
Integrative Application of Transcriptomics and Metabolomics Provides Insights into Unsynchronized Growth in Sea Cucumber (Stichopus monotuberculatus)
 , ,
, ,
Abstract
:1. Introduction
2. Results
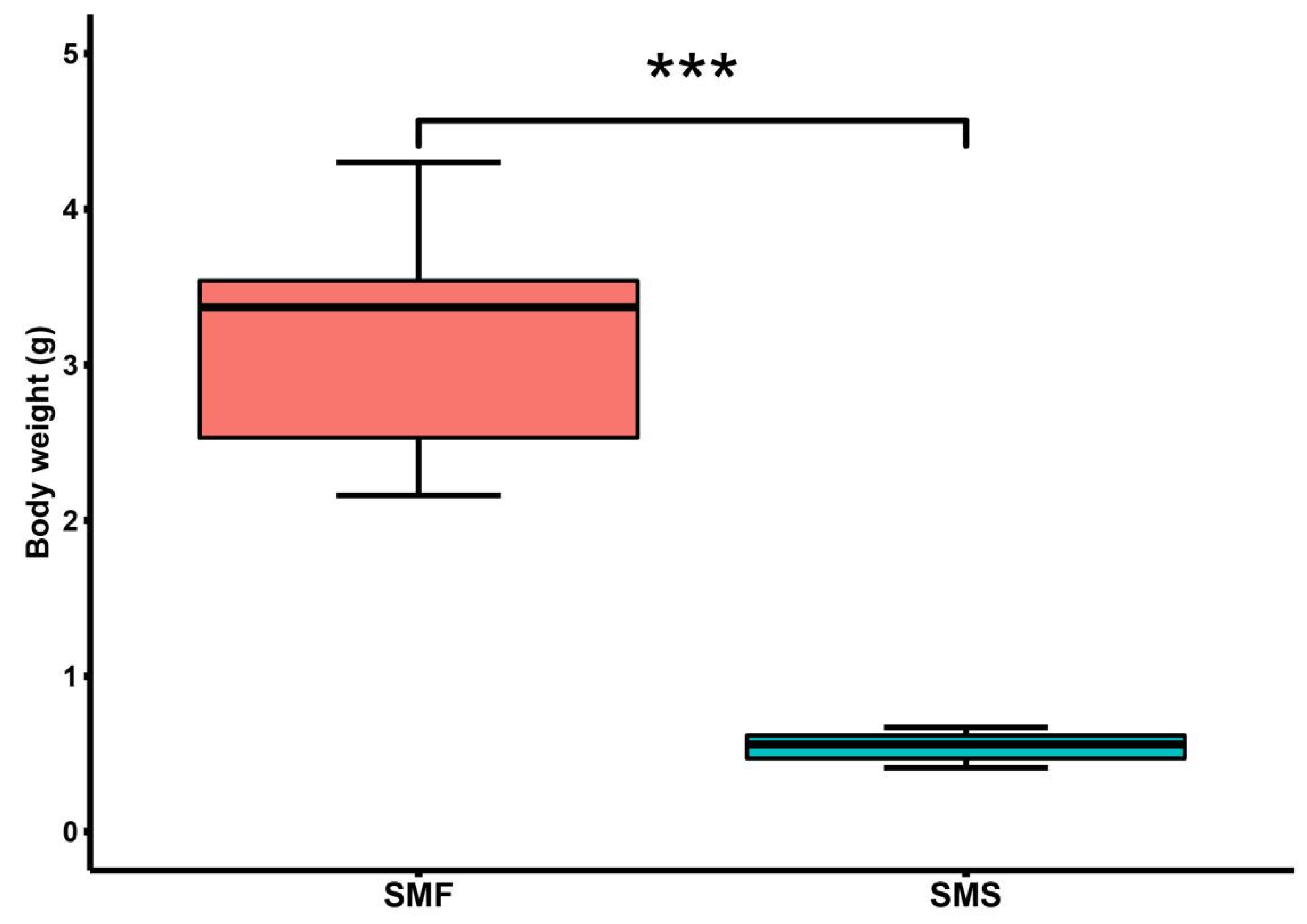
2.1. Significant Growth Performance Differences between SMF and SMS
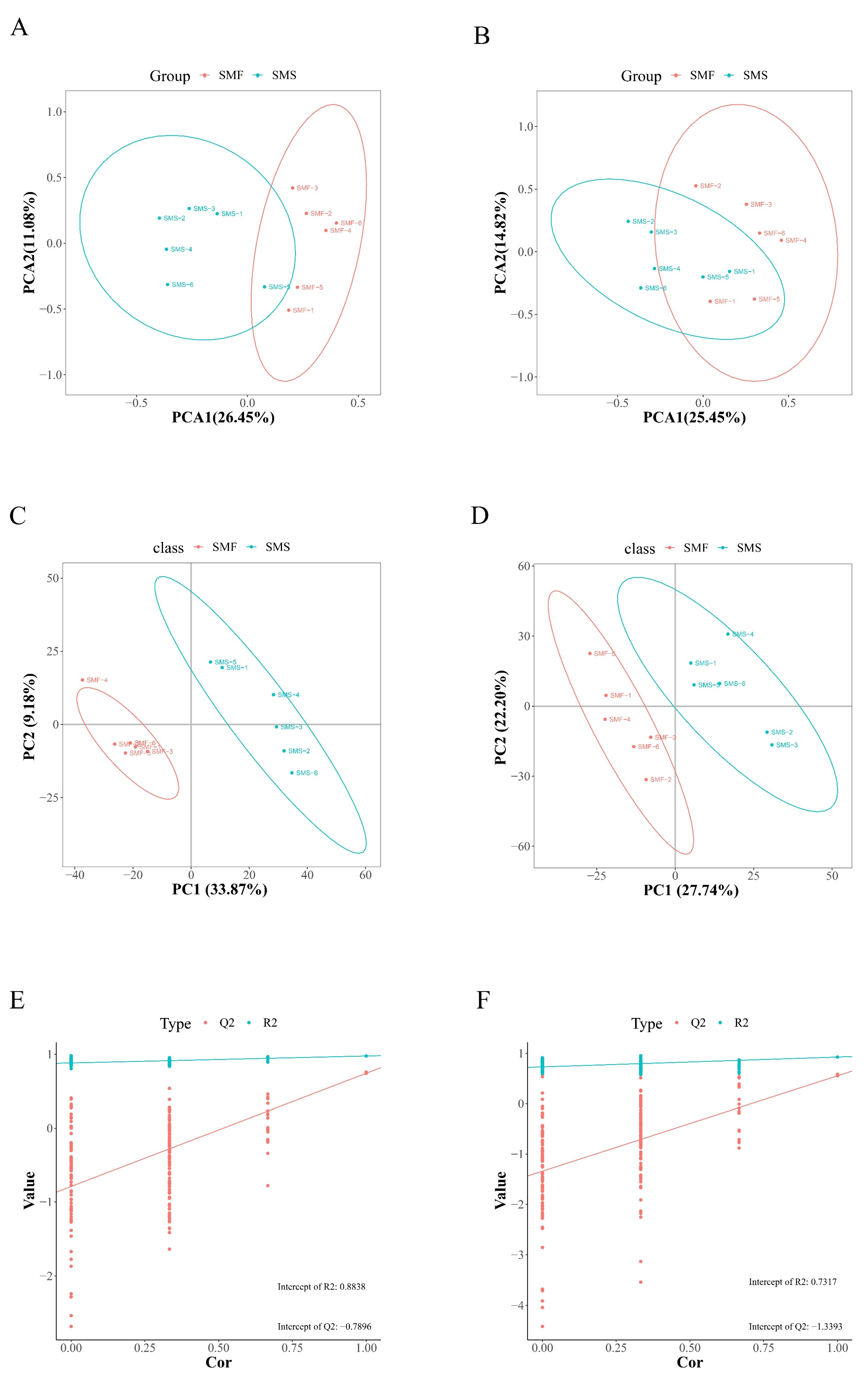
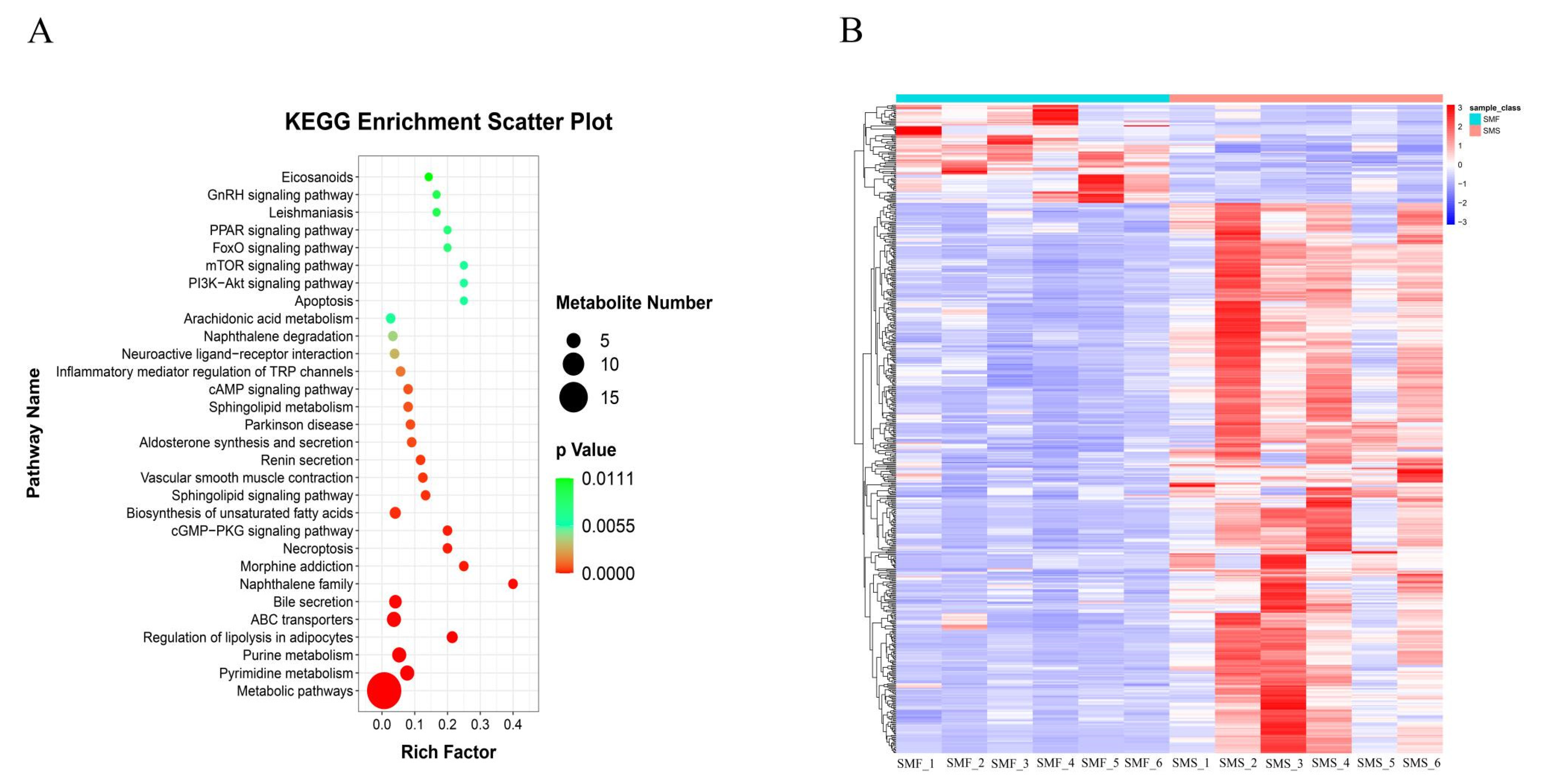
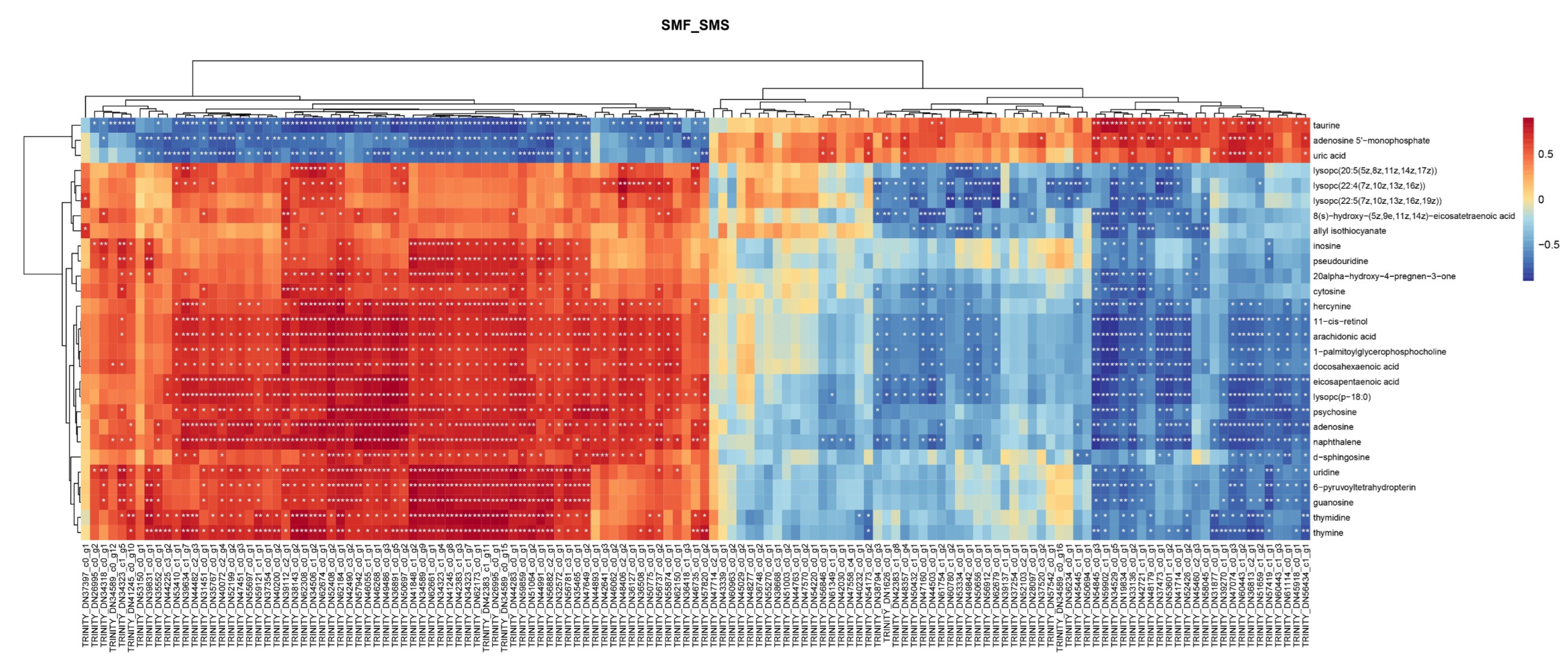
2.2. Metabolomics Analysis
2.3. Transcriptomic Analysis
2.3.1. Transcriptomic Data Sequencing and Quality Control
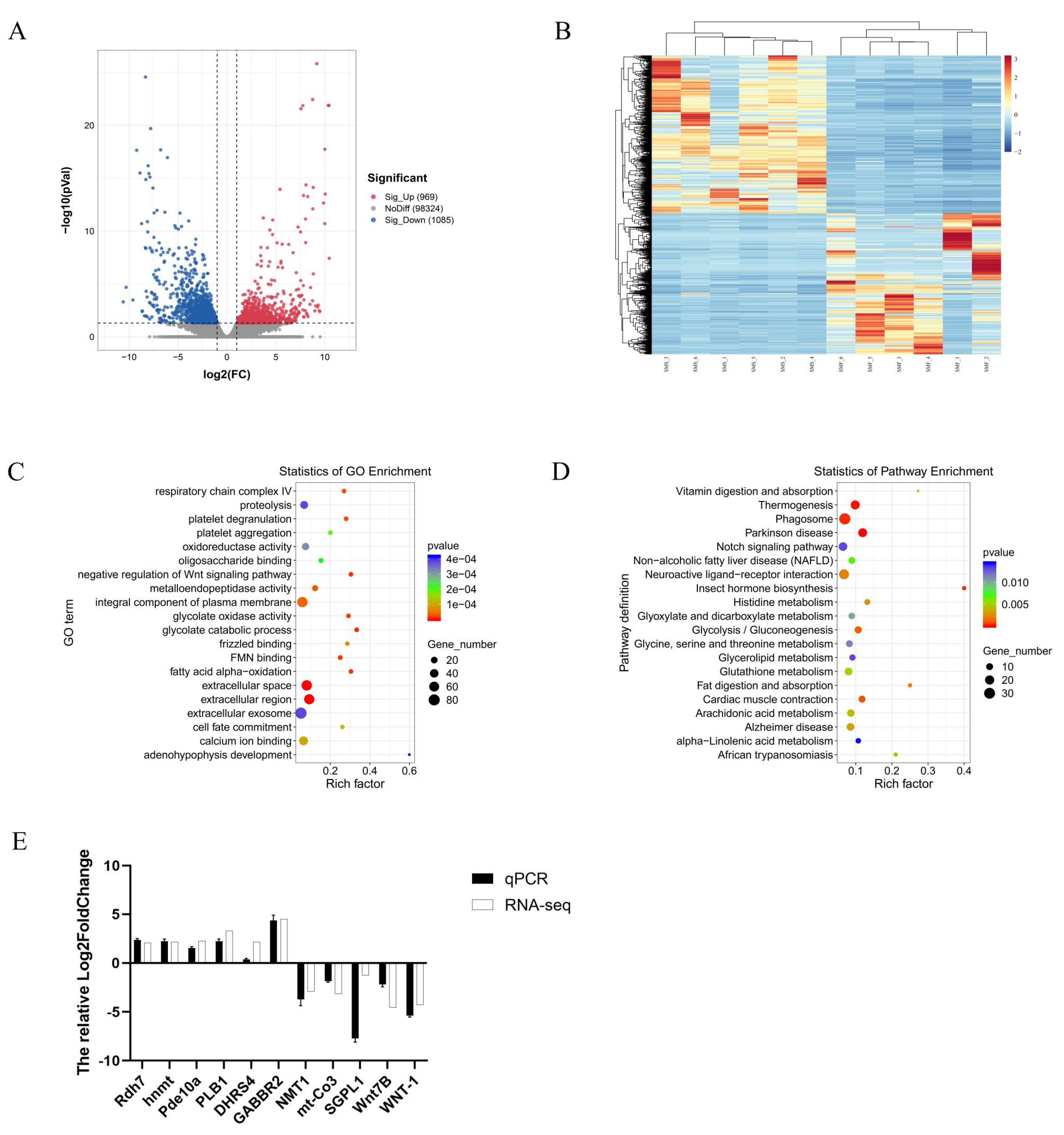
2.3.2. Identification of DEGs between the SMF and the SMS Group
2.3.3. GO and KEGG Enrichment Analysis of DEGs
2.3.4. Quantitative Real-Time PCR Validation of the DEGs
2.4. Integrative Analysis of Metabolomics and Transcriptomics
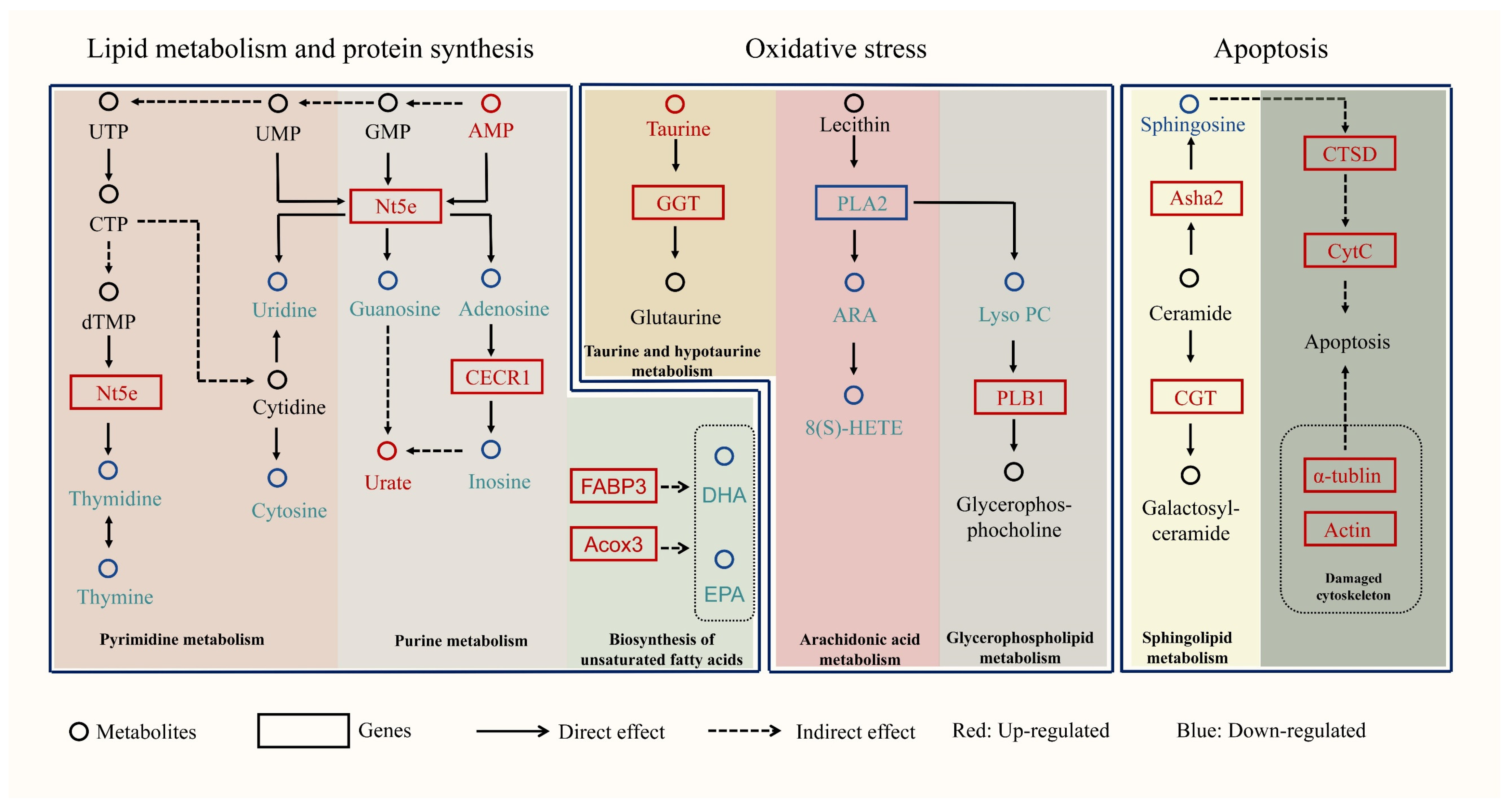
3. Discussion
3.1. The SMF Group of S. monotuberculatus Possesses a Strong Capacity for Lipid Metabolism and Protein Synthesis
3.2. The SMS Group Experienced Stronger Oxidative Stress
3.3. Apoptosis Occurred More Frequently in the SMF Group
4. Materials and Methods
4.1. Experimental Animals
4.2. Analysis of Growth Characters and Sampling
4.3. LC-MS/MS Analysis Conditions
4.4. Metabolomics Data Analysis
4.5. Transcriptomics Analysis
4.6. qRT-PCR Validation
4.7. Integrative Analysis of Metabolomics and Transcriptomics
4.8. Growth Data Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bordbar, S.; Anwar, F.; Saari, N. High-value components and bioactives from Sea Cucumbers for functional foods—A review. Mar. Drugs 2011, 9, 1761–1805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, S.J.; Feng, W.J.; Hu, S.; Liang, S.X.; An, N.N.; Mao, Y.J. Bioactive compounds of sea cucumbers and their therapeutic effects. Chin. J. Oceanol. Limnol. 2016, 34, 549–558. [Google Scholar] [CrossRef]
- Uthicke, S. Nutrient regeneration by abundant coral reef holothurians. J. Exp. Mar. Biol. Ecol. 2001, 265, 153–170. [Google Scholar] [CrossRef]
- Pandolfi, J.M.; Connolly, S.R.; Marshall, D.J.; Cohen, A.L. Projecting coral reef futures under global warming and ocean acidification. Science 2011, 333, 418–422. [Google Scholar] [CrossRef] [Green Version]
- Cramer, K.L.; Bernard, M.L.; Bernat, I.; Gutierrez, L.; Murphy, E.L.; Sangolquí, P.; Surrey, K.C.; Gerber, L.R. The Present and Future Status of Ecosystem Services for Coral Reefs; Elsevier: Amsterdam, Holland, 2022; pp. 46–54. [Google Scholar] [CrossRef]
- Slater, M.J.; Jeffs, A.G.; Sewell, M.A. Organically selective movement and deposit-feeding in juvenile sea cucumber, Australostichopus mollis determined in situ and in the laboratory. J. Exp. Mar. Biol. Ecol. 2011, 409, 315–323. [Google Scholar] [CrossRef]
- Purcell, S.W.; Conand, C.; Uthicke, S.; Byrne, M. Ecological roles of exploited sea cucumbers. In Oceanography and Marine Biology: An Annual Review; Hughes, R.N., Hughes, D.J., Smith, I.P., Dale, A.C., Eds.; CRC Press-Taylor & Francis Group: Boca Raton, FL, USA, 2016; Volume 54, pp. 367–386. [Google Scholar]
