
Unraveling the Link between Interferon-α and Systemic Lupus Erythematosus: From the Molecular Mechanisms to Target Therapies
 , , , , ,
, , , , ,  ,
,
Abstract
:1. Introduction
2. The Type-I-Interferons
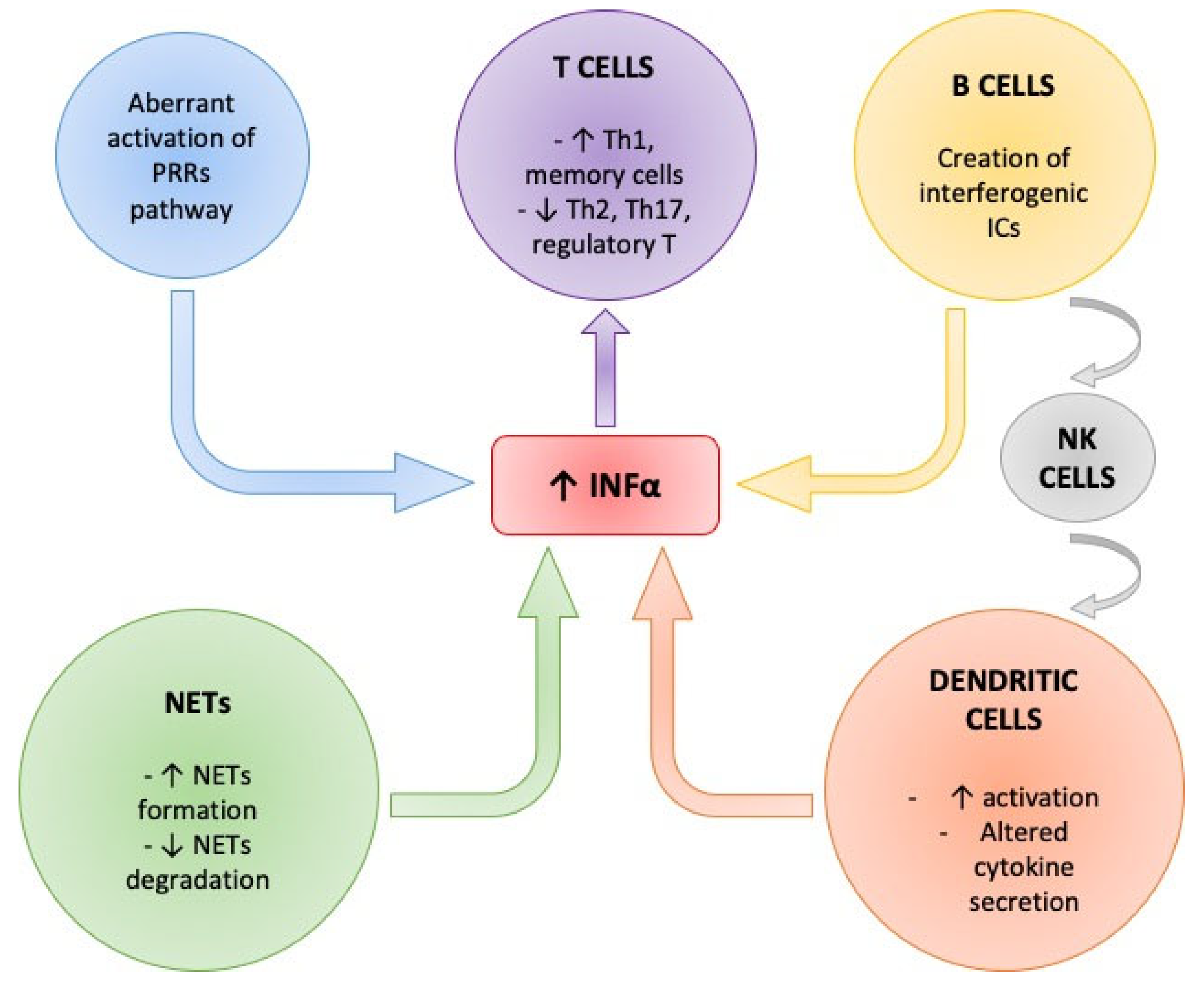
3. Role of Interferon-Alpha Production in SLE
4. Interferon-Alpha and Dendritic Cells (DCs)
5. Interferon-Alpha and B-Cells in SLE
6. Interferon-Alpha and T-Cells in SLE
7. Other Regulators of INFalpha Production
8. Environmental Factors and Interferon-Alpha Production
9. Renal Complication of SLE: The Role of the Complement System and IFNα
10. Biologic Therapy in Lupus Nephritis

{kind=link}
{kind=link}
| Therapy | Mechanism of Action | Current Development Stage | Refs |
|---|---|---|---|
| Belimumab | Anti-B lymphocyte stimulator | Phase III | [113,114,115] |
| Rituximab | Chimeric monoclonal antibody anti-CD20 | Phase II | [120] |
| Obinutuzumab | Humanized monoclonal antibody IgG1 anti-CD20 | Phase II | [122] |
| Voclosporin | Calcineurin inhibitor | Phase II | [123] |
| Sifalimumab | Fully human monoclonal antibody anti-IFNα | Phase II | [124] |
| Anifrolumab | Human monoclonal antibody anti-Type-I IFN receptor subunit 1 | Phase III | [125,126,127] |
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Eloranta, M.-L.; Rönnblom, L. Cause and Consequences of the Activated Type I Interferon System in SLE. J. Mol. Med. 2016, 94, 1103–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barber, M.R.W.; Drenkard, C.; Falasinnu, T.; Hoi, A.; Mak, A.; Kow, N.Y.; Svenungsson, E.; Peterson, J.; Clarke, A.E.; Ramsey-Goldman, R. Global Epidemiology of Systemic Lupus Erythematosus. Nat. Rev. Rheumatol. 2021, 17, 515–532. [Google Scholar] [CrossRef] [PubMed]
- Pons-Estel, G.J.; Alarcón, G.S.; Scofield, L.; Reinlib, L.; Cooper, G.S. Understanding the Epidemiology and Progression of Systemic Lupus Erythematosus. Semin. Arthritis Rheum. 2010, 39, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Rose, T.; Dörner, T. Drivers of the Immunopathogenesis in Systemic Lupus Erythematosus. Best Pract. Res. Clin. Rheumatol. 2017, 31, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Maselli, A.; Conti, F.; Alessandri, C.; Colasanti, T.; Barbati, C.; Vomero, M.; Ciarlo, L.; Patrizio, M.; Spinelli, F.R.; Ortona, E.; et al. Low Expression of Estrogen Receptor β in T Lymphocytes and High Serum Levels of Anti-Estrogen Receptor α Antibodies Impact Disease Activity in Female Patients with Systemic Lupus Erythematosus. Biol. Sex Differ. 2016, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Tsokos, G.C. Systemic Lupus Erythematosus. N. Engl. J. Med. 2011, 365, 2110–2121. [Google Scholar] [CrossRef] [Green Version]
- Gasparotto, M.; Gatto, M.; Binda, V.; Doria, A.; Moroni, G. Lupus Nephritis: Clinical Presentations and Outcomes in the 21st Century. Rheumatology 2020, 59, v39–v51. [Google Scholar] [CrossRef]
- Bernatsky, S.; Boivin, J.-F.; Joseph, L.; Manzi, S.; Ginzler, E.; Gladman, D.D.; Urowitz, M.; Fortin, P.R.; Petri, M.; Barr, S.; et al. Mortality in Systemic Lupus Erythematosus. Arthritis Rheum. 2006, 54, 2550–2557. [Google Scholar] [CrossRef]
- Honarpisheh, M.; Köhler, P.; von Rauchhaupt, E.; Lech, M. The Involvement of MicroRNAs in Modulation of Innate and Adaptive Immunity in Systemic Lupus Erythematosus and Lupus Nephritis. J. Immunol. Res. 2018, 2018, 4126106. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.-Z.; Zhou, J.; Lian, Y.; Zhang, B.; Branch, V.K.; Carr-Johnson, F.; Karp, D.R.; Mohan, C.; Wakeland, E.K.; Olsen, N.J. Interferon Signature Gene Expression Is Correlated with Autoantibody Profiles in Patients with Incomplete Lupus Syndromes. Clin. Exp. Immunol. 2010, 159, 281–291. [Google Scholar] [CrossRef]
- Baechler, E.C.; Batliwalla, F.M.; Karypis, G.; Gaffney, P.M.; Ortmann, W.A.; Espe, K.J.; Shark, K.B.; Grande, W.J.; Hughes, K.M.; Kapur, V.; et al. Interferon-Inducible Gene Expression Signature in Peripheral Blood Cells of Patients with Severe Lupus. Proc. Natl. Acad. Sci. USA 2003, 100, 2610–2615. [Google Scholar] [CrossRef] [Green Version]
- Hooks, J.J.; Moutsopoulos, H.M.; Geis, S.A.; Stahl, N.I.; Decker, J.L.; Notkins, A.L. Immune Interferon in the Circulation of Patients with Autoimmune Disease. N. Engl. J. Med. 1979, 301, 5–8. [Google Scholar] [CrossRef]
