
N-Linked Glycosylation in Chinese Hamster Ovary Cells Is Critical for Insulin-like Growth Factor 1 Signaling
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. Preparation of Cell Surface Proteins Using N-Hydroxysulfosuccinimydyl-S,S-Biotin (NHS-SS-Biotin) Based Cell Surface Protein Biotinylation and Isolation Kit
2.3. Processing of Plasma Membrane Proteins for Mass Spectrometry
2.4. Data Processing
2.5. Bioinformatics Analyses
2.6. Western Blot Analysis
2.7. In Silico Interaction Analysis of IGF-1R and IQGAP1
2.8. ELISA Analysis
2.9. Stimulation with IGF-1R Ligand
2.10. Quantitative Real-Time PCR (qRT-PCR)
2.11. Statistical Analysis
3. Results
3.1. Identification of Differential Protein Abundance in W5, Lec1, and Lec4 cells
3.2. Validation of the Proteomics Result
3.3. Functional Annotation and Enrichment of Differentially Expressed Proteins
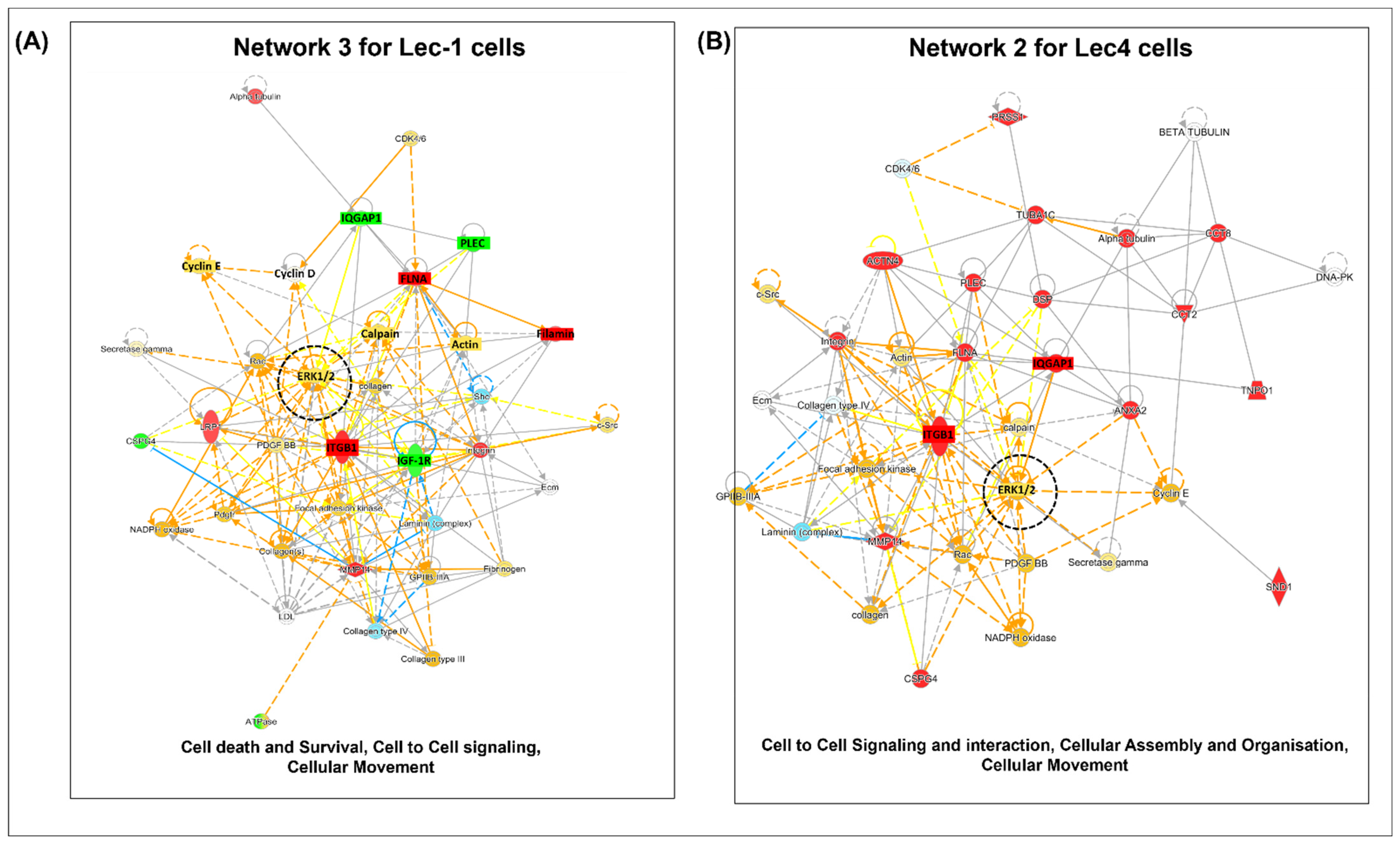
3.4. Interaction Networks of Surface Proteins Associated with Lec1/Lec4 Cells
3.5. Pathway Analysis
3.6. Integrin Regulation
3.7. Insulin Growth Factor I Receptor Expression
3.8. Effect of Altered IGF1-R on Its Downstream Molecules/Pathways
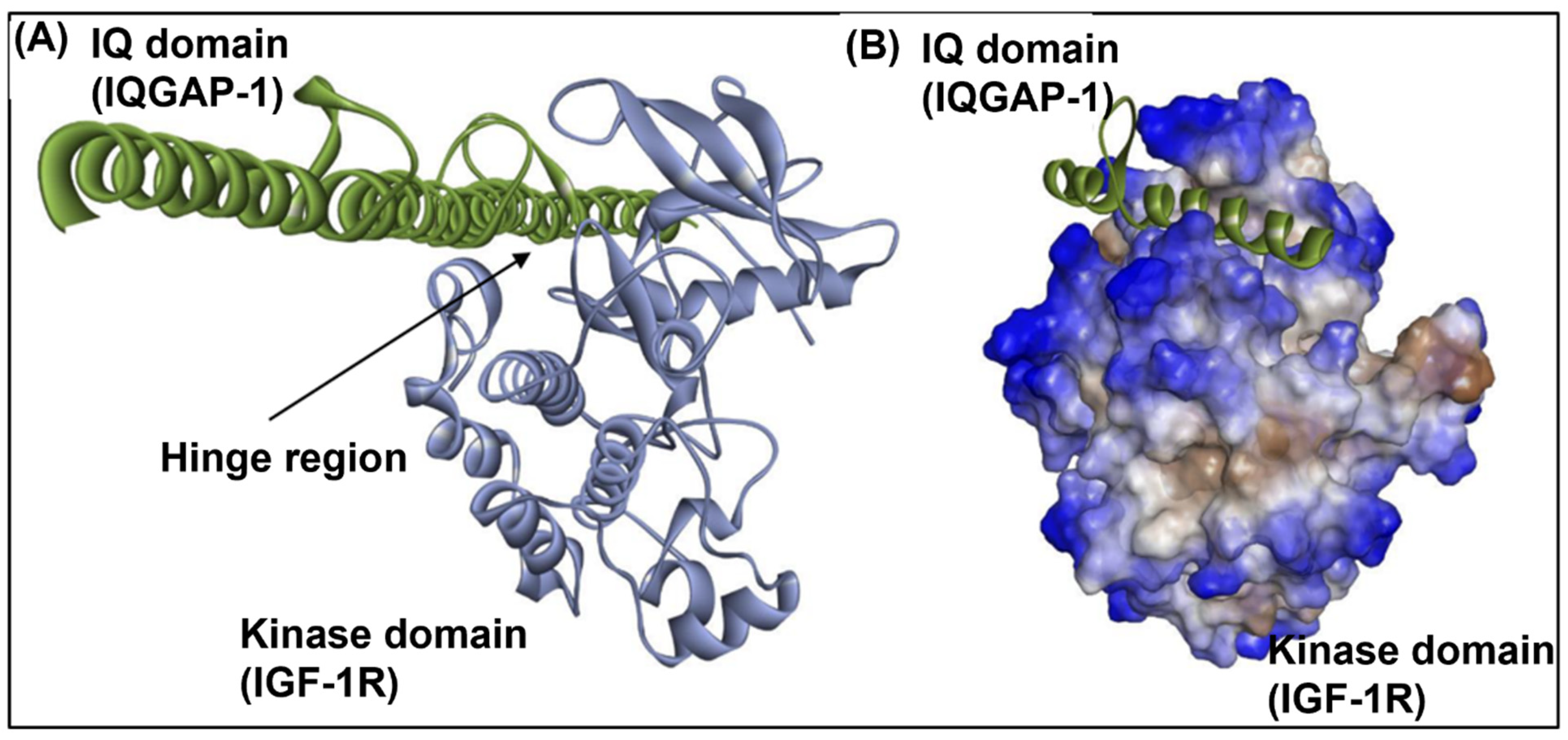
3.9. IGF1-R and IQGAP1 Interact In Silico
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ohtsubo, K.; Marth, J.D. Glycosylation in Cellular Mechanisms of Health and Disease. Cell 2006, 126, 855–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Sato, Y.; Isaji, T.; Fukuda, T.; Matsumoto, A.; Miyoshi, E.; Gu, J.; Taniguchi, N. Branched N-glycans Regulate the Biological Functions of Integrins and Cadherins. FEBS J. 2008, 275, 1939–1948. [Google Scholar] [CrossRef] [PubMed]
- Ballabio, A.; Bonifacino, J.S. Lysosomes as Dynamic Regulators of Cell and Organismal Homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 101–118. [Google Scholar] [CrossRef] [PubMed]
- Cerrato, C.P.; Künnapuu, K.; Langel, Ü. Cell-Penetrating Peptides with Intracellular Organelle Targeting. Expert Opin. Drug Deliv. 2017, 14, 245–255. [Google Scholar] [CrossRef]
- Finch, N.A.; Linser, P.J.; Ochrietor, J.D. Hydrophobic Interactions Stabilize the Basigin-MCT1 Complex. Protein J. 2009, 28, 362–368. [Google Scholar] [CrossRef]
- Lamriben, L.; Graham, J.B.; Adams, B.M.; Hebert, D.N. N-Glycan-based ER Molecular Chaperone and Protein Quality Control System: The Calnexin Binding Cycle. Traffic 2016, 17, 308–326. [Google Scholar] [CrossRef] [Green Version]
- Chandler, K.B.; Costello, C.E. Glycomics and Glycoproteomics of Membrane Proteins and Cell-Surface Receptors: Present Trends and Future Opportunities. Electrophoresis 2016, 37, 1407–1419. [Google Scholar] [CrossRef] [Green Version]
- Pucci, M.; Venturi, G.; Malagolini, N.; Chiricolo, M.; Dall’Olio, F. Glycosylation as a Main Regulator of Growth and Death Factor Receptors Signaling. Int. J. Mol. Sci. 2018, 19, 580. [Google Scholar] [CrossRef] [Green Version]
- Peixoto, A.; Relvas-Santos, M.; Azevedo, R.; Santos, L.L.; Ferreira, J.A. Protein Glycosylation and Tumor Microenvironment Alterations Driving Cancer Hallmarks. Front. Oncol. 2019, 9, 380. [Google Scholar] [CrossRef] [Green Version]
- Arey, B. The Role of Glycosylation in Receptor Signaling. In Glycosylation; InTech: London, UK, 2012. [Google Scholar] [CrossRef]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in Health and Disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef]
- Goettig, P. Effects of Glycosylation on the Enzymatic Activity and Mechanisms of Proteases. Int. J. Mol. Sci. 2016, 17, 1969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patnaik, S.K.; Stanley, P. Lectin-Resistant CHO Glycosylation Mutants. Methods Enzymol. 2006, 416, 159–182. [Google Scholar] [CrossRef]