- Rakaj, A.; Fianchini, A.; Boncagni, P.; Scardi, M.; Cataudella, S. Artificial reproduction of Holothuria polii: A new candidate for aquaculture. Aquaculture 2019, 498, 444–453. [Google Scholar] [CrossRef]
- Purcell, S.W. Value, market preferences and trade of beche-de-mer from pacific island sea cucumbers. PLoS ONE 2014, 9, e95075. [Google Scholar] [CrossRef]
- Gonzalez-Wanguemert, M.; Dominguez-Godino, J.A.; Canovas, F. The fast development of sea cucumber fisheries in the Mediterranean and NE Atlantic waters: From a new marine resource to its over-exploitation. Ocean Coast. Manag. 2018, 151, 165–177. [Google Scholar] [CrossRef]
- Yuan, L.H.; Hu, C.Q.; Zhang, L.P.; Xia, J.J. Population genetics of a tropical sea cucumber species (Stichopus monotuberculatus) in China. Conserv. Genet. 2013, 14, 1279–1284. [Google Scholar] [CrossRef]
- Xu, Q.; Wu, P.L.; Huang, D.J.; Xiao, Y.L.; Wang, X.Y.; Xia, J.Q.; Ma, W.A.; Gao, F.; Wang, A.M. Sea ranching feasibility of the hatchery-reared tropical sea cucumber Stichopus monotuberculatus in an inshore coral reef island area in south china sea (Sanya, China). Front. Mar. Sci. 2022, 9, 13. [Google Scholar] [CrossRef]
- Bell, J.D.; Purcell, S.W.; Nash, W.J. Restoring small-scale fisheries for tropical sea cucumbers. Ocean Coast. Manag. 2008, 51, 589–593. [Google Scholar] [CrossRef]
- Zhu, L.; Li, C.H.; Su, X.R.; Guo, C.Y.; Wang, Z.; Jin, C.H.; Li, Y.; Li, T.W. Identification and assessment of differentially expressed genes involved in growth regulation in Apostichopus japonicus. Genet. Mol. Res. 2013, 12, 3028–3037. [Google Scholar] [CrossRef]
- Zhang, S.C.; Yang, H.S.; Liu, S.L.; Zhang, L.B.; Ru, X.S.; Xu, D.X. The influence of genetics factor on key growth traits and quantitative genetic analysis of sea cucumber Apostichopus Japonicus (Selenka) heat-resistant and fast-growing strain. Aquac. Int. 2015, 23, 219–233. [Google Scholar] [CrossRef]
- Pell, J.M.; Stewart, C.E.H.; Bates, P.C.; Aston, R. Immunological Approaches to Modify Growth; Academic Press: Cambridge, MA, USA, 1994; pp. 303–319. [Google Scholar] [CrossRef]
- Rondon, R.; Akcha, F.; Alonso, P.; Menard, D.; Rouxel, J.; Montagnani, C.; Mitta, G.; Cosseau, C.; Grunau, C. Transcriptional changes in Crassostrea gigas oyster spat following a parental exposure to the herbicide diuron. Aquat. Toxicol. 2016, 175, 47–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, H.J.; Kim, B.M.; Hwang, I.J.; Lee, J.S.; Choi, I.Y.; Kim, Y.J.; Rhee, J.S. Thermal stress induces a distinct transcriptome profile in the Pacific oyster Crassostrea gigas. Comp. Biochem. Phys. D 2016, 19, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.L.; Li, L.; Zhu, Y.B.; Zhang, G.F.; Guo, X.M. Transcriptome analysis reveals a rich gene set related to innate immunity in the Eastern Oyster (Crassostrea virginica). Mar. Biotechnol. 2014, 16, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Li, B.S.; Song, K.; Meng, J.; Li, L.; Zhang, G.F. Integrated application of transcriptomics and metabolomics provides insights into glycogen content regulation in the Pacific oyster Crassostrea gigas. BMC Genom. 2017, 18, 14. [Google Scholar] [CrossRef] [Green Version]
