
Using Genomic Variation to Distinguish Ovarian High-Grade Serous Carcinoma from Benign Fallopian Tubes

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
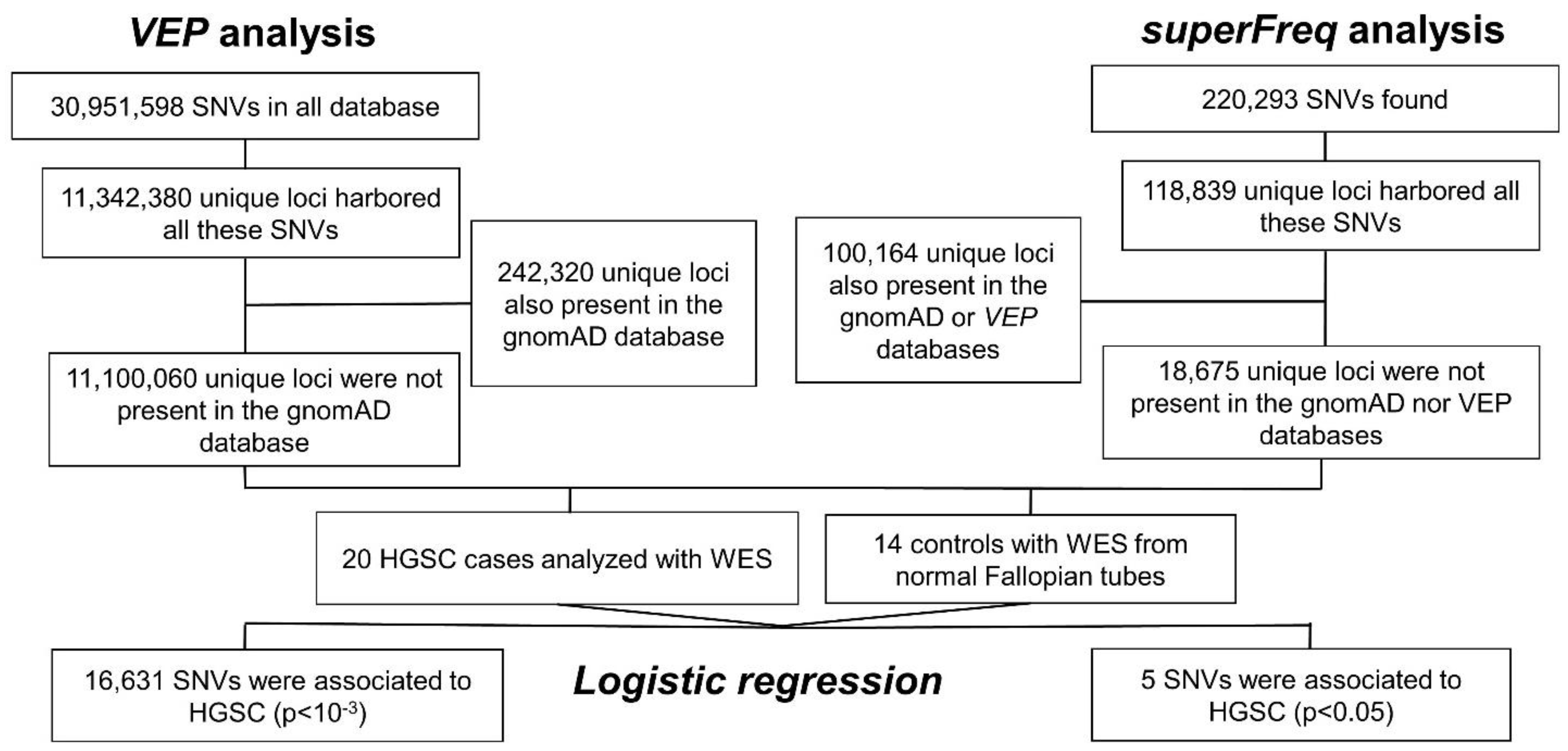
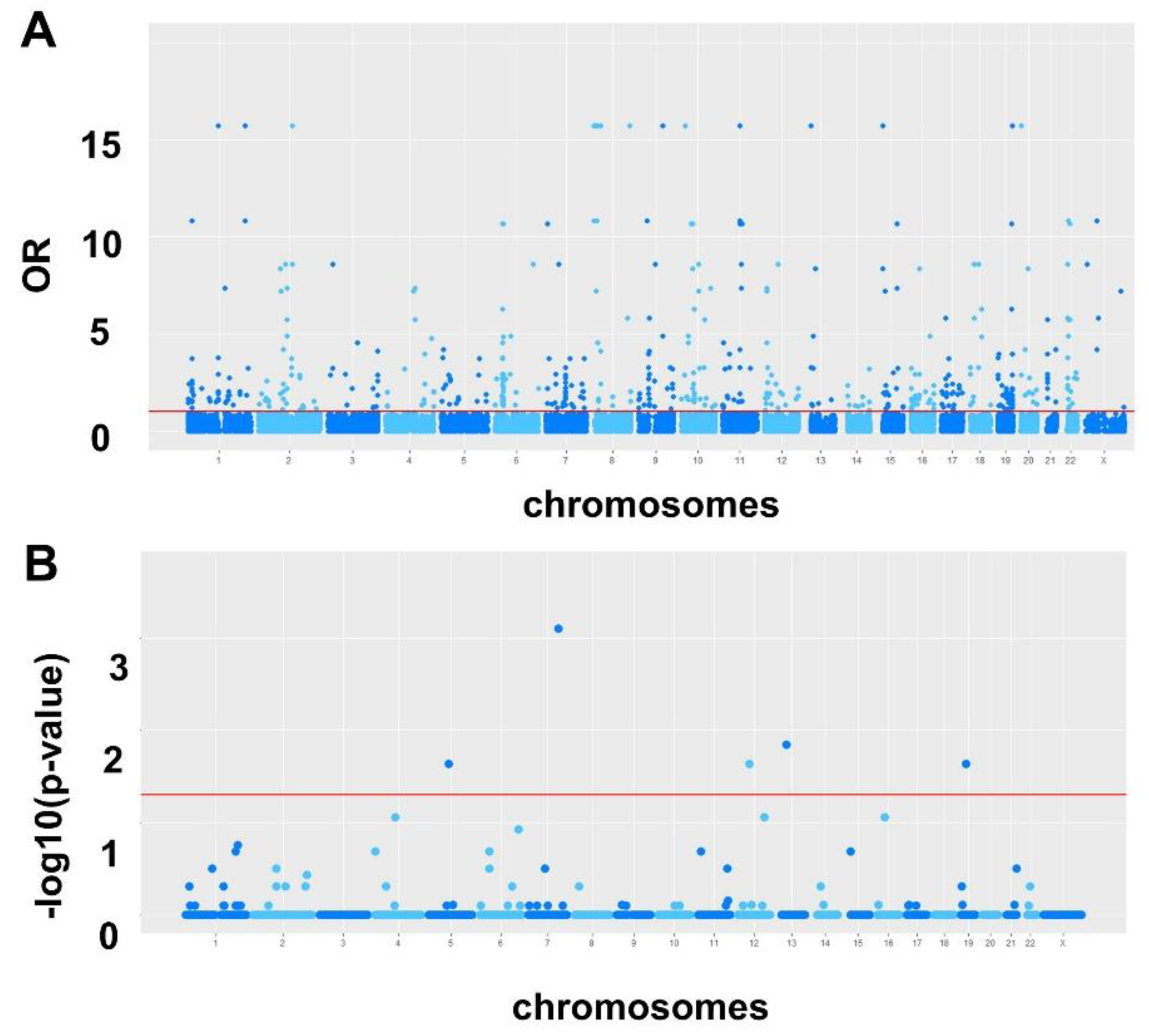
2.1. Single Nucleotide Variation
2.1.1. VEP Analysis
2.1.2. superFreq Analysis
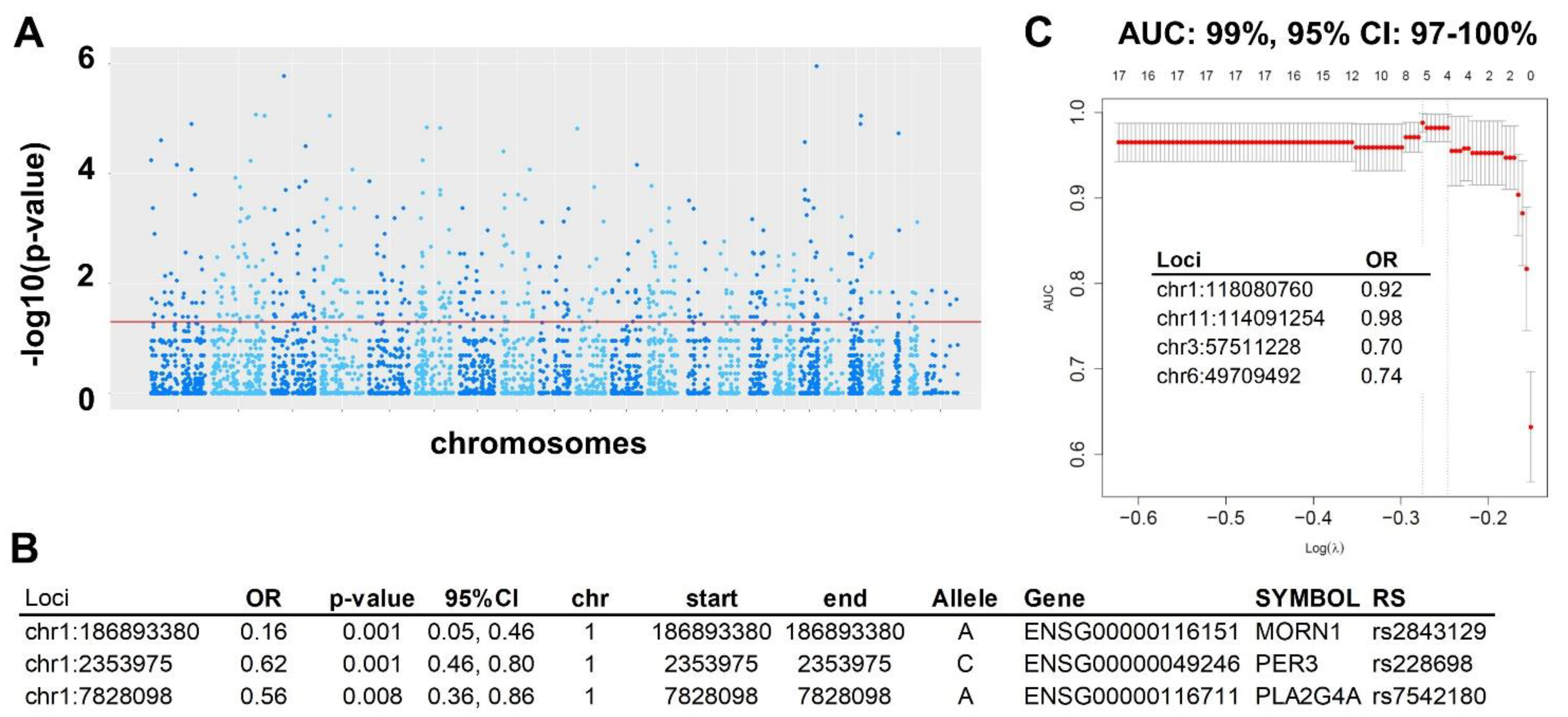
2.1.3. Prediction Modeling
2.1.4. Validation in RNA-seq
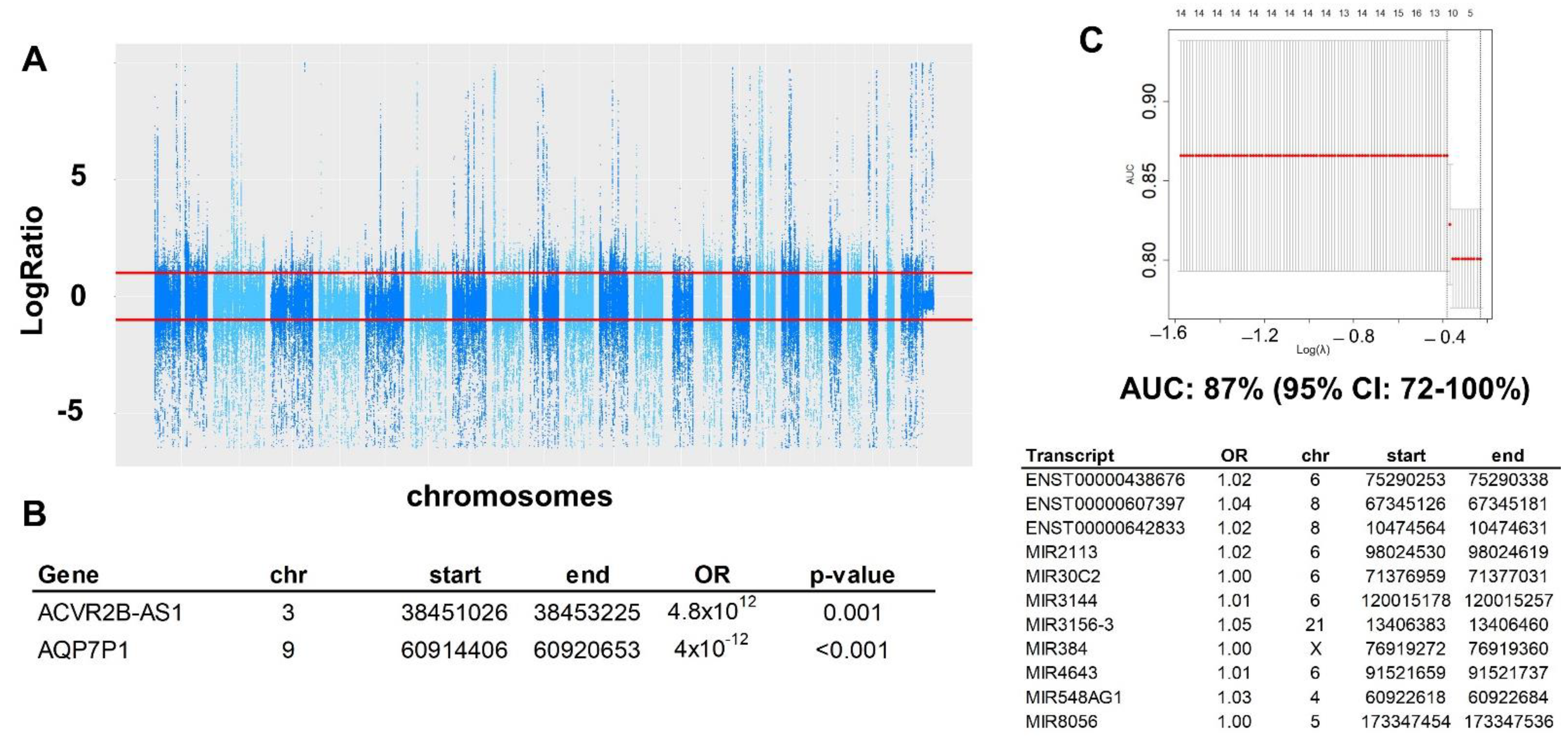
2.2. Copy Number Variation (CNV)
2.3. Structural Variation (SV)
3. Discussion
4. Materials and Methods
4.1. Specimen Acquisition
4.2. DNA Sequencing
4.3. RNA Sequencing
4.4. Single Nucleotide Variation (SNV) Analysis
4.5. Copy Number Variant (CNV) Analysis
4.6. Structural Variation (SV) Analysis
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.; Noone, A.; Krapcho, M. SEER Cancer Statistics Review 1975–2016; National Cancer Institute: Bethesda, MD, USA, 2019. [Google Scholar]
- Grossman, D.C.; Curry, S.J.; Owens, D.K.; Barry, M.J.; Davidson, K.W.; Doubeni, C.A.; Epling, J.W., Jr.; Kemper, A.R.; Krist, A.H.; Landefeld, C.S.; et al. Screening for Ovarian Cancer: US Preventive Services Task Force Recommendation Statement. JAMA 2018, 319, 588–594. [Google Scholar]