- Hall, M.; Weidner, D.; Zhu, Y.; Dayal, S.; Whitman, A.; Schwalbe, R. Predominant Expression of Hybrid N-Glycans Has Distinct Cellular Roles Relative to Complex and Oligomannose N-Glycans. Int. J. Mol. Sci. 2016, 17, 925. [Google Scholar] [CrossRef] [Green Version]
- Stanley, P.; Caillibot, V.; Siminovitch, L. Selection and Characterization of Eight Phenotypically Distinct Lines of Lectin-Resistant Chinese Hamster Ovary Cells. Cell 1975, 6, 121–128. [Google Scholar] [CrossRef]
- Pinho, S.S.; Reis, C.A. Glycosylation in Cancer: Mechanisms and Clinical Implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef]
- Cristani, M.; Speciale, A.; Saija, A.; Gangemi, S.; Lucia Minciullo, P.; Cimino, F. Circulating Advanced Oxidation Protein Products as Oxidative Stress Biomarkers and Progression Mediators in Pathological Conditions Related to Inflammation and Immune Dysregulation. Curr. Med. Chem. 2016, 23, 3862–3882. [Google Scholar] [CrossRef]
- Russo, L. Glycans and Diagnostics in Nanomedicine. In Nanomedicine; Future Medicine: London, UK, 2021; pp. 1839–1842. [Google Scholar]
- Dingerdissen, H.M.; Vora, J.; Cauley, E.; Bell, A.; King, C.H.; Mazumder, R. Differential Expression of Glycosyltransferases Identified through Comprehensive Pan-Cancer Analysis. bioRxiv 2021. [Google Scholar] [CrossRef]
- Conte, F.; van Buuringen, N.; Voermans, N.C.; Lefeber, D.J. Galactose in Human Metabolism, Glycosylation and Congenital Metabolic Diseases: Time for a Closer Look. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2021, 1865, 129898. [Google Scholar] [CrossRef] [PubMed]
- Berger, R.P.; Dookwah, M.; Steet, R.; Dalton, S. Glycosylation and Stem Cells: Regulatory Roles and Application of IPSCs in the Study of Glycosylation-related Disorders. Bioessays 2016, 38, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Lee-Sundlov, M.M.; Stowell, S.R.; Hoffmeister, K.M. Multifaceted Role of Glycosylation in Transfusion Medicine, Platelets, and Red Blood Cells. J. Thromb. Haemost. 2020, 18, 1535–1547. [Google Scholar] [CrossRef]
- Frappaolo, A.; Sechi, S.; Kumagai, T.; Karimpour-Ghahnavieh, A.; Tiemeyer, M.; Giansanti, M.G. Modeling Congenital Disorders of N-Linked Glycoprotein Glycosylation in Drosophila Melanogaster. Front. Genet. 2018, 9, 436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.-B.; Lee, I.; Kamar, M.; Akiyama, S.K.; Pierce, M. Aberrant N-Glycosylation of Β1 Integrin Causes Reduced A5β1 Integrin Clustering and Stimulates Cell Migration. Cancer Res. 2002, 62, 6837–6845. [Google Scholar]
- Zhang, Y.; Zhao, J.; Zhang, X.; Guo, H.; Liu, F.; Chen, H. Relations of the Type and Branch of Surface N-Glycans to Cell Adhesion, Migration and Integrin Expressions. Mol. Cell. Biochem. 2004, 260, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Madzharova, E.; Kastl, P.; Sabino, F.; auf dem Keller, U. Post-Translational Modification-Dependent Activity of Matrix Metalloproteinases. Int. J. Mol. Sci. 2019, 20, 3077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Dedhar, S. Integrin-Linked Kinase (ILK) and Its Interactors: A New Paradigm for the Coupling of Extracellular Matrix to Actin Cytoskeleton and Signaling Complexes. J. Cell Biol. 2001, 155, 505–510. [Google Scholar] [CrossRef]
- Gao, Y.; Liu, S.; Huang, J.; Guo, W.; Chen, J.; Zhang, L.; Zhao, B.; Peng, J.; Wang, A.; Wang, Y. The ECM-Cell Interaction of Cartilage Extracellular Matrix on Chondrocytes. Biomed. Res. Int. 2014, 2014, 648459. [Google Scholar] [CrossRef] [Green Version]