- Kotwal, S.; Kaul, S.; Sharma, P.; Gupta, M.; Shankar, R.; Jain, M.; Dhar, M.K. De novo transcriptome analysis of medicinally important Plantago ovata using RNA-Seq. PLoS ONE 2016, 11, e0150273. [Google Scholar] [CrossRef]
- Meyer, E.; Aglyamova, G.V.; Shi, W.; Buchanan-Carter, J.; Abrego, D.; Colbourne, J.K.; Willis, B.L.; Matz, M.V. Sequencing and de novo analysis of a coral larval transcriptome using 454 GSFlx. BMC Genom. 2009, 10, 219. [Google Scholar] [CrossRef] [Green Version]
- Huo, D.; Sun, L.N.; Zhang, L.B.; Ru, X.S.; Liu, S.L.; Yang, H.S. Metabolome responses of the sea cucumber Apostichopus japonicus to multiple environmental stresses: Heat and hypoxia. Mar. Pollut. Bull. 2019, 138, 407–420. [Google Scholar] [CrossRef]
- Gao, K.L.; Wang, Z.C.; Qiu, X.M.; Song, J.; Wang, H.Z.; Zhao, C.; Wang, X.L.; Chang, Y.Q. Transcriptome analysis of body wall reveals growth difference between the largest and smallest individuals in the pure and hybrid populations of Apostichopus japonicus. Comp. Biochem. Phys. D 2019, 31, 100591. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Huo, D.; Liu, S.L.; Xing, L.L.; Su, F.; Yang, H.S.; Sun, L.N. Construction of a high-density genetic linkage map for the mapping of QTL associated with growth-related traits in sea cucumber (Apostichopus japonicus). Biology 2022, 11, 50. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.H.; Sun, Y.; Cao, Y.; Han, L.S.; Li, Y.X.; Ding, B.C.; Gao, C.; Hao, P.F.; Jin, X.; Chang, Y.Q.; et al. Transcriptomic and metabolomic analyses provide insights into the growth and development advantages of triploid Apostichopus japonicus. Mar. Biotechnol. 2022, 24, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Kornthong, N.; Phanaksri, T.; Saetan, J.; Duangprom, S.; Lekskul, B.; Vivattanasarn, T.; Songkoomkrong, S.; Jattujan, P.; Cummins, S.F.; Sobhon, P.; et al. Identification and localization of growth factor genes in the sea cucumber, Holothuria scabra. Heliyon 2021, 7, e08370. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, X.; Zhao, X.; Xu, J.; Lin, C.; Jing, P.; Hu, L.; Zhao, S.; Wang, X.; Li, B. Discrimination of dried sea cucumber (Apostichopus japonicus) products from different geographical origins by sequential windowed acquisition of all theoretical fragment ion mass spectra (SWATH-MS)-based proteomic analysis and chemometrics. Food Chem. 2019, 274, 592–602. [Google Scholar] [CrossRef]
- Liu, Y.J.; Yao, M.Z.; Li, S.W.; Wei, X.F.; Ding, L.; Han, S.C.; Wang, P.; Lv, B.C.; Chen, Z.X.; Sun, Y.C. Integrated application of multi-omics approach and biochemical assays provides insights into physiological responses to saline-alkaline stress in the gills of crucian carp (Carassius auratus). Sci. Total Environ. 2022, 822, 153622. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Zhai, X.; Qu, M.; Tong, C.; Li, W. Sulfated modification of the polysaccharides from Crassostrea gigas and their antioxidant and hepatoprotective activities through metabolomics analysis. Int. J. Biol. Macromol. 2019, 129, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Yoon, D.; Kim, S.; Lee, M.; Yoon, C.; Kim, S. 1H-NMR-based metabolomic study on toxicity of methomyl and methidathion in fish. Sci. Health Part B-Pestic. Contam. Agric. Wastes 2016, 51, 824–831. [Google Scholar] [CrossRef]