- Crow, M.K. Type I Interferon in the Pathogenesis of Lupus. J. Immunol. 2014, 192, 5459–5468. [Google Scholar] [CrossRef] [Green Version]
- Absher, D.M.; Li, X.; Waite, L.L.; Gibson, A.; Roberts, K.; Edberg, J.; Chatham, W.W.; Kimberly, R.P. Genome-Wide DNA Methylation Analysis of Systemic Lupus Erythematosus Reveals Persistent Hypomethylation of Interferon Genes and Compositional Changes to CD4+ T-Cell Populations. PLoS Genet. 2013, 9, e1003678. [Google Scholar] [CrossRef] [Green Version]
- Theofilopoulos, A.N.; Baccala, R.; Beutler, B.; Kono, D.H. Type I Interferons (α/β) in Immunity and Autoimmunity. Annu. Rev. Immunol. 2005, 23, 307–335. [Google Scholar] [CrossRef]
- Gresser, I. Biologic Effects of Interferons. J. Investig. Dermatol. 1990, 95, S66–S71. [Google Scholar] [CrossRef] [Green Version]
- Gürtler, C.; Bowie, A.G. Innate Immune Detection of Microbial Nucleic Acids. Trends Microbiol. 2013, 21, 413–420. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, Interferon-like Cytokines, and Their Receptors. Immunol. Rev. 2004, 202, 8–32. [Google Scholar] [CrossRef]
- López de Padilla, C.M.; Niewold, T.B. The Type I Interferons: Basic Concepts and Clinical Relevance in Immune-Mediated Inflammatory Diseases. Gene 2016, 576, 14–21. [Google Scholar] [CrossRef]
- Stanifer, M.L.; Pervolaraki, K.; Boulant, S. Differential Regulation of Type I and Type III Interferon Signaling. Int. J. Mol. Sci. 2019, 20, 1445. [Google Scholar] [CrossRef] [Green Version]
- Hakkim, A.; Fürnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of Neutrophil Extracellular Trap Degradation Is Associated with Lupus Nephritis. Proc. Natl. Acad. Sci. USA 2010, 107, 9813–9818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leffler, J.; Ciacma, K.; Gullstrand, B.; Bengtsson, A.A.; Martin, M.; Blom, A.M. A Subset of Patients with Systemic Lupus Erythematosus Fails to Degrade DNA from Multiple Clinically Relevant Sources. Arthritis Res. 2015, 17, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chyuan, I.-T.; Tzeng, H.-T.; Chen, J.-Y. Signaling Pathways of Type I and Type III Interferons and Targeted Therapies in Systemic Lupus Erythematosus. Cells 2019, 8, 963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkon, K.B.; Wiedeman, A. Type I IFN System in the Development and Manifestations of SLE. Curr. Opin. Rheumatol. 2012, 24, 499–505. [Google Scholar] [CrossRef]
- Matsuoka, N.; Fujita, Y.; Temmoku, J.; Furuya, M.Y.; Asano, T.; Sato, S.; Matsumoto, H.; Kobayashi, H.; Watanabe, H.; Suzuki, E.; et al. Galectin-9 as a Biomarker for Disease Activity in Systemic Lupus Erythematosus. PLoS ONE 2020, 15, e0227069. [Google Scholar] [CrossRef] [Green Version]
- Chasset, F.; Arnaud, L. Targeting Interferons and Their Pathways in Systemic Lupus Erythematosus. Autoimmun. Rev. 2018, 17, 44–52. [Google Scholar] [CrossRef]
- Macri, C.; Pang, E.S.; Patton, T.; O’Keeffe, M. Dendritic Cell Subsets. Semin. Cell Dev. Biol. 2018, 84, 11–21. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. TLR Signaling. Cell Death Differ. 2006, 13, 816–825. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, A.; Ito, T.; Kibata, K.; Inagaki-Katashiba, N.; Amuro, H.; Nishizawa, T.; Son, Y.; Ozaki, Y.; Nomura, S. Serum High-Mobility Group Box 1 Is Correlated with Interferon-α and May Predict Disease Activity in Patients with Systemic Lupus Erythematosus. Lupus 2019, 28, 1120–1127. [Google Scholar] [CrossRef]
- Eloranta, M.-L.; Alm, G.V.; Rönnblom, L. Disease Mechanisms in Rheumatology-Tools and Pathways: Plasmacytoid Dendritic Cells and Their Role in Autoimmune Rheumatic Diseases. Arthritis Rheum. 2013, 65, 853–863. [Google Scholar] [CrossRef]
- Henault, J.; Riggs, J.M.; Karnell, J.L.; Liarski, V.M.; Li, J.; Shirinian, L.; Xu, L.; Casey, K.A.; Smith, M.A.; Khatry, D.B.; et al. Self-Reactive IgE Exacerbates Interferon Responses Associated with Autoimmunity. Nat. Immunol. 2016, 17, 196–203. [Google Scholar] [CrossRef] [Green Version]
- Wardowska, A. The Epigenetic Face of Lupus: Focus on Antigen-Presenting Cells. Int. Immunopharmacol. 2020, 81, 106262. [Google Scholar] [CrossRef]
- Fiore, N.; Castellano, G.; Blasi, A.; Capobianco, C.; Loverre, A.; Montinaro, V.; Netti, S.; Torres, D.; Manno, C.; Grandaliano, G.; et al. Immature Myeloid and Plasmacytoid Dendritic Cells Infiltrate Renal Tubulointerstitium in Patients with Lupus Nephritis. Mol. Immunol. 2008, 45, 259–265. [Google Scholar] [CrossRef]