- American College of Obstetricians and Gynecologists. Practice Bulletin No. 174: Evaluation and Management of Adnexal Masses. Obstet. Gynecol. 2016, 128, e210–e226. [Google Scholar] [CrossRef]
- Patel, M.D.; Ascher, S.M.; Paspulati, R.M.; Shanbhogue, A.K.; Siegelman, E.S.; Stein, M.W.; Berland, L.L. Managing incidental findings on abdominal and pelvic CT and MRI, part 1: White paper of the ACR Incidental Findings Committee II on adnexal findings. J. Am. Coll. Radiol. 2013, 10, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, D.; Planchamp, F.; Bourne, T.; Landolfo, C.; du Bois, A.; Chiva, L.; Cibula, D.; Concin, N.; Fischerova, D.; Froyman, W.; et al. ESGO/ISUOG/IOTA/ESGE Consensus Statement on pre-operative diagnosis of ovarian tumors. Int. J. Gynecol. Cancer 2021, 31, 961–982. [Google Scholar] [CrossRef]
- Amor, F.; Alcazar, J.L.; Vaccaro, H.; Leon, M.; Iturra, A. GI-RADS reporting system for ultrasound evaluation of adnexal masses in clinical practice: A prospective multicenter study. Ultrasound Obstet. Gynecol. 