- Lee, H.K.; Kim, K.; Lee, J.; Lee, J.; Lee, J.; Kim, S.; Lee, S.E.; Kim, J.H. Targeted toxicometabolomics of endosulfan sulfate in adult zebrafish (Danio rerio) using GC-MS/MS in multiple reaction monitoring mode. J. Hazard. Mater. 2020, 389, 122056. [Google Scholar] [CrossRef]
- McCombie, G.; Browning, L.M.; Titman, C.M.; Song, M.; Shockcor, J.; Jebb, S.A.; Griffin, J.L. ω-3 oil intake during weight loss in obese women results in remodelling of plasma triglyceride and fatty acids. Metabolomics 2009, 5, 363–374. [Google Scholar] [CrossRef] [Green Version]
- Palou, A.; Bonet, M.L. Controlling Lipogenesis and Thermogenesis and the Use of Ergogenic Aids for Weight Control; Woodhead: Cambridge, UK, 2007; pp. 58–103. [Google Scholar] [CrossRef]
- Stillwell, W.; Wassall, S.R. Docosahexaenoic acid: Membrane properties of a unique fatty acid. Chem. Phys. Lipids 2003, 126, 1–27. [Google Scholar] [CrossRef]
- Marrink, S.J.; de Vries, A.H.; Harroun, T.A.; Katsaras, J.; Wassall, S.R. Cholesterol shows preference for the interior of polyunsaturated lipid. J. Am. Chem. Soc. 2008, 130, 10–11. [Google Scholar] [CrossRef] [Green Version]
- Usui, K.; Hiraki, T.; Kawamoto, J.; Kurihara, T.; Nogi, Y.; Kato, C.; Abe, F. Eicosapentaenoic acid plays a role in stabilizing dynamic membrane structure in the deep-sea piezophile Shewanella violacea: A study employing high-pressure time-resolved fluorescence anisotropy measurement. Biochim. Biophys. Acta-Biomembr. 2012, 1818, 574–583. [Google Scholar] [CrossRef] [Green Version]
- Kawamoto, J.; Kurihara, T.; Yamamoto, K.; Nagayasu, M.; Tani, Y.; Mihara, H.; Hosokawa, M.; Baba, T.; Sato, S.B.; Esaki, N. Eicosapentaenoic acid plays a beneficial role in membrane organization and cell division of a cold-adapted bacterium, Shewanella livingstonensis Ac10. J. Bacteriol. 2009, 191, 632–640. [Google Scholar] [CrossRef] [Green Version]
- Li, W.Y.; Wang, J.J.; Li, J.T.; Liu, P.; Li, J.; Zhao, F.Z. Antioxidant, transcriptome and the metabolome response to dietary astaxanthin in Exopalaemon carinicauda. Front. Physiol. 2022, 13, 859305. [Google Scholar] [CrossRef]
- Chen, K.; Li, E.C.; Xu, C.; Wang, X.D.; Li, H.F.; Qin, J.G.; Chen, L.Q. Growth and metabolomic responses of Pacific white shrimp (Litopenaeus vannamei) to different dietary fatty acid sources and salinity levels. Aquaculture 2019, 499, 329–340. [Google Scholar] [CrossRef]
- Zhang, Y.F.; Gu, Z.P.; Ren, Y.D.; Wang, L.; Zhang, J.; Liang, C.W.; Tong, S.Y.; Wang, Y.T.; Xu, D.; Zhang, X.W.; et al. Integrating transcriptomics and metabolomics to characterize metabolic regulation to elevated CO2 in Chlamydomonas Reinhardtii. Mar. Biotechnol. 2021, 23, 255–275. [Google Scholar] [CrossRef]
- Zimmerman, A.W.; Veerkamp, J.H. Members of the fatty acid-binding protein family inhibit cell-free protein synthesis. FEBS Lett. 1998, 437, 183–186. [Google Scholar] [CrossRef]