- Kil, L.P.; Hendriks, R.W. Aberrant B Cell Selection and Activation in Systemic Lupus Erythematosus. Int. Rev. Immunol. 2013, 32, 445–470. [Google Scholar] [CrossRef]
- Mok, C.C.; Lau, C.S. Pathogenesis of Systemic Lupus Erythematosus. J. Clin. Pathol. 2003, 56, 481–490. [Google Scholar] [CrossRef] [Green Version]
- Fritzler, M.J. Clinical Relevance of Autoantibodies in Systemic Rheumatic Diseases. Mol. Biol. Rep. 1996, 23, 133–145. [Google Scholar] [CrossRef]
- Meffre, E. The Establishment of Early B Cell Tolerance in Humans: Lessons from Primary Immunodeficiency Diseases. Ann. N. Y. Acad. Sci. 2011, 1246, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Netti, G.S.; Infante, B.; Spadaccino, F.; Godeas, G.; Corallo, M.G.; Prisciandaro, C.; Croce, L.; Rotondi, M.; Gesualdo, L.; Stallone, G.; et al. Serum Levels of BAFF and APRIL Predict Clinical Response in Anti-PLA2R-Positive Primary Membranous Nephropathy. J. Immunol. Res. 2019, 2019, 8483650. [Google Scholar] [CrossRef] [Green Version]
- Girschick, H.J.; Grammer, A.C.; Nanki, T.; Vazquez, E.; Lipsky, P.E. Expression of Recombination Activating Genes 1 and 2 in Peripheral B Cells of Patients with Systemic Lupus Erythematosus. Arthritis Rheum. 2002, 46, 1255–1263. [Google Scholar] [CrossRef]
- López, P.; Rodríguez-Carrio, J.; Caminal-Montero, L.; Mozo, L.; Suárez, A. A Pathogenic IFNα, BLyS and IL-17 Axis in Systemic Lupus Erythematosus Patients. Sci. Rep. 2016, 6, 20651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samotij, D.; Reich, A. Biologics in the Treatment of Lupus Erythematosus: A Critical Literature Review. Biomed. Res. Int. 2019, 2019, 8142368. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, M.I.; Catalan-Dibene, J.; Zlotnik, A. B Cells Responses and Cytokine Production Are Regulated by Their Immune Microenvironment. Cytokine 2015, 74, 318–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sjöstrand, M.; Johansson, A.; Aqrawi, L.; Olsson, T.; Wahren-Herlenius, M.; Espinosa, A. The Expression of BAFF Is Controlled by IRF Transcription Factors. J. Immunol. 2016, 196, 91–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, R.; Lyons, A.B.; Roberts, D.; Wong, M.-X.; Bartley, P.A.; Jackson, D.E. Platelet Endothelial Cell Adhesion Molecule-1 (PECAM-1/CD31) Acts as a Regulator of B-Cell Development, B-Cell Antigen Receptor (BCR)–Mediated Activation, and Autoimmune Disease. Blood 2002, 100, 184–193. [Google Scholar] [CrossRef]
- Pellefigues, C.; Charles, N. The Deleterious Role of Basophils in Systemic Lupus Erythematosus. Curr. Opin. Immunol. 2013, 25, 704–711. [Google Scholar] [CrossRef] [Green Version]
- Craft, J.E. Follicular Helper T Cells in Immunity and Systemic Autoimmunity. Nat. Rev. Rheumatol. 2012, 8, 337–347. [Google Scholar] [CrossRef] [Green Version]
- Humrich, J.Y.; von Spee-Mayer, C.; Siegert, E.; Alexander, T.; Hiepe, F.; Radbruch, A.; Burmester, G.-R.; Riemekasten, G. Rapid Induction of Clinical Remission by Low-Dose Interleukin-2 in a Patient with Refractory SLE. Ann. Rheum. Dis. 2015, 74, 791–792. [Google Scholar] [CrossRef]
- Hagberg, N.; Berggren, O.; Leonard, D.; Weber, G.; Bryceson, Y.T.; Alm, G.V.; Eloranta, M.-L.; Rönnblom, L. IFN-α Production by Plasmacytoid Dendritic Cells Stimulated with RNA-Containing Immune Complexes Is Promoted by NK Cells via MIP-1β and LFA-1. J. Immunol. 2011, 186, 5085–5094. [Google Scholar] [CrossRef] [Green Version]
- Eloranta, M.-L.; Lövgren, T.; Finke, D.; Mathsson, L.; Rönnelid, J.; Kastner, B.; Alm, G.V.; Rönnblom, L. Regulation of the Interferon-α Production Induced by RNA-Containing Immune Complexes in Plasmacytoid Dendritic Cells. Arthritis Rheum. 2009, 60, 2418–2427. [Google Scholar] [CrossRef]
- González-Navajas, J.M.; Lee, J.; David, M.; Raz, E. Immunomodulatory Functions of Type I Interferons. Nat. Rev. Immunol. 2012, 12, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Dzionek, A.; Sohma, Y.; Nagafune, J.; Cella, M.; Colonna, M.; Facchetti, F.; Günther, G.; Johnston, I.; Lanzavecchia, A.; Nagasaka, T.; et al. BDCA-2, a Novel Plasmacytoid Dendritic Cell-Specific Type II C-Type Lectin, Mediates Antigen Capture and Is a Potent Inhibitor of Interferon Alpha/Beta Induction. J. Exp. Med. 2001, 194, 1823–1834. [Google Scholar] [CrossRef]
