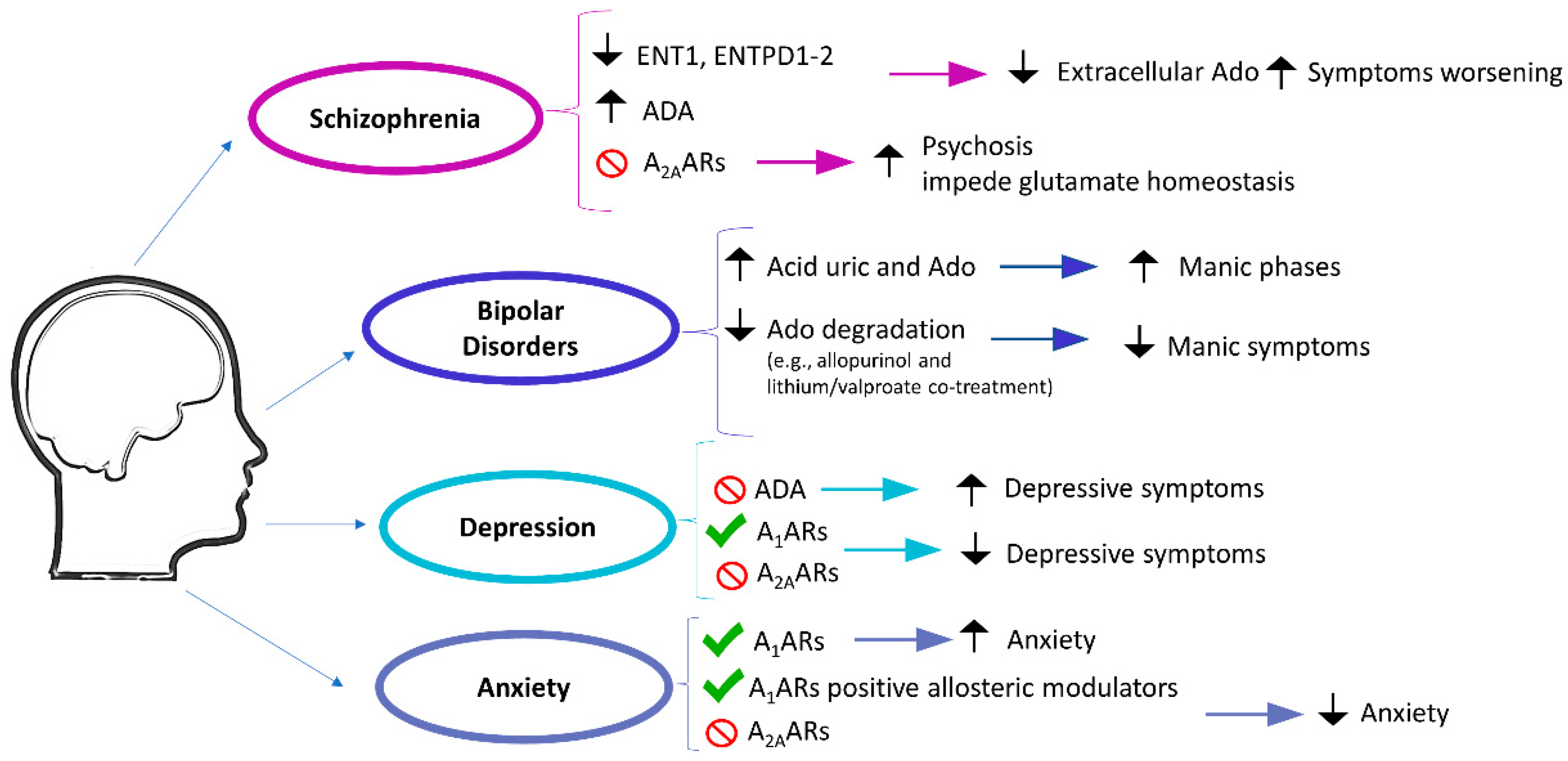
2.1. Psychotic and Mood Disorders
2.1.1. Schizophrenia
2.1.2. Bipolar Disorders
2.1.3. Depression
2.1.4. Anxiety
2.2. Neurodevelopmental Disorders
2.2.1. Autism Spectrum Disorder (ASD)
2.2.2. Fragile X Syndrome (FXS)
2.2.3. Attention-Deficit Hyperactivity Disorder (ADHD)
2.3. Neuropsychiatric Aspects in Neurodegenerative Diseases
2.3.1. Parkinson’s Disease (PD)
2.3.2. Alzheimer’s Disease (AD)
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Borea, P.A.; Gessi, S.; Merighi, S.; Varani, K. Adenosine as a Multi-Signalling Guardian Angel in Human Diseases: When, Where and How Does It Exert Its Protective Effects? Trends Pharmacol. Sci. 2016, 37, 419–434. [Google Scholar] [CrossRef]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of Adenosine Receptors: The State of the Art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef] [PubMed]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pathological Overproduction: The Bad Side of Adenosine. Br. J. Pharmacol. 2017, 174, 1945–1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincenzi, F.; Ravani, A.; Pasquini, S.; Merighi, S.; Gessi, S.; Romagnoli, R.; Baraldi, P.G.; Borea, P.A.; Varani, K. Positive Allosteric Modulation of A1 Adenosine Receptors as a Novel and Promising Therapeutic Strategy for Anxiety. Neuropharmacology 2016, 111, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Domenici, M.R.; Ferrante, A.; Martire, A.; Chiodi, V.; Pepponi, R.; Tebano, M.T.; Popoli, P. Adenosine A2A Receptor as Potential Therapeutic Target in Neuropsychiatric Disorders. Pharmacol. Res. 2019, 147, 104338. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.; Augusto, E.; Santos-Rodrigues, A.D.; Schwarzschild, M.A.; Chen, J.-F.; Cunha, R.A.; Agostinho, P. Adenosine A2A Receptors Modulate Glutamate Uptake in Cultured Astrocytes and Gliosomes. Glia 2012, 60, 702–716. [Google Scholar] [CrossRef]
- Carman, A.J.; Mills, J.H.; Krenz, A.; Kim, D.-G.; Bynoe, M.S. Adenosine Receptor Signaling Modulates Permeability of the Blood-Brain Barrier. J. Neurosci. 2011, 31, 13272–13280. [Google Scholar] [CrossRef]
- Chen, J.-F.; Lee, C.; Chern, Y. Chapter One-Adenosine Receptor Neurobiology: Overview. In International Review of Neurobiology; Mori, A., Ed.; Adenosine Receptors in Neurology and Psychiatry; Academic Press: Cambridge, MA, USA, 2014; Volume 119, pp. 1–49. [Google Scholar]
- Ferré, S.; Bonaventura, J.; Tomasi, D.; Navarro, G.; Moreno, E.; Cortés, A.; Lluís, C.; Casadó, V.; Volkow, N.D. Allosteric Mechanisms within the Adenosine A2A-Dopamine D2 Receptor Heterotetramer. Neuropharmacology 2016, 104, 154–160. [Google Scholar] [CrossRef] [Green Version]
- Fuxe, K.; Marcellino, D.; Leo, G.; Agnati, L.F. Molecular Integration via Allosteric Interactions in Receptor Heteromers. A Working Hypothesis. Curr. Opin. Pharmacol. 2010, 10, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Cristóvão-Ferreira, S.; Navarro, G.; Brugarolas, M.; Pérez-Capote, K.; Vaz, S.H.; Fattorini, G.; Conti, F.; Lluis, C.; Ribeiro, J.A.; McCormick, P.J.; et al. A1R-A2AR Heteromers Coupled to Gs and Gi/0 Proteins Modulate GABA Transport into Astrocytes. Purinergic Signal. 2013, 9, 433–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuxe, K.; Agnati, L.F.; Borroto-Escuela, D.O. The Impact of Receptor-Receptor Interactions in Heteroreceptor Complexes on Brain Plasticity. Expert Rev. Neurother. 2014, 14, 719–721. [Google Scholar] [CrossRef] [Green Version]
- Foussias, G.; Agid, O.; Fervaha, G.; Remington, G. Negative Symptoms of Schizophrenia: Clinical Features, Relevance to Real World Functioning and Specificity versus Other CNS Disorders. Eur. Neuropsychopharmacol. 2014, 24, 693–709. [Google Scholar] [CrossRef]
- Rajji, T.K.; Ismail, Z.; Mulsant, B.H. Age at Onset and Cognition in Schizophrenia: Meta-Analysis. Br. J. Psychiatry 2009, 195, 286–293. [Google Scholar] [CrossRef] [Green Version]
- Fuxe, K.; Ferré, S.; Genedani, S.; Franco, R.; Agnati, L.F. Adenosine Receptor–Dopamine Receptor Interactions in the Basal Ganglia and Their Relevance for Brain Function. Physiol. Behav. 2007, 92, 210–217. [Google Scholar] [CrossRef]
- Moghaddam, B.; Javitt, D. From Revolution to Evolution: The Glutamate Hypothesis of Schizophrenia and Its Implication for Treatment. Neuropsychopharmacology 2012, 37, 4–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howes, O.D.; Kapur, S. The Dopamine Hypothesis of Schizophrenia: Version III-The Final Common Pathway. Schizophr. Bull. 2009, 35, 549–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Field, J.R.; Walker, A.G.; Conn, P.J. Targeting Glutamate Synapses in Schizophrenia. Trends Mol. Med. 2011, 17, 689–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristiansen, L.V.; Huerta, I.; Beneyto, M.; Meador-Woodruff, J.H. NMDA Receptors and Schizophrenia. Curr. Opin. Pharmacol. 2007, 7, 48–55. [Google Scholar] [CrossRef]
- de Bartolomeis, A.; Fiore, G.; Iasevoli, F. Dopamine-Glutamate Interaction and Antipsychotics Mechanism of Action: Implication for New Pharmacological Strategies in Psychosis. Curr. Pharm. Des. 2005, 11, 3561–3594. [Google Scholar] [CrossRef]
- Javitt, D.C. Glutamate and Schizophrenia: Phencyclidine, N-Methyl-d-Aspartate Receptors, and Dopamine-Glutamate Interactions. In International Review of Neurobiology; Integrating the Neurobiology of Schizophrenia; Academic Press: Cambridge, MA, USA, 2007; Volume 78, pp. 69–108. [Google Scholar]
- Boison, D.; Singer, P.; Shen, H.-Y.; Feldon, J.; Yee, B.K. Adenosine Hypothesis of Schizophrenia-Opportunities for Pharmacotherapy. Neuropharmacology 2012, 62, 1527–1543. [Google Scholar] [CrossRef] [Green Version]