- Chmurzynska, A. The multigene family of fatty acid-binding proteins (FABPs): Function, structure and polymorphism. J. Appl. Genet. 2006, 47, 39–48. [Google Scholar] [CrossRef]
- Shan, T.Z.; Ren, Y.; Wu, T.; Liu, C.X.; Wang, Y.Z. Regulatory role of Sirt1 on the gene expression of fatty acid-binding protein 3 in cultured Porcine Adipocytes. J. Cell. Biochem. 2009, 107, 984–991. [Google Scholar] [CrossRef]
- Glatz, J.F.C.; Schaap, F.G.; Binas, B.; Bonen, A.; van der Vusse, G.J.; Luiken, J. Cytoplasmic fatty acid-binding protein facilitates fatty acid utilization by skeletal muscle. Acta Physiol. Scand. 2003, 178, 367–371. [Google Scholar] [CrossRef]
- Gapasin, R.S.J.; Duray, M.N. Effects of DHA-enriched live food on growth, survival and incidence of opercular deformities in milkfish (Chanos chanos). Aquaculture 2001, 193, 49–63. [Google Scholar] [CrossRef]
- Lee, S.M.; Lee, J.H.; Kim, K.D. Effect of dietary essential fatty acids on growth, body composition and blood chemistry of juvenile starry flounder (Platichthys stellatus). Aquaculture 2003, 225, 269–281. [Google Scholar] [CrossRef]
- Montero, D.; Socorro, J.; Tort, L.; Caballero, M.J.; Robaina, L.E.; Vergara, J.M.; Izquierdo, M.S. Glomerulonephritis and immunosuppression associated with dietary essential fatty acid deficiency in gilthead sea bream, Sparus aurata L., juveniles. J. Fish. Dis. 2004, 27, 297–306. [Google Scholar] [CrossRef]
- Poynard, T.; Imbert-Bismut, F. Laboratory Testing for Liver Disease; Elsevier: Amsterdam, Holland, 2012; pp. 201–215. [Google Scholar] [CrossRef]
- Lane, A.N.; Fan, T.W.M. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 2015, 43, 2466–2485. [Google Scholar] [CrossRef] [Green Version]
- Moffatt, B.A.; Ashihara, H. Purine and pyrimidine nucleotide synthesis and metabolism. Arab. Book 2002, 1, e0018. [Google Scholar] [CrossRef]
- Wang, J.A.; Hou, X.; Chen, X.W.; Zhang, K.J.; Wang, J.; Wang, C.H. Comprehensive analysis of metabolomics and transcriptomics provides insights into growth difference of juvenile Eriocheir sinensis during the molting cycle. Aquaculture 2021, 539, 736661. [Google Scholar] [CrossRef]
- Li, P.; Gatlin, D.M. Nucleotide nutrition in fish: Current knowledge and future applications. Aquaculture 2006, 251, 141–152. [Google Scholar] [CrossRef]
- Berlin, R.D.; Oliver, J.M. Membrane-transport of purine and pyrimidine-bases and nucleosides in animal-cells. Int. Rev. Cytol. 1975, 42, 287–336. [Google Scholar] [CrossRef] [PubMed]
- Tocher, D.R. Metabolism and functions of lipids and fatty acids in teleost fish. Rev. Fish. Sci. 2003, 11, 107–184. [Google Scholar] [CrossRef]
- Xu, H.G.; Wang, C.Q.; Zhang, Y.Q.; Wei, Y.L.; Liang, M.Q. Moderate levels of dietary arachidonic acid reduced lipid accumulation and tended to inhibit cell cycle progression in the liver of Japanese seabass Lateolabrax japonicus. Sci. Rep. 2018, 8, 10682. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.X.; Chen, W.Y.; Yuan, Y.Y.; Mao, L.Z.; Yu, S.Q.; Chen, A.J.; Ni, Y.; Roldan, E.R.S. Progesterone primes zona pellucida-induced activation of phospholipase A(2) during acrosomal exocytosis in guinea pig spermatozoa. J. Cell. Physiol. 2005, 205, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Kudo, I. Phospholipase A(2). J. Biochem. 2002, 131, 285–292. [Google Scholar] [CrossRef]