2011, 38, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Buys, S.S.; Partridge, E.; Greene, M.H.; Prorok, P.C.; Reding, D.; Riley, T.L.; Hartge, P.; Fagerstrom, R.M.; Ragard, L.R.; Chia, D.; et al. Ovarian cancer screening in the Prostate, Lung, Colorectal and Ovarian (PLCO) cancer screening trial: Findings from the initial screen of a randomized trial. Am. J. Obstet. Gynecol. 2005, 193, 1630–1639. [Google Scholar] [CrossRef]
- Earle, C.C.; Schrag, D.; Neville, B.A.; Yabroff, K.R.; Topor, M.; Fahey, A.; Trimble, E.L.; Bodurka, D.C.; Bristow, R.E.; Carney, M.; et al. Effect of surgeon specialty on processes of care and outcomes for ovarian cancer patients. J. Natl. Cancer Inst. 2006, 98, 172–180. [Google Scholar] [CrossRef]
- Elit, L.; Bondy, S.J.; Paszat, L.; Przybysz, R.; Levine, M. Outcomes in surgery for ovarian cancer. Gynecol. Oncol. 2002, 87, 260–267. [Google Scholar] [CrossRef]
- Goff, B.A.; Matthews, B.J.; Wynn, M.; Muntz, H.G.; Lishner, D.M.; Baldwin, L.M. Ovarian cancer: Patterns of surgical care across the United States. Gynecol. Oncol. 2006, 103, 383–390. [Google Scholar] [CrossRef]
- Vernooij, F.; Heintz, A.P.; Coebergh, J.W.; Massuger, L.F.; Witteveen, P.O.; van der Graaf, Y. Specialized and high-volume care leads to better outcomes of ovarian cancer treatment in the Netherlands. Gynecol. Oncol. 2009, 112, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Bodurtha Smith, A.J.; Pena, D.; Ko, E. Insurance-Mediated Disparities in Gynecologic Oncology Care. Obstet. Gynecol. 2022, 139, 305–312. [Google Scholar] [CrossRef]