- Cao, W.; Rosen, D.B.; Ito, T.; Bover, L.; Bao, M.; Watanabe, G.; Yao, Z.; Zhang, L.; Lanier, L.L.; Liu, Y.-J. Plasmacytoid Dendritic Cell-Specific Receptor ILT7-Fc EpsilonRI Gamma Inhibits Toll-like Receptor-Induced Interferon Production. J. Exp. Med. 2006, 203, 1399–1405. [Google Scholar] [CrossRef] [Green Version]
- Kemp, M.G.; Lindsey-Boltz, L.A.; Sancar, A. UV Light Potentiates STING (Stimulator of Interferon Genes)-Dependent Innate Immune Signaling through Deregulation of ULK1 (Unc51-like Kinase 1). J. Biol. Chem. 2015, 290, 12184–12194. [Google Scholar] [CrossRef] [Green Version]
- Barbhaiya, M.; Costenbader, K.H. Ultraviolet Radiation and Systemic Lupus Erythematosus. Lupus 2014, 23, 588–595. [Google Scholar] [CrossRef]
- Almaani, S.; Meara, A.; Rovin, B.H. Update on Lupus Nephritis. Clin. J. Am. Soc. Nephrol. 2017, 12, 825–835. [Google Scholar] [CrossRef] [Green Version]
- Aziz, F.; Chaudhary, K. Lupus Nephritis: A Treatment Update. Curr. Clin. Pharm. 2018, 13, 4–13. [Google Scholar] [CrossRef]
- Stokes, M.B.; D’Agati, V.D. Classification of Lupus Nephritis; Time for a Change? Adv. Chronic Kidney Dis. 2019, 26, 323–329. [Google Scholar] [CrossRef]
- Weening, J.J.; D’agati, V.D.; Schwartz, M.M.; Seshan, S.V.; Alpers, C.E.; Appel, G.B.; Balow, J.E.; Bruijn, J.A.N.A.; Cook, T.; Ferrario, F.; et al. The Classification of Glomerulonephritis in Systemic Lupus Erythematosus Revisited. Kidney Int. 2004, 65, 521–530. [Google Scholar] [CrossRef] [Green Version]
- Bajema, I.M.; Wilhelmus, S.; Alpers, C.E.; Bruijn, J.A.; Colvin, R.B.; Cook, H.T.; D’Agati, V.D.; Ferrario, F.; Haas, M.; Jennette, J.C.; et al. Revision of the International Society of Nephrology/Renal Pathology Society Classification for Lupus Nephritis: Clarification of Definitions, and Modified National Institutes of Health Activity and Chronicity Indices. Kidney Int. 2018, 93, 789–796. [Google Scholar] [CrossRef]
- Lech, M.; Anders, H.-J. The Pathogenesis of Lupus Nephritis. J. Am. Soc. Nephrol. 2013, 24, 1357–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, D.Y.H.; Lai, K.N. Pathogenesis of Renal Disease in Systemic Lupus Erythematosus--the Role of Autoantibodies and Lymphocytes Subset Abnormalities. Int. J. Mol. Sci. 2015, 16, 7917–7931. [Google Scholar] [CrossRef] [PubMed]
- Birmingham, D.J.; Hebert, L.A. The Complement System in Lupus Nephritis. Semin. Nephrol. 2015, 35, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Walport, M.J. Complement and Systemic Lupus Erythematosus. Arthritis Res. 2002, 4 (Suppl. 3), S279–S293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Łukawska, E.; Polcyn-Adamczak, M.; Niemir, Z.I. The Role of the Alternative Pathway of Complement Activation in Glomerular Diseases. Clin. Exp. Med. 2018, 18, 297–318. [Google Scholar] [CrossRef] [Green Version]
- Song, D.; Guo, W.; Wang, F.; Li, Y.; Song, Y.; Yu, F.; Zhao, M. Complement Alternative Pathway׳s Activation in Patients with Lupus Nephritis. Am. J. Med. Sci. 2017, 353, 247–257. [Google Scholar] [CrossRef]
- Edelbauer, M.; Kshirsagar, S.; Riedl, M.; Haffner, D.; Billing, H.; Tönshoff, B.; Ross, S.; Dötsch, J.; Amon, O.; Fehrenbach, H.; et al. Markers of Childhood Lupus Nephritis Indicating Disease Activity. Pediatr. Nephrol. 2010, 26, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Nauta, A.J.; Castellano, G.; Xu, W.; Woltman, A.M.; Borrias, M.C.; Daha, M.R.; van Kooten, C.; Roos, A. Opsonization with C1q and Mannose-Binding Lectin Targets Apoptotic Cells to Dendritic Cells. J. Immunol. 2004, 173, 3044–3050. [Google Scholar] [CrossRef] [Green Version]
- Turley, A.J.; Gathmann, B.; Bangs, C.; Bradbury, M.; Seneviratne, S.; Gonzalez-Granado, L.I.; Hackett, S.; Kutukculer, N.; Alachkar, H.; Hambleton, S.; et al. Spectrum and Management of Complement Immunodeficiencies (Excluding Hereditary Angioedema) Across Europe. J. Clin. Immunol. 2015, 35, 199–205. [Google Scholar] [CrossRef]
- Davidson, A. What Is Damaging the Kidney in Lupus Nephritis? Nat. Rev. Rheumatol. 2016, 12, 143–153. [Google Scholar] [CrossRef]