- Montero, D.; Mathlouthi, F.; Tort, L.; Afonso, J.M.; Torrecillas, S.; Fernandez-Vaquero, A.; Negrin, D.; Izquierdo, M.S. Replacement of dietary fish oil by vegetable oils affects humoral immunity and expression of pro-inflammatory cytokines genes in gilthead sea bream Sparus aurata. Fish Shellfish Immun. 2010, 29, 1073–1081. [Google Scholar] [CrossRef]
- Zuo, R.; Mai, K.; Xu, W.; Ai, Q. Advance of studies on the effects of fatty acids on immune responses and nutritional regulation mechanism in fish species. J. Fish. China 2015, 39, 1079–1088. [Google Scholar]
- Shimizu, T.; Wolfe, L.S. Arachidonic-acid cascade and signal transduction. J. Neurochem. 1990, 55, 1–15. [Google Scholar] [CrossRef]
- Chen, K.; Li, E.C.; Li, T.Y.; Xu, C.; Wang, X.D.; Lin, H.Z.; Qin, J.G.; Chen, L.Q. Transcriptome and Molecular Pathway Analysis of the hepatopancreas in the pacific white shrimp Litopenaeus vannamei under chronic low-salinity stress. PLoS ONE 2015, 10, e0131503. [Google Scholar] [CrossRef]
- Huang, Y.Y.; Wang, G.D.; Liu, J.S.; Zhang, L.L.; Huang, S.Y.; Wang, Y.L.; Yang, Z.W.; Ge, H. Analysis of transcriptome difference between rapid-growing and slow-growing in Penaeus vannamei. Gene 2021, 787, 145642. [Google Scholar] [CrossRef]
- Huang, M.X.; Dong, Y.F.; Zhang, Y.; Chen, Q.S.; Xie, J.; Xu, C.; Zhao, Q.; Li, E.C. Growth and lipidomic responses of juvenile pacific white shrimp Litopenaeus vannamei to low salinity. Front. Physiol. 2019, 10, 1087. [Google Scholar] [CrossRef]
- Guo, X.F.; Zhou, Y.L.; Liu, M.; Wang, Z.W.; Gui, J.F. Integrated application of Iso-seq and RNA-seq provides insights into unsynchronized growth in red swamp crayfish (Procambarus clarkii). Aquacult. Rep. 2022, 22, 101008. [Google Scholar] [CrossRef]
- Hao, Y.N.; Guo, M.; Feng, Y.W.; Dong, Q.; Cui, M. Lysophospholipids and their g-coupled protein signaling in Alzheimer’s Disease: From physiological performance to pathological impairment. Front. Molec. Neurosci. 2020, 13, 58. [Google Scholar] [CrossRef] [Green Version]
- Kouglas, P.; Chai, H.; Lin, P.H.; Lumsden, A.B.; Yao, Q.; Chen, C.Y. Lysophosphatidylcholine and secretory phospholipase A(2) in vascular disease: Mediators of endothelial dysfunction and atherosclerosis. Med. Sci. Monitor 2006, 12, RA5–RA16. [Google Scholar]
- Chi, L.; Tu, P.C.; Liu, C.W.; Lai, Y.J.; Xue, J.C.; Ru, H.Y.; Lu, K. Chronic arsenic exposure induces oxidative stress and Perturbs Serum lysolipids and fecal unsaturated fatty acid metabolism. Chem. Res. Toxicol. 2019, 32, 1204–1211. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.H.; Lee, C.H.; Tsai, M.H.; Chen, C.H.; Lin, H.F.; Hsu, C.Y.; Lai, C.L.; Chen, C.C. Activation of acid-sensing ion channel 3 by lysophosphatidylcholine 16:0 mediates psychological stress-induced fibromyalgia-like pain. Ann. Rheum. Dis. 2020, 79, 1644–1656. [Google Scholar] [CrossRef] [PubMed]