- Yee, B.K.; Singer, P.; Chen, J.-F.; Feldon, J.; Boison, D. Transgenic Overexpression of Adenosine Kinase in Brain Leads to Multiple Learning Impairments and Altered Sensitivity to Psychomimetic Drugs. Eur. J. Neurosci. 2007, 26, 3237–3252. [Google Scholar] [CrossRef]
- Coyle, J.T. Glutamate and Schizophrenia: Beyond the Dopamine Hypothesis. Cell. Mol Neurobiol. 2006, 26, 363–382. [Google Scholar] [CrossRef]
- Peng, P.-J.; Chiang, K.-T.; Liang, C.-S. Low-Dose Caffeine May Exacerbate Psychotic Symptoms in People with Schizophrenia. JNP 2014, 26, E41. [Google Scholar] [CrossRef]
- Lara, D.R.; Dall’Igna, O.P.; Ghisolfi, E.S.; Brunstein, M.G. Involvement of Adenosine in the Neurobiology of Schizophrenia and Its Therapeutic Implications. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2006, 30, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.L.; Funk, A.J.; Devine, E.; Homan, R.C.D.; Boison, D.; McCullumsmith, R.E.; O’Donovan, S.M. Adenosine Kinase Expression in the Frontal Cortex in Schizophrenia. Schizophr. Bull. 2020, 46, 690–698. [Google Scholar] [CrossRef]
- O’Donovan, S.M.; Sullivan, C.; Koene, R.; Devine, E.; Hasselfeld, K.; Moody, C.L.; McCullumsmith, R.E. Cell-Subtype-Specific Changes in Adenosine Pathways in Schizophrenia. Neuropsychopharmacology 2018, 43, 1667–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aliagas, E.; Villar-Menéndez, I.; Sévigny, J.; Roca, M.; Romeu, M.; Ferrer, I.; Martín-Satué, M.; Barrachina, M. Reduced Striatal Ecto-Nucleotidase Activity in Schizophrenia Patients Supports the “Adenosine Hypothesis”. Purinergic Signal 2013, 9, 599–608. [Google Scholar] [CrossRef] [Green Version]
- Lara, D.R.; Vianna, M.R.M.; de Paris, F.; Quevedo, J.; Oses, J.P.; Battastini, A.M.O.; Sarkis, J.J.F.; Souza, D.O. Chronic Treatment with Clozapine, but Not Haloperidol, Increases Striatal Ecto-5′-Nucleotidase Activity in Rats. NPS 2001, 44, 99–102. [Google Scholar] [CrossRef]
- Seibt, K.J.; Oliveira, R.da.L.; Rico, E.P.; Dias, R.D.; Bogo, M.R.; Bonan, C.D. Antipsychotic Drugs Inhibit Nucleotide Hydrolysis in Zebrafish (Danio Rerio) Brain Membranes. Toxicol. Vitr. 2009, 23, 78–82. [Google Scholar] [CrossRef]
- Seibt, K.J.; da Luz Oliveira, R.; Bogo, M.R.; Senger, M.R.; Bonan, C.D. Investigation into Effects of Antipsychotics on Ectonucleotidase and Adenosine Deaminase in Zebrafish Brain. Fish Physiol. Biochem. 2015, 41, 1383–1392. [Google Scholar] [CrossRef] [PubMed]
- Brunstein, M.G.; Silveira, E.M.; Chaves, L.S.; Machado, H.; Schenkel, O.; Belmonte-de-Abreu, P.; Souza, D.O.; Lara, D.R. Increased Serum Adenosine Deaminase Activity in Schizophrenic Receiving Antipsychotic Treatment. Neurosci. Lett. 2007, 414, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.-Y.; Singer, P.; Lytle, N.; Wei, C.J.; Lan, J.-Q.; Williams-Karnesky, R.L.; Chen, J.-F.; Yee, B.K.; Boison, D. Adenosine Augmentation Ameliorates Psychotic and Cognitive Endophenotypes of Schizophrenia. J. Clin. Investig. 2012, 122, 2567–2577. [Google Scholar] [CrossRef] [Green Version]
- Popoli, P.; Pèzzola, A.; de Carolis, A.S. Modulation of Striatal Adenosine A1 and A2 Receptors Induces Rotational Behaviour in Response to Dopaminergic Stimulation in Intact Rats. Eur. J. Pharmacol. 1994, 257, 21–25. [Google Scholar] [CrossRef]
- Borroto-Escuela, D.O.; Romero-Fernandez, W.; Tarakanov, A.O.; Gómez-Soler, M.; Corrales, F.; Marcellino, D.; Narvaez, M.; Frankowska, M.; Flajolet, M.; Heintz, N.; et al. Characterization of the A2AR-D2R Interface: Focus on the Role of the C-Terminal Tail and the Transmembrane Helices. Biochem. Biophys. Res. Commun. 2010, 402, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Borroto-Escuela, D.O.; Pintsuk, J.; Schäfer, T.; Friedland, K.; Ferraro, L.; Tanganelli, S.; Liu, F.; Fuxe, K. Multiple D2 Heteroreceptor Complexes: New Targets for Treatment of Schizophrenia. Ther. Adv. Psychopharmacol. 2016, 6, 77–94. [Google Scholar] [CrossRef] [Green Version]
- Borroto-Escuela, D.O.; Marcellino, D.; Narvaez, M.; Flajolet, M.; Heintz, N.; Agnati, L.; Ciruela, F.; Fuxe, K. A Serine Point Mutation in the Adenosine A2AR C-Terminal Tail Reduces Receptor Heteromerization and Allosteric Modulation of the Dopamine D2R. Biochem. Biophys. Res. Commun. 2010, 394, 222–227. [Google Scholar] [CrossRef]
- Filip, M.; Zaniewska, M.; Frankowska, M.; Wydra, K.; Fuxe, K. The Importance of the Adenosine A2A Receptor-Dopamine D2 Receptor Interaction in Drug Addiction. Curr. Med. Chem. 2011, 19, 317–355. [Google Scholar] [CrossRef]
- Matute, C.; Melone, M.; Vallejo-Illarramendi, A.; Conti, F. Increased Expression of the Astrocytic Glutamate Transporter GLT-1 in the Prefrontal Cortex of Schizophrenics. Glia 2005, 49, 451–455. [Google Scholar] [CrossRef] [Green Version]
- Matos, M.; Shen, H.-Y.; Augusto, E.; Wang, Y.; Wei, C.J.; Wang, Y.T.; Agostinho, P.; Boison, D.; Cunha, R.A.; Chen, J.-F. Deletion of Adenosine A2A Receptors from Astrocytes Disrupts Glutamate Homeostasis Leading to Psychomotor and Cognitive Impairment: Relevance to Schizophrenia. Biol. Psychiatry 2015, 78, 763–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vieta, E.; Berk, M.; Schulze, T.G.; Carvalho, A.F.; Suppes, T.; Calabrese, J.R.; Gao, K.; Miskowiak, K.W.; Grande, I. Bipolar Disorders. Nat. Rev. Dis. Primers 2018, 4, 18008. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, R.S.; Berk, M.; Brietzke, E.; Goldstein, B.I.; López-Jaramillo, C.; Kessing, L.V.; Malhi, G.S.; Nierenberg, A.A.; Rosenblat, J.D.; Majeed, A.; et al. Bipolar Disorders. Lancet 2020, 396, 1841–1856. [Google Scholar] [CrossRef]