- Mitchell, D.A.; Pickering, M.C.; Warren, J.; Fossati-Jimack, L.; Cortes-Hernandez, J.; Cook, H.T.; Botto, M.; Walport, M.J. C1q Deficiency and Autoimmunity: The Effects of Genetic Background on Disease Expression. J. Immunol. 2002, 168, 2538–2543. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Hu, Q.; Madri, J.A.; Rollins, S.A.; Chodera, A.; Matis, L.A. Amelioration of Lupus-like Autoimmune Disease in NZB/WF1 Mice after Treatment with a Blocking Monoclonal Antibody Specific for Complement Component C5. Proc. Natl. Acad. Sci. USA 1996, 93, 8563–8568. [Google Scholar] [CrossRef] [Green Version]
- Castellano, G.; Trouw, L.A.; Fiore, N.; Daha, M.R.; Schena, F.P.; van Kooten, C. Infiltrating Dendritic Cells Contribute to Local Synthesis of C1q in Murine and Human Lupus Nephritis. Mol. Immunol. 2010, 47, 2129–2137. [Google Scholar] [CrossRef]
- Thurman, J.M.; Kulik, L.; Orth, H.; Wong, M.; Renner, B.; Sargsyan, S.A.; Mitchell, L.M.; Hourcade, D.E.; Hannan, J.P.; Kovacs, J.M.; et al. Detection of Complement Activation Using Monoclonal Antibodies against C3d. J. Clin. Investig. 2013, 123, 2218–2230. [Google Scholar] [CrossRef] [Green Version]
- Sciascia, S.; Radin, M.; Yazdany, J.; Tektonidou, M.; Cecchi, I.; Roccatello, D.; Dall’Era, M. Expanding the Therapeutic Options for Renal Involvement in Lupus: Eculizumab, Available Evidence. Rheumatol. Int. 2017, 37, 1249–1255. [Google Scholar] [CrossRef]
- Oke, V.; Gunnarsson, I.; Dorschner, J.; Eketjäll, S.; Zickert, A.; Niewold, T.B.; Svenungsson, E. High Levels of Circulating Interferons Type I, Type II and Type III Associate with Distinct Clinical Features of Active Systemic Lupus Erythematosus. Arthritis Res. 2019, 21, 107. [Google Scholar] [CrossRef] [Green Version]
- Niewold, T.B.; Hua, J.; Lehman, T.J.A.; Harley, J.B.; Crow, M.K. High Serum IFN-Alpha Activity Is a Heritable Risk Factor for Systemic Lupus Erythematosus. Genes Immun. 2007, 8, 492–502. [Google Scholar] [CrossRef]
- Qin, L.; Lv, J.; Zhou, X.; Hou, P.; Yang, H.; Zhang, H. Association of IRF5 Gene Polymorphisms and Lupus Nephritis in a Chinese Population. Nephrology 2010, 15, 710–713. [Google Scholar] [CrossRef]
- Ko, K.; Franek, B.S.; Marion, M.; Kaufman, K.M.; Langefeld, C.D.; Harley, J.B.; Niewold, T.B. Genetic Ancestry, Serum Interferon-α Activity, and Autoantibodies in Systemic Lupus Erythematosus. J. Rheumatol. 2012, 39, 1238–1240. [Google Scholar] [CrossRef] [Green Version]
- Yung, S.; Chan, T.M. Anti-DsDNA Antibodies and Resident Renal Cells—Their Putative Roles in Pathogenesis of Renal Lesions in Lupus Nephritis. Clin. Immunol. 2017, 185, 40–50. [Google Scholar] [CrossRef]
- Murayama, G.; Furusawa, N.; Chiba, A.; Yamaji, K.; Tamura, N.; Miyake, S. Enhanced IFN-α Production Is Associated with Increased TLR7 Retention in the Lysosomes of Palasmacytoid Dendritic Cells in Systemic Lupus Erythematosus. Arthritis Res. 2017, 19, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakata, K.; Nakayamada, S.; Miyazaki, Y.; Kubo, S.; Ishii, A.; Nakano, K.; Tanaka, Y. Up-Regulation of TLR7-Mediated IFN-α Production by Plasmacytoid Dendritic Cells in Patients with Systemic Lupus Erythematosus. Front Immunol. 2018, 9, 1957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patole, P.S.; Gröne, H.-J.; Segerer, S.; Ciubar, R.; Belemezova, E.; Henger, A.; Kretzler, M.; Schlöndorff, D.; Anders, H.-J. Viral Double-Stranded RNA Aggravates Lupus Nephritis through Toll-Like Receptor 3 on Glomerular Mesangial Cells and Antigen-Presenting Cells. J. Am. Soc. Nephrol. 2005, 16, 1326–1338. [Google Scholar] [CrossRef] [PubMed]
- Patole, P.S.; Pawar, R.D.; Lech, M.; Zecher, D.; Schmidt, H.; Segerer, S.; Ellwart, A.; Henger, A.; Kretzler, M.; Anders, H.-J. Expression and Regulation of Toll-like Receptors in Lupus-like Immune Complex Glomerulonephritis of MRL-Fas(Lpr) Mice. Nephrol. Dial. Transplant. 2006, 21, 3062–3073. [Google Scholar] [CrossRef] [PubMed]
- Benigni, A.; Caroli, C.; Longaretti, L.; Gagliardini, E.; Zoja, C.; Galbusera, M.; Moioli, D.; Romagnani, P.; Tincani, A.; Andreoli, L.; et al. Involvement of Renal Tubular Toll-like Receptor 9 in the Development of Tubulointerstitial Injury in Systemic Lupus. Arthritis Rheum. 2007, 56, 1569–1578. [Google Scholar] [CrossRef]
- Ciferska, H.; Horak, P.; Konttinen, Y.T.; Krejci, K.; Tichy, T.; Hermanova, Z.; Zadrazil, J. Expression of Nucleic Acid Binding Toll-like Receptors in Control, Lupus and Transplanted Kidneys—A Preliminary Pilot Study. Lupus 2008, 17, 580–585. [Google Scholar] [CrossRef]