- Aruoma, O.I.; Halliwell, B.; Hoey, B.M.; Butler, J. The antioxidant action of taurine, hypotaurine and their metabolic precursors. Biochem. J. 1988, 256, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Peter, N.; Pradhan, C.; Dileep, N.; Musharraf, M.; Vasunambisan, S.T. Dietary taurine improved growth performance, nutrient utilization, and antioxidant enzyme activities in pangasius Pangasianodon hypophthalmus. J. World. Aquacult. Soc. 2022, 53, 106–121. [Google Scholar] [CrossRef]
- Banks, M.A.; Porter, D.W.; Martin, W.G.; Castranova, V. Taurine protects against oxidant injury to rat alveolar pneumocytes. In Waltham Symp on Taurine and Cat Nutrition/International Taurine Symp: New Dimensions on Its Mechanisms of Action; 315; Plenum Press Div Plenum Publishing Corp: Orange Beach, AL, USA, 1991; pp. 341–354. [Google Scholar]
- Ceccotti, C.; Al-Sulaivany, B.S.A.; Al-Habbib, O.A.M.; Saroglia, M.; Rimoldi, S.; Terova, G. Protective Effect of Dietary Taurine from ROS production in European Seabass under conditions of forced swimming. Animals 2019, 9, 607. [Google Scholar] [CrossRef] [Green Version]
- Gunathilaka, G.L.B.E.; Kim, M.-G.; Chorong, L.; Shin, J.; Lee, B.-J.; Lee, K.-J. Effects of taurine supplementation in low fish meal diets for red seabream (Pagrus major) in low water temperature season. Fish. Aquatic Sci. 2019, 22, 23. [Google Scholar] [CrossRef]
- Maslinska, D. Apoptosis: Physiological cell death and its role in pathogenesis of diseases. Neurol. Neurochir. Pol. 2003, 37, 315–326. [Google Scholar]
- Jia, R.; Han, C.; Lei, J.L.; Liu, B.L.; Huang, B.; Huo, H.H.; Yin, S.T. Effects of nitrite exposure on haematological parameters, oxidative stress and apoptosis in juvenile turbot (Scophthalmus maximus). Aquat. Toxicol. 2015, 169, 1–9. [Google Scholar] [CrossRef]
- Fulda, S.; Debatin, K.M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuura, K.; Canfield, K.; Feng, W.; Kurokawa, M. Metabolic Regulation of Apoptosis in Cancer. In International Review of Cell and Molecular Biology; Jeon, K.W., Galluzzi, L., Eds.; Elsevier Academic Press Inc.: San Diego, CA, USA, 2016; Volume 327, pp. 43–87. [Google Scholar] [CrossRef] [Green Version]
- Guicciardi, M.E.; Leist, M.; Gores, G.J. Lysosomes in cell death. Oncogene 2004, 23, 2881–2890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emert-Sedlak, L.; Shangary, S.; Rabinovitz, A.; Miranda, M.B.; Delach, S.M.; Johnson, D.E. Involvement of cathepsin D in chemotherapy-induced cytochrome c release, caspase activation, and cell death. Mol. Cancer Ther. 2005, 4, 733–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitaker, J.N.; Rhodes, R.H. The distribution of cathepsin-d in rat-tissues determined by immunocytochemistry. Am. J. Anat. 1983, 166, 417–428. [Google Scholar] [CrossRef]
- Marks, N.; Lajtha, A. Separation of acid and neutral proteinases of brain. Biochem. J. 1965, 97, 74–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberg, K.; Kagedal, K.; Ollinger, K. Microinjection of cathepsin D induces caspase-dependent apoptosis in fibroblasts. Am. J. Pathol. 2002, 161, 89–96. [Google Scholar] [CrossRef] [Green Version]