- Saint Pierre, A.; Genin, E. How important are rare variants in common disease? Brief. Funct. Genom. 2014, 13, 353–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuk, O.; Schaffner, S.F.; Samocha, K.; Do, R.; Hechter, E.; Kathiresan, S.; Daly, M.J.; Neale, B.M.; Sunyaev, S.R.; Lander, E.S. Searching for missing heritability: Designing rare variant association studies. Proc. Natl. Acad. Sci. USA 2014, 111, E455–E464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martignetti, J.A.; Camacho-Vanegas, O.; Priedigkeit, N.; Camacho, C.; Pereira, E.; Lin, L.; Garnar-Wortzel, L.; Miller, D.; Losic, B.; Shah, H.; et al. Personalized ovarian cancer disease surveillance and detection of candidate therapeutic drug target in circulating tumor DNA. Neoplasia 2014, 16, 97–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oikkonen, J.; Zhang, K.; Salminen, L.; Schulman, I.; Lavikka, K.; Andersson, N.; Ojanpera, E.; Hietanen, S.; Grenman, S.; Lehtonen, R.; et al. Prospective Longitudinal ctDNA Workflow Reveals Clinically Actionable Alterations in Ovarian Cancer. JCO Precis. Oncol. 2019, 3, PO-18. [Google Scholar] [CrossRef] [PubMed]