- Salvadore, G.; Viale, C.I.; Luckenbaugh, D.A.; Zanatto, V.C.; Portela, L.V.; Souza, D.O.; Zarate, C.A.; Machado-Vieira, R. Increased Uric Acid Levels in Drug-Naïve Subjects with Bipolar Disorder during a First Manic Episode. Prog. Neuropsychopharmacol. Biol. Psychiatry 2010, 34, 819–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartoli, F.; Crocamo, C.; Mazza, M.G.; Clerici, M.; Carrà, G. Uric Acid Levels in Subjects with Bipolar Disorder: A Comparative Meta-Analysis. J. Psychiatr. Res. 2016, 81, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Bartoli, F.; Crocamo, C.; Clerici, M.; Carrà, G. Allopurinol as Add-on Treatment for Mania Symptoms in Bipolar Disorder: Systematic Review and Meta-Analysis of Randomised Controlled Trials. Br. J. Psychiatry 2017, 210, 10–15. [Google Scholar] [CrossRef]
- Bartoli, F.; Crocamo, C.; Dakanalis, A.; Brosio, E.; Miotto, A.; Capuzzi, E.; Clerici, M.; Carrà, G. Purinergic System Dysfunctions in Subjects with Bipolar Disorder: A Comparative Cross-Sectional Study. Compr. Psychiatry 2017, 73, 1–6. [Google Scholar] [CrossRef]
- Machado-Vieira, R.; Soares, J.C.; Lara, D.R.; Luckenbaugh, D.A.; Busnello, J.V.; Marca, G.; Cunha, A.; Souza, D.O.; Zarate, C.A.; Kapczinski, F. A Double-Blind, Randomized, Placebo-Controlled 4-Week Study on the Efficacy and Safety of the Purinergic Agents Allopurinol and Dipyridamole Adjunctive to Lithium in Acute Bipolar Mania. J. Clin. Psychiatry 2008, 69, 1237–1245. [Google Scholar] [CrossRef]
- Jahangard, L.; Soroush, S.; Haghighi, M.; Ghaleiha, A.; Bajoghli, H.; Holsboer-Trachsler, E.; Brand, S. In a Double-Blind, Randomized and Placebo-Controlled Trial, Adjuvant Allopurinol Improved Symptoms of Mania in in-Patients Suffering from Bipolar Disorder. Eur. Neuropsychopharmacol. 2014, 24, 1210–1221. [Google Scholar] [CrossRef]
- Weiser, M.; Burshtein, S.; Gershon, A.A.; Marian, G.; Vlad, N.; Grecu, I.G.; Tocari, E.; Tiugan, A.; Hotineanu, M.; Davis, J.M. Allopurinol for Mania: A Randomized Trial of Allopurinol versus Placebo as Add-on Treatment to Mood Stabilizers and/or Antipsychotic Agents in Manic Patients with Bipolar Disorder. Bipolar Disord. 2014, 16, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, R.; Ulrich, H.; Zarate, C.A.; Machado-Vieira, R. Purinergic System Dysfunction in Mood Disorders: A Key Target for Developing Improved Therapeutics. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2015, 57, 117–131. [Google Scholar] [CrossRef] [Green Version]
- Gubert, C.; Moritz, C.E.J.; Vasconcelos-Moreno, M.P.; dos Santos, B.T.M.Q.; Sartori, J.; Fijtman, A.; Kauer-Sant’Anna, M.; Kapczinski, F.; Battastini, A.M.O.; da Silva Magalhães, P.V. Peripheral Adenosine Levels in Euthymic Patients with Bipolar Disorder. Psychiatry Res. 2016, 246, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.S.; Gordon-Smith, K.; Forty, L.; Di Florio, A.; Craddock, N.; Jones, L.; Jones, I. Sleep Loss as a Trigger of Mood Episodes in Bipolar Disorder: Individual Differences Based on Diagnostic Subtype and Gender. Br. J. Psychiatry 2017, 211, 169–174. [Google Scholar] [CrossRef] [Green Version]
- van Calker, D.; Biber, K.; Domschke, K.; Serchov, T. The Role of Adenosine Receptors in Mood and Anxiety Disorders. J. Neurochem. 2019, 151, 11–27. [Google Scholar] [CrossRef] [Green Version]
- Chand, S.P.; Arif, H. Depression. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Serchov, T.; Clement, H.-W.; Schwarz, M.K.; Iasevoli, F.; Tosh, D.K.; Idzko, M.; Jacobson, K.A.; de Bartolomeis, A.; Normann, C.; Biber, K.; et al. Increased Signaling via Adenosine A1 Receptors, Sleep Deprivation, Imipramine, and Ketamine Inhibit Depressive-like Behavior via Induction of Homer1a. Neuron 2015, 87, 549–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallaspezia, S.; Benedetti, F. Sleep Deprivation Therapy for Depression. In Sleep, Neuronal Plasticity and Brain Function; Meerlo, P., Benca, R.M., Abel, T., Eds.; Current Topics in Behavioral Neurosciences; Springer: Berlin/Heidelberg, Germany, 2015; pp. 483–502. ISBN 978-3-662-46878-4. [Google Scholar]
- Coelho, J.E.; Alves, P.; Canas, P.M.; Valadas, J.S.; Shmidt, T.; Batalha, V.L.; Ferreira, D.G.; Ribeiro, J.A.; Bader, M.; Cunha, R.A.; et al. Overexpression of Adenosine A2A Receptors in Rats: Effects on Depression, Locomotion, and Anxiety. Front. Psychiatry 2014, 5, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crema, L.M.; Pettenuzzo, L.F.; Schlabitz, M.; Diehl, L.; Hoppe, J.; Mestriner, R.; Laureano, D.; Salbego, C.; Dalmaz, C.; Vendite, D. The Effect of Unpredictable Chronic Mild Stress on Depressive-like Behavior and on Hippocampal A1 and Striatal A2A Adenosine Receptors. Physiol. Behav. 2013, 109, 1–7. [Google Scholar] [CrossRef]
- Kaster, M.P.; Machado, N.J.; Silva, H.B.; Nunes, A.; Ardais, A.P.; Santana, M.; Baqi, Y.; Müller, C.E.; Rodrigues, A.L.S.; Porciúncula, L.O.; et al. Caffeine Acts through Neuronal Adenosine A2A Receptors to Prevent Mood and Memory Dysfunction Triggered by Chronic Stress. Proc. Natl. Acad. Sci. USA 2015, 112, 7833–7838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szopa, A.; Bogatko, K.; Serefko, A.; Wyska, E.; Wośko, S.; Świąder, K.; Doboszewska, U.; Wlaź, A.; Wróbel, A.; Wlaź, P.; et al. Agomelatine and Tianeptine Antidepressant Activity in Mice Behavioral Despair Tests Is Enhanced by DMPX, a Selective Adenosine A2A Receptor Antagonist, but Not DPCPX, a Selective Adenosine A1 Receptor Antagonist. Pharmacol. Rep. 2019, 71, 676–681. [Google Scholar] [CrossRef]
- Batalha, V.L.; Pego, J.M.; Fontinha, B.M.; Costenla, A.R.; Valadas, J.S.; Baqi, Y.; Radjainia, H.; Müller, C.E.; Sebastião, A.M.; Lopes, L.V. Adenosine A2A Receptor Blockade Reverts Hippocampal Stress-Induced Deficits and Restores Corticosterone Circadian Oscillation. Mol. Psychiatry 2013, 18, 320–331. [Google Scholar] [CrossRef] [Green Version]