- Brown, N.H.; Gregory, S.L.; Martin-Bermudo, M.D. Integrins as Mediators of Morphogenesis in Drosophila. Dev. Biol. 2000, 223, 1–16. [Google Scholar] [CrossRef]
- Lee, J.W.; Juliano, R. Mitogenic Signal Transduction by Integrin-and Growth Factor Receptor-Mediated Pathways. Mol. Cells (Springer Sci. Bus. Media BV) 2004, 17, 188–202. [Google Scholar]
- Xynos, I.D.; Edgar, A.J.; Buttery, L.D.K.; Hench, L.L.; Polak, J.M. Gene-expression Profiling of Human Osteoblasts Following Treatment with the Ionic Products of Bioglass® 45S5 Dissolution. J. Biomed. Mater. Res. Off. J. Soc. Biomater. Jpn. Soc. Biomater. Aust. Soc. Biomater. Korean Soc. Biomater. 2001, 55, 151–157. [Google Scholar] [CrossRef]
- St-Pierre, Y.; Themsche, C.V.; Estève, P.-O. Emerging Features in the Regulation of MMP-9 Gene Expression for the Development of Novel Molecular Targets and Therapeutic Strategies. Curr. Drug Targets-Inflamm. Allergy 2003, 2, 206–215. [Google Scholar] [CrossRef] [Green Version]
- Seguin, L.; Desgrosellier, J.S.; Weis, S.M.; Cheresh, D.A. Integrins and Cancer: Regulators of Cancer Stemness, Metastasis, and Drug Resistance. Trends Cell Biol. 2015, 25, 234–240. [Google Scholar] [CrossRef]
- Hamidi, H.; Ivaska, J. Every Step of the Way: Integrins in Cancer Progression and Metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef] [Green Version]
- Weis, S.M.; Cheresh, D.A. Tumor Angiogenesis: Molecular Pathways and Therapeutic Targets. Nat. Med. 2011, 17, 1359–1370. [Google Scholar] [CrossRef]
- Wrigley, S.; Arafa, D.; Tropea, D. Insulin-Like Growth Factor 1: At the Crossroads of Brain Development and Aging. Front. Cell Neurosci. 2017, 11, 14. [Google Scholar] [CrossRef] [Green Version]
- Cabail, M.Z.; Li, S.; Lemmon, E.; Bowen, M.E.; Hubbard, S.R.; Miller, W.T. The Insulin and IGF1 Receptor Kinase Domains Are Functional Dimers in the Activated State. Nat. Commun. 2015, 6, 6406. [Google Scholar] [CrossRef] [Green Version]
- Adams, T.E.; Epa, V.C.; Garrett, T.P.J. Structure and Function of the Type 1 Insulin-like Growth Factor Receptor. Cell. Mol. Life Sci. 2000, 57, 1050–1093. [Google Scholar] [CrossRef]
- Gao, Y.; Luan, X.; Melamed, J.; Brockhausen, I. Role of Glycans on Key Cell Surface Receptors That Regulate Cell Proliferation and Cell Death. Cells 2021, 10, 1252. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girnita, L.; Wang, M.; Xie, Y.; Nilsson, G.; Dricu, A.; Wejde, J.; Larsson, O. Inhibition of N-Linked Glycosylation down-Regulates Insulin-like Growth Factor-1 Receptor at the Cell Surface and Kills Ewing’s Sarcoma Cells: Therapeutic Implications. Anticancer Drug Des. 2000, 15, 67–72. [Google Scholar] [PubMed]
- Ulanet, D.B.; Ludwig, D.L.; Kahn, C.R.; Hanahan, D. Insulin Receptor Functionally Enhances Multistage Tumor Progression and Conveys Intrinsic Resistance to IGF-1R Targeted Therapy. Proc. Natl. Acad. Sci. USA 2010, 107, 10791–10798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.; Yee, D. Disrupting Insulin and IGF Receptor Function in Cancer. Int. J. Mol. Sci. 2021, 22, 555. [Google Scholar] [CrossRef] [PubMed]
- Vewinger, N.; Huprich, S.; Seidmann, L.; Russo, A.; Alt, F.; Bender, H.; Sommer, C.; Samuel, D.; Lehmann, N.; Backes, N.; et al. IGF1R Is a Potential New Therapeutic Target for HGNET-BCOR Brain Tumor Patients. Int. J. Mol. Sci. 2019, 20, 3027. [Google Scholar] [CrossRef] [Green Version]
- Carlberg, M.; Dricu, A.; Blegen, H.; Wang, M.; Hjertman, M.; Zickert, P.; Höög, A.; Larsson, O. Mevalonic Acid Is Limiting for N-Linked Glycosylation and Translocation of the Insulin-like Growth Factor-1 Receptor to the Cell Surface: Evidence for a New Link between 3-Hydroxy-3-Methylglutaryl-Coenzyme a Reductase and Cell Growth. J. Biol. Chem. 1996, 271, 17453–17462. [Google Scholar] [CrossRef] [Green Version]