- Vanderstichele, A.; Busschaert, P.; Smeets, D.; Landolfo, C.; Van Nieuwenhuysen, E.; Leunen, K.; Neven, P.; Amant, F.; Mahner, S.; Braicu, E.I.; et al. Chromosomal Instability in Cell-Free DNA as a Highly Specific Biomarker for Detection of Ovarian Cancer in Women with Adnexal Masses. Clin. Cancer Res. 2017, 23, 2223–2231. [Google Scholar] [CrossRef] [Green Version]
- Nakabayashi, M.; Kawashima, A.; Yasuhara, R.; Hayakawa, Y.; Miyamoto, S.; Iizuka, C.; Sekizawa, A. Massively parallel sequencing of cell-free DNA in plasma for detecting gynaecological tumour-associated copy number alteration. Sci. Rep. 2018, 8, 11205. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Hu, G.; Yang, Q.; Dong, R.; Xie, X.; Ma, D.; Shen, K.; Kong, B. A multiplex methylation-specific PCR assay for the detection of early-stage ovarian cancer using cell-free serum DNA. Gynecol. Oncol. 2013, 130, 132–139. [Google Scholar] [CrossRef]
- Gupta, R.; Othman, T.; Chen, C.; Sandhu, J.; Ouyang, C.; Fakih, M. Guardant360 Circulating Tumor DNA Assay Is Concordant with FoundationOne Next-Generation Sequencing in Detecting Actionable Driver Mutations in Anti-EGFR Naive Metastatic Colorectal Cancer. Oncologist 2020, 25, 235–243. [Google Scholar] [CrossRef] [Green Version]