- Papadimitraki, E.D.; Tzardi, M.; Bertsias, G.; Sotsiou, E.; Boumpas, D.T. Glomerular Expression of Toll-like Receptor-9 in Lupus Nephritis but Not in Normal Kidneys: Implications for the Amplification of the Inflammatory Response. Lupus 2009, 18, 831–835. [Google Scholar] [CrossRef]
- Liu, Z.; Bethunaickan, R.; Huang, W.; Ramanujam, M.; Madaio, M.P.; Davidson, A. IFN-α Confers Resistance of Systemic Lupus Erythematosus Nephritis to Therapy in NZB/W F1 Mice. J. Immunol. 2011, 187, 1506–1513. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, C.; Tsumiyama, K.; Uchimura, C.; Honda, E.; Miyazaki, Y.; Sakurai, K.; Miura, Y.; Hashiramoto, A.; Felsher, D.W.; Shiozawa, S. Conditional Upregulation of IFN-α Alone Is Sufficient to Induce Systemic Lupus Erythematosus. J. Immunol. 2019, 203, 835–843. [Google Scholar] [CrossRef]
- Santiago-Raber, M.-L.; Baccala, R.; Haraldsson, K.M.; Choubey, D.; Stewart, T.A.; Kono, D.H.; Theofilopoulos, A.N. Type-I Interferon Receptor Deficiency Reduces Lupus-like Disease in NZB Mice. J. Exp. Med. 2003, 197, 777–788. [Google Scholar] [CrossRef]
- Braun, D. Type I Interferon Controls the Onset and Severity of Autoimmune Manifestations in Lpr Mice. J. Autoimmun. 2003, 20, 15–25. [Google Scholar] [CrossRef]
- Tada, Y.; Kondo, S.; Aoki, S.; Koarada, S.; Inoue, H.; Suematsu, R.; Ohta, A.; Mak, T.W.; Nagasawa, K. Interferon Regulatory Factor 5 Is Critical for the Development of Lupus in MRL/Lpr Mice. Arthritis Rheum. 2011, 63, 738–748. [Google Scholar] [CrossRef]
- Yasuda, K.; Watkins, A.A.; Kochar, G.S.; Wilson, G.E.; Laskow, B.; Richez, C.; Bonegio, R.G.; Rifkin, I.R. Interferon Regulatory Factor-5 Deficiency Ameliorates Disease Severity in the MRL/Lpr Mouse Model of Lupus in the Absence of a Mutation in DOCK2. PLoS ONE 2014, 9, e103478. [Google Scholar] [CrossRef]
- Dai, C.; Wang, H.; Sung, S.-S.J.; Sharma, R.; Kannapell, C.; Han, W.; Wang, Q.; Davidson, A.; Gaskin, F.; Fu, S.M. Interferon Alpha on NZM2328.Lc1R27: Enhancing Autoimmunity and Immune Complex-Mediated Glomerulonephritis without End Stage Renal Failure. Clin. Immunol. 2014, 154, 66–71. [Google Scholar] [CrossRef] [Green Version]
- Kirou, K.A.; Lee, C.; George, S.; Louca, K.; Peterson, M.G.E.; Crow, M.K. Activation of the Interferon-α Pathway Identifies a Subgroup of Systemic Lupus Erythematosus Patients with Distinct Serologic Features and Active Disease. Arthritis Rheum. 2005, 52, 1491–1503. [Google Scholar] [CrossRef]
- Feng, X.; Wu, H.; Grossman, J.M.; Hanvivadhanakul, P.; FitzGerald, J.D.; Park, G.S.; Dong, X.; Chen, W.; Kim, M.H.; Weng, H.H.; et al. Association of Increased Interferon-Inducible Gene Expression with Disease Activity and Lupus Nephritis in Patients with Systemic Lupus Erythematosus. Arthritis Rheum. 2006, 54, 2951–2962. [Google Scholar] [CrossRef]
- Peterson, K.S.; Huang, J.-F.; Zhu, J.; D’Agati, V.; Liu, X.; Miller, N.; Erlander, M.G.; Jackson, M.R.; Winchester, R.J. Characterization of Heterogeneity in the Molecular Pathogenesis of Lupus Nephritis from Transcriptional Profiles of Laser-Captured Glomeruli. J. Clin. Investig. 2004, 113, 1722–1733. [Google Scholar] [CrossRef] [Green Version]
- Tucci, M.; Quatraro, C.; Lombardi, L.; Pellegrino, C.; Dammacco, F.; Silvestris, F. Glomerular Accumulation of Plasmacytoid Dendritic Cells in Active Lupus Nephritis: Role of Interleukin-18. Arthritis Rheum. 2007, 58, 251–262. [Google Scholar] [CrossRef]
- Qi, Y.-Y.; Zhou, X.-J.; Cheng, F.-J.; Hou, P.; Ren, Y.-L.; Wang, S.-X.; Zhao, M.-H.; Yang, L.; Martinez, J.; Zhang, H. Increased Autophagy Is Cytoprotective against Podocyte Injury Induced by Antibody and Interferon-α in Lupus Nephritis. Ann. Rheum. Dis. 2018, 77, 1799–1809. [Google Scholar] [CrossRef]
- Migliorini, A.; Angelotti, M.L.; Mulay, S.R.; Kulkarni, O.O.; Demleitner, J.; Dietrich, A.; Sagrinati, C.; Ballerini, L.; Peired, A.; Shankland, S.J.; et al. The Antiviral Cytokines IFN-α and IFN-β Modulate Parietal Epithelial Cells and Promote Podocyte Loss. Am. J. Pathol. 2013, 183, 431–440. [Google Scholar] [CrossRef]