- Nagae, M.; Kizuka, Y.; Mihara, E.; Kitago, Y.; Hanashima, S.; Ito, Y.; Takagi, J.; Taniguchi, N.; Yamaguchi, Y. Structure and Mechanism of Cancer-Associated N-Acetylglucosaminyltransferase-V. Nat. Commun. 2018, 9, 3380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; ten Dijke, P.; Wuhrer, M.; Zhang, T. Role of Glycosylation in TGF-β Signaling and Epithelial-to-Mesenchymal Transition in Cancer. Protein Cell 2021, 12, 89–106. [Google Scholar] [CrossRef]
- Kizuka, Y.; Taniguchi, N. Enzymes for N-Glycan Branching and Their Genetic and Nongenetic Regulation in Cancer. Biomolecules 2016, 6, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markowska, A.I.; Jefferies, K.C.; Panjwani, N. Galectin-3 Protein Modulates Cell Surface Expression and Activation of Vascular Endothelial Growth Factor Receptor 2 in Human Endothelial Cells. J. Biol. Chem. 2011, 286, 29913–29921. [Google Scholar] [CrossRef] [Green Version]
- Erdemir, H.H.; Li, Z.; Sacks, D.B. IQGAP1 Binds to Estrogen Receptor-α and Modulates Its Function. J. Biol. Chem. 2014, 289, 9100–9112. [Google Scholar] [CrossRef] [Green Version]
- Kohno, T.; Urao, N.; Ashino, T.; Sudhahar, V.; Inomata, H.; Yamaoka-Tojo, M.; McKinney, R.D.; Fukai, T.; Ushio-Fukai, M. IQGAP1 Links PDGF Receptor-β Signal to Focal Adhesions Involved in Vascular Smooth Muscle Cell Migration: Role in Neointimal Formation after Vascular Injury. Am. J. Physiol.-Cell Physiol. 2013, 305, C591–C600. [Google Scholar] [CrossRef] [Green Version]
- McNulty, D.E.; Li, Z.; White, C.D.; Sacks, D.B.; Annan, R.S. MAPK Scaffold IQGAP1 Binds the EGF Receptor and Modulates Its Activation. J. Biol. Chem. 2011, 286, 15010–15021. [Google Scholar] [CrossRef] [Green Version]
- White, C.D.; Li, Z.; Dillon, D.A.; Sacks, D.B. IQGAP1 Protein Binds Human Epidermal Growth Factor Receptor 2 (HER2) and Modulates Trastuzumab Resistance. J. Biol. Chem. 2011, 286, 29734–29747. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka-Tojo, M.; Ushio-Fukai, M.; Hilenski, L.; Dikalov, S.I.; Chen, Y.E.; Tojo, T.; Fukai, T.; Fujimoto, M.; Patrushev, N.A.; Wang, N.; et al. IQGAP1, a Novel Vascular Endothelial Growth Factor Receptor Binding Protein, Is Involved in Reactive Oxygen Species—Dependent Endothelial Migration and Proliferation. Circ. Res. 2004, 95, 276–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, M.; Li, Z.; Sacks, D.B. IQGAP1 Is a Scaffold for Mitogen-Activated Protein Kinase Signaling. Mol. Cell Biol. 2005, 25, 7940–7952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, M.D.; Sacks, D.B. IQGAP1 in Cellular Signaling: Bridging the GAP. Trends Cell Biol. 2006, 16, 242–249. [Google Scholar] [CrossRef]
- Abel, A.M.; Schuldt, K.M.; Rajasekaran, K.; Hwang, D.; Riese, M.J.; Rao, S.; Thakar, M.S.; Malarkannan, S. IQGAP1: Insights into the Function of a Molecular Puppeteer. Mol. Immunol. 2015, 65, 336–349. [Google Scholar] [CrossRef] [Green Version]
- Weissbach, L.; Settleman, J.; Kalady, M.F.; Snijders, A.J.; Murthy, A.E.; Yan, Y.-X.; Bernards, A. Identification of a Human RasGAP-Related Protein Containing Calmodulin-Binding Motifs. J. Biol. Chem. 1994, 269, 20517–20521. [Google Scholar] [CrossRef]
- Kuroda, S.; Fukata, M.; Kobayashi, K.; Nakafuku, M.; Nomura, N.; Iwamatsu, A.; Kaibuchi, K. Identification of IQGAP as a Putative Target for the Small GTPases, Cdc42 and Rac1. J. Biol. Chem. 1996, 271, 23363–23367. [Google Scholar] [CrossRef] [Green Version]