- Björkholm, C.; Monteggia, L.M. BDNF-a Key Transducer of Antidepressant Effects. Neuropharmacology 2016, 102, 72–79. [Google Scholar] [CrossRef] [Green Version]
- van Calker, D.; Serchov, T.; Normann, C.; Biber, K. Recent Insights into Antidepressant Therapy: Distinct Pathways and Potential Common Mechanisms in the Treatment of Depressive Syndromes. Neurosci. Biobehav. Rev. 2018, 88, 63–72. [Google Scholar] [CrossRef]
- Rombo, D.M.; Ribeiro, J.A.; Sebastião, A.M. Hippocampal GABAergic Transmission: A New Target for Adenosine Control of Excitability. J. Neurochem. 2016, 139, 1056–1070. [Google Scholar] [CrossRef] [Green Version]
- Stockwell, J.; Jakova, E.; Cayabyab, F.S. Adenosine A1 and A2A Receptors in the Brain: Current Research and Their Role in Neurodegeneration. Molecules 2017, 22, 676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serchov, T.; Atas, H.-C.; Normann, C.; van Calker, D.; Biber, K. Genetically Controlled Upregulation of Adenosine A1 Receptor Expression Enhances the Survival of Primary Cortical Neurons. Mol. Neurobiol. 2012, 46, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Serchov, T.; Heumann, R.; van Calker, D.; Biber, K. Signaling Pathways Regulating Homer1a Expression: Implications for Antidepressant Therapy. Biol. Chem. 2016, 397, 207–214. [Google Scholar] [CrossRef]
- Holz, A.; Mülsch, F.; Schwarz, M.K.; Hollmann, M.; Döbrössy, M.D.; Coenen, V.A.; Bartos, M.; Normann, C.; Biber, K.; van Calker, D.; et al. Enhanced MGlu5 Signaling in Excitatory Neurons Promotes Rapid Antidepressant Effects via AMPA Receptor Activation. Neuron 2019, 104, 338–352.e7. [Google Scholar] [CrossRef]
- Chand, S.P.; Marwaha, R. Anxiety. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Munir, S.; Takov, V. Generalized Anxiety Disorder. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- McLean, C.P.; Asnaani, A.; Litz, B.T.; Hofmann, S.G. Gender Differences in Anxiety Disorders: Prevalence, Course of Illness, Comorbidity and Burden of Illness. J. Psychiatr. Res. 2011, 45, 1027–1035. [Google Scholar] [CrossRef] [Green Version]
- Barth, M.; Kriston, L.; Klostermann, S.; Barbui, C.; Cipriani, A.; Linde, K. Efficacy of Selective Serotonin Reuptake Inhibitors and Adverse Events: Meta-Regression and Mediation Analysis of Placebo-Controlled Trials. Br. J. Psychiatry 2016, 208, 114–119. [Google Scholar] [CrossRef] [Green Version]
- Yamada, K.; Kobayashi, M.; Kanda, T. Chapter Fifteen-Involvement of Adenosine A2A Receptors in Depression and Anxiety. In International Review of Neurobiology; Mori, A., Ed.; Adenosine Receptors in Neurology and Psychiatry; Academic Press: Cambridge, MA, USA, 2014; Volume 119, pp. 373–393. [Google Scholar]
- Gorwood, P.; Richard-Devantoy, S.; Baylé, F.; Cléry-Melun, M.L. Psychomotor Retardation Is a Scar of Past Depressive Episodes, Revealed by Simple Cognitive Tests. Eur. Neuropsychopharmacol. 2014, 24, 1630–1640. [Google Scholar] [CrossRef]
- Huang, Z.; Liu, R.; Bai, X.; Zhao, G.; Song, J.; Wu, S.; Du, G. Protective Effects of the Novel Adenosine Derivative WS0701 in a Mouse Model of Posttraumatic Stress Disorder. Acta Pharmacol. Sin. 2014, 35, 24–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, C.J.; Augusto, E.; Gomes, C.A.; Singer, P.; Wang, Y.; Boison, D.; Cunha, R.A.; Yee, B.K.; Chen, J.-F. Regulation of Fear Responses by Striatal and Extrastriatal Adenosine A2A Receptors in Forebrain. Biol. Psychiatry 2014, 75, 855–863. [Google Scholar] [CrossRef] [Green Version]
- Hohoff, C.; Mullings, E.L.; Heatherley, S.V.; Freitag, C.M.; Neumann, L.C.; Domschke, K.; Krakowitzky, P.; Rothermundt, M.; Keck, M.E.; Erhardt, A.; et al. Adenosine A2A Receptor Gene: Evidence for Association of Risk Variants with Panic Disorder and Anxious Personality. J. Psychiatr. Res. 2010, 44, 930–937. [Google Scholar] [CrossRef] [PubMed]
- Chiu, G.S.; Darmody, P.T.; Walsh, J.P.; Moon, M.L.; Kwakwa, K.A.; Bray, J.K.; McCusker, R.H.; Freund, G.G. Adenosine through the A2A Adenosine Receptor Increases IL-1β in the Brain Contributing to Anxiety. Brain Behav. Immun. 2014, 41, 218–231. [Google Scholar] [CrossRef] [Green Version]
- Caetano, L.; Pinheiro, H.; Patrício, P.; Mateus-Pinheiro, A.; Alves, N.D.; Coimbra, B.; Baptista, F.I.; Henriques, S.N.; Cunha, C.; Santos, A.R.; et al. Adenosine A2A Receptor Regulation of Microglia Morphological Remodeling-Gender Bias in Physiology and in a Model of Chronic Anxiety. Mol. Psychiatry 2017, 22, 1035–1043. [Google Scholar] [CrossRef]
- Chen, J.-F.; Eltzschig, H.K.; Fredholm, B.B. Adenosine Receptors as Drug Targets-What Are the Challenges? Nat. Rev. Drug Discov. 2013, 12, 265–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, B.; Halldner, L.; Dunwiddie, T.V.; Masino, S.A.; Poelchen, W.; Giménez-Llort, L.; Escorihuela, R.M.; Fernández-Teruel, A.; Wiesenfeld-Hallin, Z.; Xu, X.-J.; et al. Hyperalgesia, Anxiety, and Decreased Hypoxic Neuroprotection in Mice Lacking the Adenosine A1 Receptor. Proc. Natl. Acad. Sci. USA 2001, 98, 9407–9412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giménez-Llort, L.; Fernández-Teruel, A.; Escorihuela, R.M.; Fredholm, B.B.; Tobeña, A.; Pekny, M.; Johansson, B. Mice Lacking the Adenosine A1 Receptor Are Anxious and Aggressive, but Are Normal Learners with Reduced Muscle Strength and Survival Rate. Eur. J. Neurosci. 2002, 16, 547–550. [Google Scholar] [CrossRef] [PubMed]
- Lang, U.E.; Lang, F.; Richter, K.; Vallon, V.; Lipp, H.-P.; Schnermann, J.; Wolfer, D.P. Emotional Instability but Intact Spatial Cognition in Adenosine Receptor 1 Knock out Mice. Behav. Brain Res. 2003, 145, 179–188. [Google Scholar] [CrossRef]