- Holzinger, D.; Jorns, C.; Stertz, S.; Boisson-Dupuis, S.; Thimme, R.; Weidmann, M.; Casanova, J.-L.; Haller, O.; Kochs, G. Induction of MxA Gene Expression by Influenza A Virus Requires Type I or Type III Interferon Signaling. J. Virol. 2007, 81, 7776–7785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellano, G.; Cafiero, C.; Divella, C.; Sallustio, F.; Gigante, M.; Pontrelli, P.; de Palma, G.; Rossini, M.; Grandaliano, G.; Gesualdo, L. Local Synthesis of Interferon-Alpha in Lupus Nephritis Is Associated with Type I Interferons Signature and LMP7 Induction in Renal Tubular Epithelial Cells. Arthritis Res. 2015, 17, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arazi, A.; Rao, D.A.; Berthier, C.C.; Davidson, A.; Liu, Y.; Hoover, P.J.; Chicoine, A.; Eisenhaure, T.M.; Jonsson, A.H.; Li, S.; et al. The Immune Cell Landscape in Kidneys of Patients with Lupus Nephritis. Nat. Immunol. 2019, 20, 902–914. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Sun, S.-C. NF-ΚB in Inflammation and Renal Diseases. Cell Biosci. 2015, 5, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.; Sinniah, R.; Hsu, S.I.-H. Pathogenic Role of NF-KappaB Activation in Tubulointerstitial Inflammatory Lesions in Human Lupus Nephritis. J. Histochem. Cytochem. 2008, 56, 517–529. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Sinniah, R.; Hsu, S.I.-H. Renal Cell Apoptosis and Proliferation May Be Linked to Nuclear Factor–ΚB Activation and Expression of Inducible Nitric Oxide Synthase in Patients with Lupus Nephritis. Hum. Pathol. 2006, 37, 637–647. [Google Scholar] [CrossRef]
- Izquierdo, M.C.; Perez-Gomez, M.V.; Sanchez-Nino, M.D.; Sanz, A.B.; Ruiz-Andres, O.; Poveda, J.; Moreno, J.A.; Egido, J.; Ortiz, A. Klotho, Phosphate and Inflammation/Ageing in Chronic Kidney Disease. Nephrol. Dial. Transplant. 2012, 27, iv6–iv10. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Fergusson, M.M.; Castilho, R.M.; Liu, J.; Cao, L.; Chen, J.; Malide, D.; Rovira, I.I.; Schimel, D.; Kuo, C.J.; et al. Augmented Wnt Signaling in a Mammalian Model of Accelerated Aging. Science (1979) 2007, 317, 803–806. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, H.; Yoshida, T.; Shiohira, S.; Kohei, J.; Mitobe, M.; Kurosu, H.; Kuro-o, M.; Nitta, K.; Tsuchiya, K. Reduced Klotho Expression Level in Kidney Aggravates Renal Interstitial Fibrosis. Am. J. Physiol. Ren. Physiol. 2012, 302, F1252–F1264. [Google Scholar] [CrossRef]
- Satoh, M.; Nagasu, H.; Morita, Y.; Yamaguchi, T.P.; Kanwar, Y.S.; Kashihara, N. Klotho Protects against Mouse Renal Fibrosis by Inhibiting Wnt Signaling. Am. J. Physiol. Ren. Physiol. 2012, 303, F1641–F1651. [Google Scholar] [CrossRef]
- Gatto, M.; Zen, M.; Iaccarino, L.; Doria, A. New Therapeutic Strategies in Systemic Lupus Erythematosus Management. Nat. Rev. Rheumatol. 2018, 15, 30–48. [Google Scholar] [CrossRef]
- Klavdianou, K.; Lazarini, A.; Fanouriakis, A. Targeted Biologic Therapy for Systemic Lupus Erythematosus: Emerging Pathways and Drug Pipeline. BioDrugs 2020, 34, 133–147. [Google Scholar] [CrossRef]
- Furie, R.; Petri, M.; Zamani, O.; Cervera, R.; Wallace, D.J.; Tegzová, D.; Sanchez-Guerrero, J.; Schwarting, A.; Merrill, J.T.; Chatham, W.W.; et al. A Phase III, Randomized, Placebo-Controlled Study of Belimumab, a Monoclonal Antibody That Inhibits B Lymphocyte Stimulator, in Patients with Systemic Lupus Erythematosus. Arthritis Rheum. 2011, 63, 3918–3930. [Google Scholar] [CrossRef] [Green Version]
- Navarra, S.V.; Guzmán, R.M.; Gallacher, A.E.; Hall, S.; Levy, R.A.; Jimenez, R.E.; Li, E.K.-M.; Thomas, M.; Kim, H.-Y.; León, M.G.; et al. Efficacy and Safety of Belimumab in Patients with Active Systemic Lupus Erythematosus: A Randomised, Placebo-Controlled, Phase 3 Trial. Lancet 2011, 377, 721–731. [Google Scholar] [CrossRef]
- Furie, R.; Rovin, B.H.; Houssiau, F.; Malvar, A.; Teng, Y.K.O.; Contreras, G.; Amoura, Z.; Yu, X.; Mok, C.-C.; Santiago, M.B.; et al. Two-Year, Randomized, Controlled Trial of Belimumab in Lupus Nephritis. N. Engl. J. Med. 2020, 383, 1117–1128. [Google Scholar] [CrossRef]
- Rovin, B.H.; Furie, R.; Latinis, K.; Looney, R.J.; Fervenza, F.C.; Sanchez-Guerrero, J.; Maciuca, R.; Zhang, D.; Garg, J.P.; Brunetta, P.; et al. Efficacy and Safety of Rituximab in Patients with Active Proliferative Lupus Nephritis: The Lupus Nephritis Assessment with Rituximab Study. Arthritis Rheum. 2012, 64, 1215–1226. [Google Scholar] [CrossRef]