- Zoheir, K.; Abd-Rabou, A.A.; Harisa, G.I.; Kumar, A.; Ahmad, S.F.; Ansari, M.A.; Abd-Allah, A.R. IQGAP1 Gene Silencing Induces Apoptosis and Decreases the Invasive Capacity of Human Hepatocellular Carcinoma Cells. Tumor Biol. 2016, 37, 13927–13939. [Google Scholar] [CrossRef]
- Chen, M.; Choi, S.; Jung, O.; Wen, T.; Baum, C.; Thapa, N.; Lambert, P.F.; Rapraeger, A.C.; Anderson, R.A. The Specificity of EGF-Stimulated IQGAP1 Scaffold towards the PI3K-Akt Pathway Is Defined by the IQ3 Motif. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-MTOR Pathways: Cross-Talk and Compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaney, W.; Sundaram, S.; Friedman, N.; Stanley, P. The Lec4A CHO Glycosylation Mutant Arises from Miscompartmentalization of a Golgi Glycosyltransferase. J. Cell Biol. 1989, 109, 2089–2096. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, J.; Sundaram, S.; Wang, X.; Delgado, D.; Basu, R.; Stanley, P. A Point Mutation Causes Mistargeting of Golgi GlcNAc-TV in the Lec4A Chinese Hamster Ovary Glycosylation Mutant. J. Biol. Chem. 1996, 271, 27462–27469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro Web Server for Protein–Protein Docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for Rigid and Symmetric Docking. Nucleic Acids Res. 2005, 33 (Suppl. S2), W363–W367. [Google Scholar] [CrossRef] [Green Version]
- Ho, W.-L.; Hsu, W.-M.; Huang, M.-C.; Kadomatsu, K.; Nakagawara, A. Protein Glycosylation in Cancers and Its Potential Therapeutic Applications in Neuroblastoma. J. Hematol. Oncol. 2016, 9, 100. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Zhang, H. Characterization of Disease-Associated N -Linked Glycoproteins. Proteomics 2013, 13, 504–511. [Google Scholar] [CrossRef] [Green Version]
- Galonić, D.P.; Gin, D.Y. Chemical Glycosylation in the Synthesis of Glycoconjugate Antitumour Vaccines. Nature 2007, 446, 1000–1007. [Google Scholar] [CrossRef] [Green Version]
- Tang, F.; Zhou, M.; Qin, K.; Shi, W.; Yashinov, A.; Yang, Y.; Yang, L.; Guan, D.; Zhao, L.; Tang, Y. Selective N-Glycan Editing on Living Cell Surfaces to Probe Glycoconjugate Function. Nat. Chem. Biol. 2020, 16, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Pan, D.; Bellis, S.L.; Song, Y. Effect of Altered Glycosylation on the Structure of the I-like Domain of Β1 Integrin: A Molecular Dynamics Study. Proteins Struct. Funct. Bioinform. 2008, 73, 989–1000. [Google Scholar] [CrossRef] [PubMed]
- Marsico, G.; Russo, L.; Quondamatteo, F.; Pandit, A. Glycosylation and Integrin Regulation in Cancer. Trends Cancer 2018, 4, 537–552. [Google Scholar] [CrossRef]
- Bellis, S.L. Variant Glycosylation: An Underappreciated Regulatory Mechanism for Β1 Integrins. Biochim. Biophys. Acta (BBA)-Biomembr. 2004, 1663, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Stawikowski, M.J.; Aukszi, B.; Stawikowska, R.; Cudic, M.; Fields, G.B. Glycosylation Modulates Melanoma Cell A2β1 and A3β1 Integrin Interactions with Type IV Collagen. J. Biol. Chem. 2014, 289, 21591–21604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klaver, E.; Zhao, P.; May, M.; Flanagan-Steet, H.; Freeze, H.H.; Gilmore, R.; Wells, L.; Contessa, J.; Steet, R. Selective Inhibition of N-Linked Glycosylation Impairs Receptor Tyrosine Kinase Processing. Dis. Model Mech. 2019, 12, dmm039602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beattie, J.; McIntosh, L.; van der Walle, C.F. Cross-talk between the Insulin-like Growth Factor (IGF) Axis and Membrane Integrins to Regulate Cell Physiology. J. Cell. Physiol. 2010, 224, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Sarker, F.A.; Prior, V.G.; Bax, S.; O’Neill, G.M. Forcing a Growth Factor Response–Tissue-Stiffness Modulation of Integrin Signaling and Crosstalk with Growth Factor Receptors. J. Cell Sci. 2020, 133, jcs242461. [Google Scholar] [CrossRef]