- Romagnoli, R.; Baraldi, P.G.; Tabrizi, M.A.; Gessi, S.; Borea, P.A.; Merighi, S. Allosteric Enhancers of A1 Adenosine Receptors: State of the Art and New Horizons for Drug Development. Curr. Med. Chem. 2010, 17, 3488–3502. [Google Scholar] [CrossRef]
- Kiesman, W.F.; Elzein, E.; Zablocki, J. A1 Adenosine Receptor Antagonists, Agonists, and Allosteric Enhancers. In Adenosine Receptors in Health and Disease; Wilson, C.N., Mustafa, S.J., Eds.; Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2009; pp. 25–58. ISBN 978-3-540-89615-9. [Google Scholar]
- Childers, S.R.; Li, X.; Xiao, R.; Eisenach, J.C. Allosteric Modulation of Adenosine A1 Receptor Coupling to G-Proteins in Brain. J. Neurochem. 2005, 93, 715–723. [Google Scholar] [CrossRef]
- Gao, Z.-G.; Kim, S.-K.; IJzerman, A.P.; Jacobson, K.A. Allosteric Modulation of the Adenosine Family of Receptors. Mini Rev. Med. Chem. 2005, 5, 545–553. [Google Scholar] [CrossRef] [Green Version]
- Romagnoli, R.; Baraldi, P.G.; Carrion, M.D.; Cara, C.L.; Cruz-Lopez, O.; Iaconinoto, M.A.; Preti, D.; Shryock, J.C.; Moorman, A.R.; Vincenzi, F.; et al. Synthesis and Biological Evaluation of 2-Amino-3-(4-Chlorobenzoyl)-4-[N-(Substituted) Piperazin-1-Yl] Thiophenes as Potent Allosteric Enhancers of the A1 Adenosine Receptor. J. Med. Chem. 2008, 51, 5875–5879. [Google Scholar] [CrossRef]
- Romagnoli, R.; Baraldi, P.G.; IJzerman, A.P.; Massink, A.; Cruz-Lopez, O.; Lopez-Cara, L.C.; Saponaro, G.; Preti, D.; Tabrizi, M.A.; Baraldi, S.; et al. Synthesis and Biological Evaluation of Novel Allosteric Enhancers of the A1 Adenosine Receptor Based on 2-Amino-3-(4′-Chlorobenzoyl)-4-Substituted-5-Arylethynyl Thiophene. J. Med. Chem. 2014, 57, 7673–7686. [Google Scholar] [CrossRef]
- Vincenzi, F.; Targa, M.; Romagnoli, R.; Merighi, S.; Gessi, S.; Baraldi, P.G.; Borea, P.A.; Varani, K. TRR469, a Potent A1 Adenosine Receptor Allosteric Modulator, Exhibits Anti-Nociceptive Properties in Acute and Neuropathic Pain Models in Mice. Neuropharmacology 2014, 81, 6–14. [Google Scholar] [CrossRef]
- Lord, C.; Elsabbagh, M.; Baird, G.; Veenstra-Vanderweele, J. Autism Spectrum Disorder. Lancet 2018, 392, 508–520. [Google Scholar] [CrossRef]
- Varghese, M.; Keshav, N.; Jacot-Descombes, S.; Warda, T.; Wicinski, B.; Dickstein, D.L.; Harony-Nicolas, H.; De Rubeis, S.; Drapeau, E.; Buxbaum, J.D.; et al. Autism Spectrum Disorder: Neuropathology and Animal Models. Acta Neuropathol. 2017, 134, 537–566. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.R.; Gonda, X.; Tarazi, F.I. Autism Spectrum Disorder: Classification, Diagnosis and Therapy. Pharmacol. Ther. 2018, 190, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Behmanesh, H.; Moghaddam, H.S.; Mohammadi, M.-R.; Akhondzadeh, S. Risperidone Combination Therapy with Propentofylline for Treatment of Irritability in Autism Spectrum Disorders: A Randomized, Double-Blind, Placebo-Controlled Clinical Trial. Clin. Neuropharmacol. 2019, 42, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Masino, S.A.; Kawamura, M.; Plotkin, L.M.; Svedova, J.; DiMario, F.J.; Eigsti, I.-M. The Relationship between the Neuromodulator Adenosine and Behavioral Symptoms of Autism. Neurosci. Lett. 2011, 500, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Freitag, C.M.; Agelopoulos, K.; Huy, E.; Rothermundt, M.; Krakowitzky, P.; Meyer, J.; Deckert, J.; von Gontard, A.; Hohoff, C. Adenosine A(2A) Receptor Gene (ADORA2A) Variants May Increase Autistic Symptoms and Anxiety in Autism Spectrum Disorder. Eur. Child Adolesc. Psychiatry 2010, 19, 67–74. [Google Scholar] [CrossRef]
- Poleszak, E.; Malec, D. Influence of Adenosine Receptor Agonists and Antagonists on Amphetamine-Induced Stereotypy in Rats. Pol. J. Pharmacol. 2000, 52, 423–429. [Google Scholar]
- Muehlmann, A.; Edington, G.; Mihalik, A.; Buchwald, Z.; Koppuzha, D.; Korah, M.; Lewis, M. Further Characterization of Repetitive Behavior in C58 Mice: Developmental Trajectory and Effects of Environmental Enrichment. Behav. Brain Res. 2012, 235, 143–149. [Google Scholar] [CrossRef] [Green Version]
- Lewis, M.H.; Rajpal, H.; Muehlmann, A.M. Reduction of Repetitive Behavior by Co-Administration of Adenosine Receptor Agonists in C58 Mice. Pharmacol. Biochem. Behav. 2019, 181, 110–116. [Google Scholar] [CrossRef]
- Tanimura, Y.; Vaziri, S.; Lewis, M.H. Indirect Basal Ganglia Pathway Mediation of Repetitive Behavior: Attenuation by Adenosine Receptor Agonists. Behav. Brain Res. 2010, 210, 116–122. [Google Scholar] [CrossRef] [Green Version]
- Amodeo, D.A.; Cuevas, L.; Dunn, J.T.; Sweeney, J.A.; Ragozzino, M.E. The Adenosine A2A Receptor Agonist, CGS 21680, Attenuates a Probabilistic Reversal Learning Deficit and Elevated Grooming Behavior in BTBR Mice. Autism Res. 2018, 11, 223–233. [Google Scholar] [CrossRef]
- Ashwood, P.; Krakowiak, P.; Hertz-Picciotto, I.; Hansen, R.; Pessah, I.N.; Van de Water, J. Altered T Cell Responses in Children with Autism. Brain Behav. Immun. 2011, 25, 840–849. [Google Scholar] [CrossRef] [Green Version]
- Garbett, K.; Ebert, P.J.; Mitchell, A.; Lintas, C.; Manzi, B.; Mirnics, K.; Persico, A.M. Immune Transcriptome Alterations in the Temporal Cortex of Subjects with Autism. Neurobiol. Dis. 2008, 30, 303–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, S.F.; Ansari, M.A.; Nadeem, A.; Bakheet, S.A.; Almutairi, M.M.; Attia, S.M. Adenosine A2A Receptor Signaling Affects IL-21/IL-22 Cytokines and GATA3/T-Bet Transcription Factor Expression in CD4+ T Cells from a BTBR T+ Itpr3tf/J Mouse Model of Autism. J. Neuroimmunol. 2017, 311, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.F.; Ansari, M.A.; Nadeem, A.; Bakheet, S.A.; Al-Ayadhi, L.Y.; Attia, S.M. Toll-like Receptors, NF-ΚB, and IL-27 Mediate Adenosine A2A Receptor Signaling in BTBR T+ Itpr3tf/J Mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 79, 184–191. [Google Scholar] [CrossRef]