- Cohen, G.M. Caspases: The executioners of apoptosis. Biochem. J. 1997, 326, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.Y.; Dou, J.; Wu, Q.Y.; Ye, Y.F.; Song, C.B.; Mu, C.K.; Wang, C.L.; Ren, Z.M.; Shi, C. Investigation of the light intensity effect on growth, molting, hemolymph lipid, and antioxidant capacity of juvenile swimming crab Portunus trituberculatus. Front. Mar. Sci. 2022, 9, 922021. [Google Scholar] [CrossRef]
- Hu, C.Q.; Li, H.P.; Xia, J.J.; Zhang, L.P.; Luo, P.; Fan, S.G.; Peng, P.F.; Yang, H.P.; Wen, J. Spawning, larval development and juvenile growth of the sea cucumber Stichopus horrens. Aquaculture 2013, 404, 47–54. [Google Scholar] [CrossRef]
- Kechin, A.; Boyarskikh, U.; Kel, A.; Filipenko, M. cutPrimers: A new tool for accurate cutting of primers from reads of targeted next generation sequencing. J. Comput. Biol. 2017, 24, 1138–1143. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Arocho, A.; Chen, B.Y.; Ladanyi, M.; Pan, Q.L. Validation of the 2(-Delta Delta Ct) calculation as an alternate method of data analysis for quantitative PCR of BCR-ABL P210 transcripts. Diagn. Mol. Pathol. 2006, 15, 56–61. [Google Scholar] [CrossRef]
- Rodgers, J.; Nicewander, A. Thirteen Ways to Look at the Correlation Coefficient. Am. Stat. 1988, 42, 59–66. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Primer (5′→3′) |
|---|---|
| Rdh7 | F: ACTACGCAATACAGGTTGTT R: CCCATTGAAAGCTGCTTATG |
| Pde10a | F: GGTACCTGCTAACTTTGACA R: ATCAGGACCAAAAAGGTTGT |
| hnmt | F: TATATCCCCAGAAGGTCGTT R: TGGTTCTATGACTGTGTTGG |
| PLB1 | F: GACTTCACCATTCTGAGGGA R: TAATCGCTCAGCCATGTACT |
| DHRS4 | F: CACTGCCTACTTGTTACTGT R: GAGCTCAGTGTACGAATCAT |
| GABBR2 | F: CGGACAATGGTTTATTGGCT R: TTAAACATTGGAGGGGACCA |
| NMT1 | F: TATTTGGCGAAGGAGTTTGA R: TTACAGCCATCTCCTGTCTA |
| mt-Co3 | F: TTTTCAGCCCTCCTTCTAAC R: TCCTTCACGAATTACGTCTC |
| SGPL1 | F: CGACTGAGAAAGAACAAGGA R: GGAACTGCAAGGAGTTTAGA |
| Wnt7B | F: TGTGACAAAAGGTATCCCGA R: AGCATCTACGAACTCTCTGG |
| WNT-1 | F: ACGAGCCTGTTCGGTAA R: CCCACTCCCAGTCTTCA |
| β-actin | F: GAGGTCTGCAATACCTGCGATT R: TTTTGTGTGGGGTGTGGTTG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, B.; Liu, Y.; Pan, W.; Li, Z.; Ren, C.; Hu, C.; Luo, P. Integrative Application of Transcriptomics and Metabolomics Provides Insights into Unsynchronized Growth in Sea Cucumber (Stichopus monotuberculatus). Int. J. Mol. Sci. 2022, 23, 15478. https://doi.org/10.3390/ijms232415478
Ma B, Liu Y, Pan W, Li Z, Ren C, Hu C, Luo P. Integrative Application of Transcriptomics and Metabolomics Provides Insights into Unsynchronized Growth in Sea Cucumber (Stichopus monotuberculatus). International Journal of Molecular Sciences. 2022; 23(24):15478. https://doi.org/10.3390/ijms232415478
Chicago/Turabian StyleMa, Bo, Yang Liu, Wenjie Pan, Zhuobo Li, Chunhua Ren, Chaoqun Hu, and Peng Luo. 2022. "Integrative Application of Transcriptomics and Metabolomics Provides Insights into Unsynchronized Growth in Sea Cucumber (Stichopus monotuberculatus)" International Journal of Molecular Sciences 23, no. 24: 15478. https://doi.org/10.3390/ijms232415478