- Takada, Y.; Takada, Y.K.; Fujita, M. Crosstalk between Insulin-like Growth Factor (IGF) Receptor and Integrins through Direct Integrin Binding to IGF1. Cytokine Growth Factor Rev. 2017, 34, 67–72. [Google Scholar] [CrossRef] [Green Version]
- White, C.D.; Brown, M.D.; Sacks, D.B. IQGAPs in Cancer: A Family of Scaffold Proteins Underlying Tumorigenesis. FEBS Lett. 2009, 583, 1817–1824. [Google Scholar] [CrossRef] [Green Version]
- Jadeski, L.; Mataraza, J.M.; Jeong, H.-W.; Li, Z.; Sacks, D.B. IQGAP1 Stimulates Proliferation and Enhances Tumorigenesis of Human Breast Epithelial Cells. J. Biol. Chem. 2008, 283, 1008–1017. [Google Scholar] [CrossRef] [Green Version]
- Benseñor, L.B.; Kan, H.-M.; Wang, N.; Wallrabe, H.; Davidson, L.A.; Cai, Y.; Schafer, D.A.; Bloom, G.S. IQGAP1 Regulates Cell Motility by Linking Growth Factor Signaling to Actin Assembly. J. Cell Sci. 2007, 120, 658–669. [Google Scholar] [CrossRef] [Green Version]
- Hedman, A.C.; Smith, J.M.; Sacks, D.B. The Biology of IQGAP Proteins: Beyond the Cytoskeleton. EMBO Rep. 2015, 16, 427–446. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.M.; Canniff, N.P.; Guay, K.P.; Larsen, I.S.B.; Hebert, D.N. Quantitative Glycoproteomics Reveals Cellular Substrate Selectivity of the ER Protein Quality Control Sensors UGGT1 and UGGT2. Elife 2020, 9, e63997. [Google Scholar] [CrossRef] [PubMed]
- Kutbi, E.; Alsaif, H.; AlOtaiby, S.; Baradwan, S. Association of Chromosome 6 Open Reading Frame 106 in Different Cancers. Front. Biosci. (Landmark Ed.) 2021, 26, 360–369. [Google Scholar] [PubMed]
- Wei, J.; Huang, K.; Chen, Z.; Hu, M.; Bai, Y.; Lin, S.; Du, H. Characterization of Glycolysis-Associated Molecules in the Tumor Microenvironment Revealed by Pan-Cancer Tissues and Lung Cancer Single Cell Data. Cancers 2020, 12, 1788. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession | Description | q-Value | Peptides | Abundances (Grouped) W5 | Abundances (Grouped) Lec1 | Abundances (Grouped) Lec4 |
|---|---|---|---|---|---|---|
| P53690 | Matrix metalloproteinase-14 OS = Mus musculus OX = 10090 GN = Mmp14 PE = 2 SV = 3 | 0 | 4 | 21.7 | 280.9 | 62.6 |
| P09055 | Integrin beta-1 OS = Mus musculus OX = 10090 GN = Itgb1 PE = 1 SV = 1 | 0 | 10 | 19.1 | 191.5 | 102.3 |
| Q71LX8 | Heat shock protein 84b OS = Mus musculus OX = 10090 GN = Hsp90ab1 PE = 1 SV = 1 | 0 | 16 | 22.8 | 130.6 | 113.1 |
| Q6IFX2 | Keratin, type I cytoskeletal 42 OS = Mus musculus OX = 10090 GN = Krt42 PE = 1 SV = 1 | 0 | 13 | 47.9 | 173.8 | 100.4 |
| A0A0R4J0I9 | Low density lipoprotein receptor-related protein 1 OS = Mus musculus OX = 10090 GN = Lrp1 PE = 1 SV = 1 | 0 | 8 | 59.9 | 173.1 | 66.8 |
| Q3UFT0 | Uncharacterized protein (Fragment) OS = Mus musculus OX = 10090 GN = Myh9 PE = 2 SV = 1 | 0 | 14 | 55.4 | 160.8 | 62.3 |
| Q3TIZ0 | Tubulin alpha chain OS = Mus musculus OX = 10090 GN = Tuba1c PE = 2 SV = 1 | 0 | 16 | 58.9 | 185.6 | 70.8 |
| E9QNN1 | ATP-dependent RNA helicase A OS = Mus musculus OX = 10090 GN = Dhx9 PE = 1 SV = 1 | 0 | 11 | 79.9 | 85.7 | 86.6 |
| Q8BTM8 | Filamin-A OS = Mus musculus OX = 10090 GN = Flna PE = 1 SV = 5 | 0 | 13 | 50 | 50.2 | 125.1 |
| Q3U7R1 | Extended synaptotagmin-1 OS = Mus musculus OX = 10090 GN = Esyt1 PE = 1 SV = 2 | 0 | 5 | 45.7 | 47.9 | 205.6 |
| A8IP69 | 14-3-3 protein gamma subtype OS = Mus musculus OX = 10090 GN = Ywhag PE = 1 SV = 1 | 0 | 2 | 185.6 | 185.9 | 25.2 |