- Ansari, M.A.; Nadeem, A.; Attia, S.M.; Bakheet, S.A.; Raish, M.; Ahmad, S.F. Adenosine A2A Receptor Modulates Neuroimmune Function through Th17/Retinoid-Related Orphan Receptor Gamma t (RORγt) Signaling in a BTBR T+ Itpr3tf/J Mouse Model of Autism. Cell Signal 2017, 36, 14–24. [Google Scholar] [CrossRef]
- Ansari, M.A.; Attia, S.M.; Nadeem, A.; Bakheet, S.A.; Raish, M.; Khan, T.H.; Al-Shabanah, O.A.; Ahmad, S.F. Activation of Adenosine A2A Receptor Signaling Regulates the Expression of Cytokines Associated with Immunologic Dysfunction in BTBR T+ Itpr3tf/J Mice. Mol. Cell Neurosci. 2017, 82, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.F.; Ansari, M.A.; Nadeem, A.; Bakheet, S.A.; Mohammad, R.; Attia, S.M. Immune Alterations in CD8+ T Cells Are Associated with Neuronal C-C and C-X-C Chemokine Receptor Regulation Through Adenosine A2A Receptor Signaling in a BTBR T+ Itpr3tf/J Autistic Mouse Model. Mol. Neurobiol. 2018, 55, 2603–2616. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.J.; Berry-Kravis, E.; Hazlett, H.C.; Bailey, D.B.; Moine, H.; Kooy, R.F.; Tassone, F.; Gantois, I.; Sonenberg, N.; Mandel, J.L.; et al. Fragile X Syndrome. Nat. Rev. Dis. Primers 2017, 3, 17065. [Google Scholar] [CrossRef] [PubMed]
- Pop, A.S.; Gomez-Mancilla, B.; Neri, G.; Willemsen, R.; Gasparini, F. Fragile X Syndrome: A Preclinical Review on Metabotropic Glutamate Receptor 5 (MGluR5) Antagonists and Drug Development. Psychopharmacology 2014, 231, 1217–1226. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Hagerman, P.J. Fragile X Syndrome: Lessons Learned and What New Treatment Avenues Are on the Horizon. Annu. Rev. Pharmacol. Toxicol. 2021, 62, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, A.; Boussadia, Z.; Borreca, A.; Mallozzi, C.; Pedini, G.; Pacini, L.; Pezzola, A.; Armida, M.; Vincenzi, F.; Varani, K.; et al. Adenosine A2A Receptor Inhibition Reduces Synaptic and Cognitive Hippocampal Alterations in Fmr1 KO Mice. Transl. Psychiatry 2021, 11, 112. [Google Scholar] [CrossRef]
- Tripp, G.; Wickens, J.R. Neurobiology of ADHD. Neuropharmacology 2009, 57, 579–589. [Google Scholar] [CrossRef]
- Grimm, O.; Kranz, T.M.; Reif, A. Genetics of ADHD: What Should the Clinician Know? Curr. Psychiatry Rep. 2020, 22, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinberg, E.E.; Boivin, J.R.; Saunders, B.T.; Witten, I.B.; Deisseroth, K.; Janak, P.H. Positive Reinforcement Mediated by Midbrain Dopamine Neurons Requires D1 and D2 Receptor Activation in the Nucleus Accumbens. PLoS ONE 2014, 9, e94771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golmirzaei, J.; Mahboobi, H.; Yazdanparast, M.; Mushtaq, G.; Kamal, M.A.; Hamzei, E. Psychopharmacology of Attention-Deficit Hyperactivity Disorder: Effects and Side Effects. Curr. Pharm. Des. 2016, 22, 590–594. [Google Scholar] [CrossRef]
- Asherson, P.; Gurling, H. Quantitative and Molecular Genetics of ADHD. Curr. Top. Behav. Neurosci. 2012, 9, 239–272. [Google Scholar] [CrossRef]
- Salamone, J.D.; Correa, M.; Ferrigno, S.; Yang, J.H.; Rotolo, R.A.; Presby, R.E. The Psychopharmacology of Effort-Related Decision Making: Dopamine, Adenosine, and Insights into the Neurochemistry of Motivation. Pharmacol. Rev. 2018, 70, 747–762. [Google Scholar] [CrossRef] [Green Version]
- Molero, Y.; Gumpert, C.; Serlachius, E.; Lichtenstein, P.; Walum, H.; Johansson, D.; Anckarsäter, H.; Westberg, L.; Eriksson, E.; Halldner, L. A Study of the Possible Association between Adenosine A2A Receptor Gene Polymorphisms and Attention-Deficit Hyperactivity Disorder Traits. Genes Brain Behav. 2013, 12, 305–310. [Google Scholar] [CrossRef]
- Fraporti, T.T.; Contini, V.; Tovo-Rodrigues, L.; Recamonde-Mendoza, M.; Rovaris, D.L.; Rohde, L.A.; Hutz, M.H.; Salatino-Oliveira, A.; Genro, J.P. Synergistic Effects between ADORA2A and DRD2 Genes on Anxiety Disorders in Children with ADHD. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 93, 214–220. [Google Scholar] [CrossRef]
- Meneses, A.; Perez-Garcia, G.; Ponce-Lopez, T.; Tellez, R.; Gallegos-Cari, A.; Castillo, C. Spontaneously Hypertensive Rat (SHR) as an Animal Model for ADHD: A Short Overview. Rev. Neurosci. 2011, 22, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Pires, V.A.; Pamplona, F.A.; Pandolfo, P.; Fernandes, D.; Prediger, R.D.S.; Takahashi, R.N. Adenosine Receptor Antagonists Improve Short-Term Object-Recognition Ability of Spontaneously Hypertensive Rats: A Rodent Model of Attention-Deficit Hyperactivity Disorder. Behav. Pharmacol. 2009, 20, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Pandolfo, P.; Machado, N.J.; Köfalvi, A.; Takahashi, R.N.; Cunha, R.A. Caffeine Regulates Frontocorticostriatal Dopamine Transporter Density and Improves Attention and Cognitive Deficits in an Animal Model of Attention Deficit Hyperactivity Disorder. Eur. Neuropsychopharmacol. 2013, 23, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Leffa, D.T.; Ferreira, S.G.; Machado, N.J.; Souza, C.M.; da Rosa, F.; de Carvalho, C.; Kincheski, G.C.; Takahashi, R.N.; Porciúncula, L.O.; Souza, D.O.; et al. Caffeine and Cannabinoid Receptors Modulate Impulsive Behavior in an Animal Model of Attentional Deficit and Hyperactivity Disorder. Eur. J. Neurosci. 2019, 49, 1673–1683. [Google Scholar] [CrossRef] [PubMed]
- Duncan, G.W.; Khoo, T.K.; Yarnall, A.J.; O’Brien, J.T.; Coleman, S.Y.; Brooks, D.J.; Barker, R.A.; Burn, D.J. Health-Related Quality of Life in Early Parkinson’s Disease: The Impact of Nonmotor Symptoms. Mov. Disord. 2014, 29, 195–202. [Google Scholar] [CrossRef]