- Merrill, J.T.; Neuwelt, C.M.; Wallace, D.J.; Shanahan, J.C.; Latinis, K.M.; Oates, J.C.; Utset, T.O.; Gordon, C.; Isenberg, D.A.; Hsieh, H.-J.; et al. Efficacy and Safety of Rituximab in Moderately-to-Severely Active Systemic Lupus Erythematosus: The Randomized, Double-Blind, Phase II/III Systemic Lupus Erythematosus Evaluation of Rituximab Trial. Arthritis Rheum. 2010, 62, 222–233. [Google Scholar] [CrossRef] [Green Version]
- Almaani, S.; Rovin, B.H. B-Cell Therapy in Lupus Nephritis: An Overview. Nephrol. Dial. Transpl. 2019, 34, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Atisha-Fregoso, Y.; Malkiel, S.; Harris, K.M.; Byron, M.; Ding, L.; Kanaparthi, S.; Barry, W.T.; Gao, W.; Ryker, K.; Tosta, P.; et al. Phase II Randomized Trial of Rituximab Plus Cyclophosphamide Followed by Belimumab for the Treatment of Lupus Nephritis. Arthritis Rheumatol. 2021, 73, 121–131. [Google Scholar] [CrossRef]
- Kraaij, T.; Arends, E.J.; van Dam, L.S.; Kamerling, S.W.A.; van Daele, P.L.A.; Bredewold, O.W.; Ray, A.; Bakker, J.A.; Scherer, H.U.; Huizinga, T.J.W.; et al. Long-Term Effects of Combined B-Cell Immunomodulation with Rituximab and Belimumab in Severe, Refractory Systemic Lupus Erythematosus: 2-Year Results. Nephrol. Dial. Transpl. 2021, 36, 1474–1483. [Google Scholar] [CrossRef]
- Marcus, R.; Davies, A.; Ando, K.; Klapper, W.; Opat, S.; Owen, C.; Phillips, E.; Sangha, R.; Schlag, R.; Seymour, J.F.; et al. Obinutuzumab for the First-Line Treatment of Follicular Lymphoma. N. Engl. J. Med. 2017, 377, 1331–1344. [Google Scholar] [CrossRef] [PubMed]
- Furie, R.A.; Aroca, G.; Cascino, M.D.; Garg, J.P.; Rovin, B.H.; Alvarez, A.; Fragoso-Loyo, H.; Zuta-Santillan, E.; Schindler, T.; Brunetta, P.; et al. B-Cell Depletion with Obinutuzumab for the Treatment of Proliferative Lupus Nephritis: A Randomised, Double-Blind, Placebo-Controlled Trial. Ann. Rheum. Dis. 2022, 81, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Kostopoulou, M.; Pitsigavdaki, S.; Bertsias, G. Lupus Nephritis: Improving Treatment Options. Drugs 2022, 82, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Kalunian, K.C.; Merrill, J.T.; Maciuca, R.; McBride, J.M.; Townsend, M.J.; Wei, X.; Davis, J.C.; Kennedy, W.P. A Phase II Study of the Efficacy and Safety of Rontalizumab (RhuMAb Interferon-α) in Patients with Systemic Lupus Erythematosus (ROSE). Ann. Rheum. Dis. 2015, 75, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Morand, E.F.; Furie, R.; Tanaka, Y.; Bruce, I.N.; Askanase, A.D.; Richez, C.; Bae, S.-C.; Brohawn, P.Z.; Pineda, L.; Berglind, A.; et al. Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N. Engl. J. Med. 2020, 382, 211–221. [Google Scholar] [CrossRef]
- Furie, R.; Khamashta, M.; Merrill, J.T.; Werth, V.P.; Kalunian, K.; Brohawn, P.; Illei, G.G.; Drappa, J.; Wang, L.; Yoo, S.; et al. Anifrolumab, an Anti-Interferon-α Receptor Monoclonal Antibody, in Moderate-to-Severe Systemic Lupus Erythematosus. Arthritis Rheumatol. 2017, 69, 376–386. [Google Scholar] [CrossRef] [Green Version]
- Loncharich, M.F.; Anderson, C.W. Interferon Inhibition for Lupus with Anifrolumab: Critical Appraisal of the Evidence Leading to FDA Approval. ACR Open Rheumatol. 2022, 4, 486–491. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Infante, B.; Mercuri, S.; Dello Strologo, A.; Franzin, R.; Catalano, V.; Troise, D.; Cataldo, E.; Pontrelli, P.; Alfieri, C.; Binda, V.; et al. Unraveling the Link between Interferon-α and Systemic Lupus Erythematosus: From the Molecular Mechanisms to Target Therapies. Int. J. Mol. Sci. 2022, 23, 15998. https://doi.org/10.3390/ijms232415998
Infante B, Mercuri S, Dello Strologo A, Franzin R, Catalano V, Troise D, Cataldo E, Pontrelli P, Alfieri C, Binda V, et al. Unraveling the Link between Interferon-α and Systemic Lupus Erythematosus: From the Molecular Mechanisms to Target Therapies. International Journal of Molecular Sciences. 2022; 23(24):15998. https://doi.org/10.3390/ijms232415998
Chicago/Turabian StyleInfante, Barbara, Silvia Mercuri, Andrea Dello Strologo, Rossana Franzin, Valeria Catalano, Dario Troise, Emanuela Cataldo, Paola Pontrelli, Carlo Alfieri, Valentina Binda, and et al. 2022. "Unraveling the Link between Interferon-α and Systemic Lupus Erythematosus: From the Molecular Mechanisms to Target Therapies" International Journal of Molecular Sciences 23, no. 24: 15998. https://doi.org/10.3390/ijms232415998