| A0A0A0MQF6 | Glyceraldehyde-3-phosphate dehydrogenase OS = Mus musculus OX = 10090 GN = Gapdh PE = 1 SV = 1 | 0 | 6 | 77.8 | 76.9 | 106.7 |
| P07901 | Heat shock protein HSP 90-alpha OS = Mus musculus OX = 10090 GN = Hsp90aa1 PE = 1 SV = 4 | 0 | 11 | 130.8 | 102.9 | 62 |
| P02535 | Keratin, type I cytoskeletal 10 OS = Mus musculus OX = 10090 GN = Krt10 PE = 1 SV = 3 | 0 | 9 | 45.1 | 28.2 | 146.9 |
| Q6P5E4 | UDP-glucose: glycoprotein glucosyltransferase 1 OS = Mus musculus OX = 10090 GN = Uggt1 PE = 1 SV = 4 | 0 | 3 | 244.4 | 149.1 | 2.8 |
| Q61543 | Golgi apparatus protein 1 OS = Mus musculus OX = 10090 GN = Glg1 PE = 1 SV = 1 | 0 | 4 | 120.6 | 73.5 | 137.3 |
| P62806 | Histone H4 OS = Mus musculus OX = 10090 GN = Hist1h4a PE = 1 SV = 2 | 0 | 2 | 310.2 | 89.8 | - |
| Q8VHY0 | Chondroitin sulfate proteoglycan 4 OS = Mus musculus OX = 10090 GN = Cspg4 PE = 1 SV = 3 | 0 | 21 | 279.3 | 83.4 | 18.2 |
| Q9D8N0 | Elongation factor 1-gamma OS = Mus musculus OX = 10090 GN = Eef1g PE = 1 SV = 3 | 0 | 4 | 200.9 | 60.9 | 91.9 |
| Q3UV17 | Keratin, type II cytoskeletal 2 oral OS = Mus musculus OX = 10090 GN = Krt76 PE = 1 SV = 1 | 0 | 9 | 337.3 | 60.6 | 2 |
| F8WHL2 | Coatomer subunit alpha OS = Mus musculus OX = 10090 GN = Copa PE = 1 SV = 1 | 0 | 6 | 29.5 | 9 | 161.7 |
| A0A2R8VHP3 | Predicted pseudogene 5478 OS = Mus musculus OX = 10090 GN = Gm5478 PE = 1 SV = 1 | 0 | 9 | 25.1 | 6.3 | 193.7 |
| Q6NZJ6 | Eukaryotic translation initiation factor 4 gamma 1 OS = Mus musculus OX = 10090 GN = Eif4g1 PE = 1 SV = 1 | 0 | 5 | 229 | 50.6 | 35 |
| Q9QXS1 | Plectin OS = Mus musculus OX = 10090 GN = Plec PE = 1 SV = 3 | 0 | 53 | 54.5 | 9.8 | 18.1 |
| B2RU65 | Deubiquitinating enzyme 1 OS = Mus musculus OX = 10090 GN = Usp17la PE = 2 SV = 1 | 0.008 | 1 | 352.1 | 47.4 | 0.3 |
| P20029 | Endoplasmic reticulum chaperone BiP OS = Mus musculus OX = 10090 GN = Hspa5 PE = 1 SV = 3 | 0 | 22 | 134.5 | 18.3 | 155.1 |
| E9Q557 | Desmoplakin OS = Mus musculus OX = 10090 GN = Dsp PE = 1 SV = 1 | 0 | 7 | 178.6 | 29.6 | 133.1 |
| Q6A0F1 | MKIAA0002 protein (Fragment) OS = Mus musculus OX = 10090 GN = Cct8 PE = 2 SV = 1 | 0 | 7 | 199.9 | 29.9 | 58.5 |
| P70168 | Importin subunit beta-1 OS = Mus musculus OX = 10090 GN = Kpnb1 PE = 1 SV = 2 | 0 | 7 | 203.3 | 27.6 | 82.3 |
| Q7TMK9 | Heterogeneous nuclear ribonucleoprotein Q OS = Mus musculus OX = 10090 GN = Syncrip PE = 1 SV = 2 | 0 | 3 | 98.5 | 15.3 | 65.1 |
| P17751 | Triosephosphate isomerase OS = Mus musculus OX = 10090 GN = Tpi1 PE = 1 SV = 4 | 0.003 | 1 | 342.5 | 44.1 | 7.4 |
| Q60751 | Insulin-like growth factor 1 receptor OS = Mus musculus OX = 10090 GN = IGF-1R PE = 1 SV = 3 | 0.001 | 1 | 358.9 | 41.1 | - |
| Q542X7 | Chaperonin subunit 2 (Beta), isoform CRAa OS = Mus musculus OX = 10090 GN = Cct2 PE = 1 SV = 1 | 0 | 3 | 214.5 | 23 | 90 |
| Q8BFY9 | Transportin-1 OS = Mus musculus OX = 10090 GN = Tnpo1 PE = 1 SV = 2 | 0 | 1 | 329.1 | 27 | 17.2 |
| Q3UCD3 | Annexin OS = Mus musculus OX = 10090 GN = Anxa2 PE = 2 SV = 1 | 0 | 3 | 299.4 | 11.4 | 45.1 |
| Q3TRW3 | Uncharacterized protein OS = Mus musculus OX = 10090 GN = Snd1 PE = 2 SV = 1 | 0 | 7 | 182.6 | 4.8 | 102.3 |
| Q3TH04 | Uncharacterized protein (Fragment) OS = Mus musculus OX = 10090 GN = Hspa8 PE = 2 SV = 1 | 0 | 9 | 369.3 | 7.2 | 4.8 |
| Q6ZQK2 | MKIAA0051 protein (Fragment) OS = Mus musculus OX = 10090 GN = IQGAP1 PE = 2 SV = 1 | 0 | 7 | 307.7 | 1.6 | 28.4 |
| B2CY77 | Laminin receptor (Fragment) OS = Mus musculus OX = 10090 GN = Rpsa PE = 2 SV = 1 | 0 | 2 | 111.2 | - | 86.4 |
| A0A1L1SV25 | Alpha-actinin-4 OS = Mus musculus OX = 10090 GN = Actn4 PE = 1 SV = 1 | 0 | 3 | 194.8 | - | 88.3 |