- Martinez-Martin, P.; Rodriguez-Blazquez, C.; Kurtis, M.M.; Chaudhuri, K.R. The Impact of Non-Motor Symptoms on Health-Related Quality of Life of Patients with Parkinson’s Disease. Mov. Disord. 2011, 26, 399–406. [Google Scholar] [CrossRef]
- Prakash, K.M.; Nadkarni, N.V.; Lye, W.-K.; Yong, M.-H.; Tan, E.-K. The Impact of Non-Motor Symptoms on the Quality of Life of Parkinson’s Disease Patients: A Longitudinal Study. Eur. J. Neurol. 2016, 23, 854–860. [Google Scholar] [CrossRef]
- Kelberman, M.A.; Vazey, E.M. New Pharmacological Approaches to Treating Non-Motor Symptoms of Parkinson’s Disease. Curr. Pharmacol. Rep. 2016, 2, 253–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadastik-Eerme, L.; Muldmaa, M.; Lilles, S.; Rosenthal, M.; Taba, N.; Taba, P. Nonmotor Features in Parkinson’s Disease: What Are the Most Important Associated Factors? Park. Dis. 2016, 2016, 4370674. [Google Scholar] [CrossRef] [Green Version]
- Pinna, A.; Serra, M.; Marongiu, J.; Morelli, M. Pharmacological Interactions between Adenosine A2A Receptor Antagonists and Different Neurotransmitter Systems. Parkinsonism Relat. Disord. 2020, 80, S37–S44. [Google Scholar] [CrossRef]
- Nazario, L.R.; da Silva, R.S.; Bonan, C.D. Targeting Adenosine Signaling in Parkinson’s Disease: From Pharmacological to Non-Pharmacological Approaches. Front. Neurosci. 2017, 11, 658. [Google Scholar] [CrossRef] [Green Version]
- Mori, A. Chapter Four-Mode of Action of Adenosine A2A Receptor Antagonists as Symptomatic Treatment for Parkinson’s Disease. In International Review of Neurobiology; Mori, A., Ed.; Adenosine Receptors in Neurology and Psychiatry; Academic Press: Cambridge, MA, USA, 2014; Volume 119, pp. 87–116. [Google Scholar]
- Witt, K.; Daniels, C.; Herzog, J.; Lorenz, D.; Volkmann, J.; Reiff, J.; Mehdorn, M.; Deuschl, G.; Krack, P. Differential Effects of L-Dopa and Subthalamic Stimulation on Depressive Symptoms and Hedonic Tone in Parkinson’s Disease. J. Neuropsychiatry Clin. Neurosci. 2006, 18, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Kobayashi, M.; Mori, A.; Jenner, P.; Kanda, T. Antidepressant-like Activity of the Adenosine A2A Receptor Antagonist, Istradefylline (KW-6002), in the Forced Swim Test and the Tail Suspension Test in Rodents. Pharmacol. Biochem. Behav. 2013, 114–115, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Nagayama, H.; Kano, O.; Murakami, H.; Ono, K.; Hamada, M.; Toda, T.; Sengoku, R.; Shimo, Y.; Hattori, N. Effect of Istradefylline on Mood Disorders in Parkinson’s Disease. J. Neurol. Sci. 2019, 396, 78–83. [Google Scholar] [CrossRef]
- Schapira, A.H.V.; Chaudhuri, K.R.; Jenner, P. Non-Motor Features of Parkinson Disease. Nat. Rev. Neurosci. 2017, 18, 435–450. [Google Scholar] [CrossRef]
- Lazarus, M.; Chen, J.-F.; Huang, Z.-L.; Urade, Y.; Fredholm, B.B. Adenosine and Sleep. In Sleep-Wake Neurobiology and Pharmacology; Landolt, H.-P., Dijk, D.-J., Eds.; Handbook of Experimental Pharmacology; Springer International Publishing: Cham, Switzerland, 2019; pp. 359–381. ISBN 978-3-030-11272-1. [Google Scholar]
- Suzuki, K.; Miyamoto, M.; Miyamoto, T.; Uchiyama, T.; Watanabe, Y.; Suzuki, S.; Kadowaki, T.; Fujita, H.; Matsubara, T.; Sakuramoto, H.; et al. Istradefylline Improves Daytime Sleepiness in Patients with Parkinson’s Disease: An Open-Label, 3-Month Study. J. Neurol. Sci. 2017, 380, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, K.; Kajikawa, H.; Tabei, K.-I.; Satoh, M.; Kida, H.; Nakamura, N.; Tomimoto, H. The Effectiveness of Istradefylline for the Treatment of Gait Deficits and Sleepiness in Patients with Parkinson’s Disease. Neurosci. Lett. 2018, 662, 158–161. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Kobayashi, M.; Shiozaki, S.; Ohta, T.; Mori, A.; Jenner, P.; Kanda, T. Antidepressant Activity of the Adenosine A2A Receptor Antagonist, Istradefylline (KW-6002) on Learned Helplessness in Rats. Psychopharmacology 2014, 231, 2839–2849. [Google Scholar] [CrossRef]
- Jenner, P.; Mori, A.; Kanda, T. Can Adenosine A2A Receptor Antagonists Be Used to Treat Cognitive Impairment, Depression or Excessive Sleepiness in Parkinson’s Disease? Parkinsonism Relat. Disord. 2020, 80, S28–S36. [Google Scholar] [CrossRef] [PubMed]
- Mangialasche, F.; Solomon, A.; Winblad, B.; Mecocci, P.; Kivipelto, M. Alzheimer’s Disease: Clinical Trials and Drug Development. Lancet Neurol. 2010, 9, 702–716. [Google Scholar] [CrossRef]
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s Disease. Lancet 2011, 377, 1019–1031. [Google Scholar] [CrossRef]
- Meraz-Ríos, M.A.; Lira-De León, K.I.; Campos-Peña, V.; De Anda-Hernández, M.A.; Mena-López, R. Tau Oligomers and Aggregation in Alzheimer’s Disease. J. Neurochem. 2010, 112, 1353–1367. [Google Scholar] [CrossRef]
- Cummings, J.L.; Tong, G.; Ballard, C. Treatment Combinations for Alzheimer’s Disease: Current and Future Pharmacotherapy Options. J. Alzheimers Dis. 2019, 67, 779–794. [Google Scholar] [CrossRef] [Green Version]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, E5789. [Google Scholar] [CrossRef]
- Li, X.-L.; Hu, N.; Tan, M.-S.; Yu, J.-T.; Tan, L. Behavioral and Psychological Symptoms in Alzheimer’s Disease. BioMed Res. Int. 2014, 2014, 927804. [Google Scholar] [CrossRef]
- Lyketsos, C.G.; Carrillo, M.C.; Ryan, J.M.; Khachaturian, A.S.; Trzepacz, P.; Amatniek, J.; Cedarbaum, J.; Brashear, R.; Miller, D.S. Neuropsychiatric Symptoms in Alzheimer’s Disease. Alzheimers Dement. 2011, 7, 532–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dam, D.; Vermeiren, Y.; Dekker, A.D.; Naudé, P.J.W.; De Deyn, P.P. Neuropsychiatric Disturbances in Alzheimer’s Disease: What Have We Learned from Neuropathological Studies? Curr. Alzheimer Res. 2016, 13, 1145–1164. [Google Scholar] [CrossRef] [PubMed]
- Merighi, S.; Borea, P.A.; Varani, K.; Vincenzi, F.; Jacobson, K.A.; Gessi, S. A2A Adenosine Receptor Antagonists in Neurodegenerative Diseases. Curr. Med. Chem. 2021, 28, 1. [Google Scholar] [CrossRef]
- Eskelinen, M.H.; Kivipelto, M. Caffeine as a Protective Factor in Dementia and Alzheimer’s Disease. J. Alzheimers Dis. 2010, 20 (Suppl. 1), S167–S174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dall’Igna, O.P.; Fett, P.; Gomes, M.W.; Souza, D.O.; Cunha, R.A.; Lara, D.R. Caffeine and Adenosine A2a Receptor Antagonists Prevent β-Amyloid (25–35)-Induced Cognitive Deficits in Mice. Exp. Neurol. 2007, 203, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Canas, P.M.; Porciúncula, L.O.; Cunha, G.M.A.; Silva, C.G.; Machado, N.J.; Oliveira, J.M.A.; Oliveira, C.R.; Cunha, R.A. Adenosine A2A Receptor Blockade Prevents Synaptotoxicity and Memory Dysfunction Caused by β-Amyloid Peptides via P38 Mitogen-Activated Protein Kinase Pathway. J. Neurosci. 2009, 29, 14741–14751. [Google Scholar] [CrossRef]
- Franco, R.; Rivas-Santisteban, R.; Casanovas, M.; Lillo, A.; Saura, C.A.; Navarro, G. Adenosine A2A Receptor Antagonists Affects NMDA Glutamate Receptor Function. Potential to Address Neurodegeneration in Alzheimer’s Disease. Cells 2020, 9, 1075. [Google Scholar] [CrossRef]
- Laurent, C.; Eddarkaoui, S.; Derisbourg, M.; Leboucher, A.; Demeyer, D.; Carrier, S.; Schneider, M.; Hamdane, M.; Müller, C.E.; Buée, L.; et al. Beneficial Effects of Caffeine in a Transgenic Model of Alzheimer’s Disease-like Tau Pathology. Neurobiol. Aging 2014, 35, 2079–2090. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, K.; Faivre, E.; Pietrowski, M.J.; Marques, X.; Gomez-Murcia, V.; Deleau, A.; Huin, V.; Hansen, J.N.; Kozlov, S.; Danis, C.; et al. Exacerbation of C1q Dysregulation, Synaptic Loss and Memory Deficits in Tau Pathology Linked to Neuronal Adenosine A2A Receptor. Brain 2019, 142, 3636–3654. [Google Scholar] [CrossRef]
- Merighi, S.; Poloni, T.E.; Pelloni, L.; Pasquini, S.; Varani, K.; Vincenzi, F.; Borea, P.A.; Gessi, S. An Open Question: Is the A2A Adenosine Receptor a Novel Target for Alzheimer’s Disease Treatment? Front. Pharmacol. 2021, 12, 652455. [Google Scholar] [CrossRef] [PubMed]
- Albasanz, J.L.; Perez, S.; Barrachina, M.; Ferrer, I.; Martín, M. Up-Regulation of Adenosine Receptors in the Frontal Cortex in Alzheimer’s Disease. Brain Pathol. 2008, 18, 211–219. [Google Scholar] [CrossRef]
- Canas, P.M.; Duarte, J.M.N.; Rodrigues, R.J.; Köfalvi, A.; Cunha, R.A. Modification upon Aging of the Density of Presynaptic Modulation Systems in the Hippocampus. Neurobiol. Aging 2009, 30, 1877–1884. [Google Scholar] [CrossRef] [Green Version]
- Merighi, S.; Battistello, E.; Casetta, I.; Gragnaniello, D.; Poloni, T.E.; Medici, V.; Cirrincione, A.; Varani, K.; Vincenzi, F.; Borea, P.A.; et al. Upregulation of Cortical A2A Adenosine Receptors Is Reflected in Platelets of Patients with Alzheimer’s Disease. J. Alzheimers Dis. 2021, 80, 1105–1117. [Google Scholar] [CrossRef] [PubMed]
- Merighi, S.; Poloni, T.E.; Terrazzan, A.; Moretti, E.; Gessi, S.; Ferrari, D. Alzheimer and Purinergic Signaling: Just a Matter of Inflammation? Cells 2021, 10, 1267. [Google Scholar] [CrossRef] [PubMed]
- Cerri, A.P.; Arosio, B.; Viazzoli, C.; Confalonieri, R.; Teruzzi, F.; Annoni, G. -308(G/A) TNF-Alpha Gene Polymorphism and Risk of Depression Late in the Life. Arch. Gerontol. Geriatr. 2009, 49 (Suppl. 1), 29–34. [Google Scholar] [CrossRef]
- Banerjee, A.; Khemka, V.K.; Roy, D.; Dhar, A.; Roy, T.K.S.; Biswas, A.; Mukhopadhyay, B.; Chakrabarti, S. Role of Pro-Inflammatory Cytokines and Vitamin D in Probable Alzheimer’s Disease with Depression. Aging Dis. 2017, 8, 267–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmgren, S.; Hjorth, E.; Schultzberg, M.; Lärksäter, M.; Frenkel, D.; Tysen-Bäckström, A.C.; Aarsland, D.; Freund-Levi, Y. Neuropsychiatric Symptoms in Dementia-a Role for Neuroinflammation? Brain Res. Bull. 2014, 108, 88–93. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Receptor Subtype | CNS Effects and Interactions | Pharmacological Strategy | Therapeutic Potential in Neuropsychiatric Diseases |
|---|---|---|---|
| A1ARs | Inhibition of neurotransmitter release Reduction of dopamine D1 signaling Reduction of neuronal excitability Increase of Homer1a expression | Activation | Depression Anxiety |
| A2AARs | Reduction of dopamine D2 signaling Increase of excitatory neurotransmitter release Increase of mGLUR5 signaling Regulation of neuroinflammation | Activation | Schizophrenia Autism spectrum disorder |
| Inhibition | Depression Fragile X syndrome Attention-deficit hyperactivity disorder Parkinson’s disease Alzheimer’s disease |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pasquini, S.; Contri, C.; Merighi, S.; Gessi, S.; Borea, P.A.; Varani, K.; Vincenzi, F. Adenosine Receptors in Neuropsychiatric Disorders: Fine Regulators of Neurotransmission and Potential Therapeutic Targets. Int. J. Mol. Sci. 2022, 23, 1219. https://doi.org/10.3390/ijms23031219
Pasquini S, Contri C, Merighi S, Gessi S, Borea PA, Varani K, Vincenzi F. Adenosine Receptors in Neuropsychiatric Disorders: Fine Regulators of Neurotransmission and Potential Therapeutic Targets. International Journal of Molecular Sciences. 2022; 23(3):1219. https://doi.org/10.3390/ijms23031219
Chicago/Turabian StylePasquini, Silvia, Chiara Contri, Stefania Merighi, Stefania Gessi, Pier Andrea Borea, Katia Varani, and Fabrizio Vincenzi. 2022. "Adenosine Receptors in Neuropsychiatric Disorders: Fine Regulators of Neurotransmission and Potential Therapeutic Targets" International Journal of Molecular Sciences 23, no. 3: 1219. https://doi.org/10.3390/ijms23031219