| G5E8F8 | Integrin beta OS = Mus musculus OX = 10090 GN = Itgb5 PE = 1 SV = 1 | 0 | 4 | 259 | - | 73.3 |
| B7ZWC0 | N-myc downstream regulated gene 1 OS = Mus musculus OX = 10090 GN = Ndrg1 PE = 2 SV = 1 | 0 | 1 | 307.5 | - | 49.2 |
| G5E866 | Splicing factor 3B subunit 1 OS = Mus musculus OX = 10090 GN = Sf3b1 PE = 1 SV = 1 | 0 | 2 | 339.9 | - | 24.9 |
| Q9DCL9 | Multifunctional protein ADE2 OS = Mus musculus OX = 10090 GN = Paics PE = 1 SV = 4 | 0 | 375.4 | - | 10.5 | |
| Q3U900 | Dolichyl-diphosphooligosaccharide--protein glycosyltransferase subunit 1 OS = Mus musculus OX = 10090 GN = Rpn1 PE = 2 SV = 1 | 0.005 | 1 | 273 | - | 12.9 |
| Q792Z1 | MCG140784 OS = Mus musculus OX = 10090 GN = Try10 PE = 1 SV = 1 | 0 | 1 | 391 | - | 4 |
| A0JNY7 | Eukaryotic translation initiation factor 4, gamma 2 OS = Mus musculus OX = 10090 GN = Eif4g2 PE = 2 SV = 1 | 0 | 1 | 398.2 | - | 0.5 |
| P29341 | Polyadenylate-binding protein 1 OS = Mus musculus OX = 10090 GN = Pabpc1 PE = 1 SV = 2 | 0 | 4 | 178.9 | 10 | 63.6 |
| Lec1 Cells | ||
|---|---|---|
| Proteins | Protein Function and Activity | Regulation |
| MMP14 | Collagenolytic activity | Upregulated |
| KRT42, TUBA1C | Microtubule association | Upregulated |
| ITGB1 | Cytoskeleton and the extracellular matrix interaction. | Upregulated |
| HSP90AB1 | Chaperone-related activity | Upregulated |
| LRP9 | Internalization/signal transduction | Upregulated |
| IQGAP1 | GTPase activating protein | Downregulated |
| IGF1-R | Cell growth proliferation and survival control | Downregulated |
| Lec4 Cells | ||
| Proteins | Protein Function and Activity | Regulation |
| MMP14 | Collagenolytic activity | Upregulated |
| KRT42, KRT10, TUBA1C, GM5478, FLNA | Microtubule association | Upregulated |
| ITGB1 | Cytoskeleton and the extracellular matrix interaction. | Upregulated |
| HSP90AB1 | Chaperone-related activity | Upregulated |
| COPA, EYST1 | Protein transport | Upregulated |
| GAPDH | Glycolysis | Upregulated |
| IQGAP-1 | GTPase activating protein. | Downregulated |
| Gene | Nucleotide Sequence from 5′to 3′ | Product Size in bp | |
|---|---|---|---|
| IGF-1R | Forward Reverse | GAGCCAAGACCCGAAAACTC AGTTCCCTGGGTTTAGACGG | 181 |
| HSPA5 | Forward Reverse | TATTCCTGCGTCGGTGTGTT ATTCCAAGTGCGTCCGATGA | 198 |
| HSPA8 | Forward Reverse | TGATCGGGCGTAGGTTTGAT CGCTTCTGCAATTTCCTTCA | 202 |
| UGGT1 | Forward Reverse | TACGATGCCGTATTGGAAGC ACGACTTGCACCCTTCTGGT | 221 |
| IQGAP1 | Forward Reverse | GGCAGAACGTGGCTTATGAA TTTCTTCAGGGACACCACTTTG | 209 |
| MMP14 | Forward Reverse | CCAAGGCAGCAACTTCAGC CAAATCAGCCTTGCCTGTCA | 212 |
| ITGB1 | Forward Reverse | AATGCCAAATCTTGCGGAGA TATGTCACTTGGCTGGCAAC | 221 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salvi, R.; Kumar, C.; Brahmbhatt, K.; Subedi, R.; Idicula-Thomas, S.; Madan, T.; Biswas, B. N-Linked Glycosylation in Chinese Hamster Ovary Cells Is Critical for Insulin-like Growth Factor 1 Signaling. Int. J. Mol. Sci. 2022, 23, 14952. https://doi.org/10.3390/ijms232314952
Salvi R, Kumar C, Brahmbhatt K, Subedi R, Idicula-Thomas S, Madan T, Biswas B. N-Linked Glycosylation in Chinese Hamster Ovary Cells Is Critical for Insulin-like Growth Factor 1 Signaling. International Journal of Molecular Sciences. 2022; 23(23):14952. https://doi.org/10.3390/ijms232314952
Chicago/Turabian StyleSalvi, Rupashree, Chandan Kumar, Krupanshi Brahmbhatt, Rambhadur Subedi, Susan Idicula-Thomas, Taruna Madan, and Barnali Biswas. 2022. "N-Linked Glycosylation in Chinese Hamster Ovary Cells Is Critical for Insulin-like Growth Factor 1 Signaling" International Journal of Molecular Sciences 23, no. 23: 14952. https://doi.org/10.3390/ijms232314952