- Rolfo, C.; Mack, P.; Scagliotti, G.V.; Aggarwal, C.; Arcila, M.E.; Barlesi, F.; Bivona, T.; Diehn, M.; Dive, C.; Dziadziuszko, R.; et al. Liquid Biopsy for Advanced NSCLC: A Consensus Statement From the International Association for the Study of Lung Cancer. J. Thorac. Oncol. 2021, 16, 1647–1662. [Google Scholar] [CrossRef]
- Minato, T.; Ito, S.; Li, B.; Fujimori, H.; Mochizuki, M.; Yamaguchi, K.; Tamai, K.; Shimada, M.; Tokunaga, H.; Shigeta, S.; et al. Liquid biopsy with droplet digital PCR targeted to specific mutations in plasma cell-free tumor DNA can detect ovarian cancer recurrence earlier than CA125. Gynecol. Oncol. Rep. 2021, 38, 100847. [Google Scholar] [CrossRef]
- Romero, A.; Serna-Blasco, R.; Calvo, V.; Provencio, M. Use of Liquid Biopsy in the Care of Patients with Non-Small Cell Lung Cancer. Curr. Treat. Options Oncol. 2021, 22, 86. [Google Scholar] [CrossRef] [PubMed]
- Erickson, B.K.; Conner, M.G.; Landen, C.N., Jr. The role of the fallopian tube in the origin of ovarian cancer. Am. J. Obstet. Gynecol. 2013, 209, 409–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes, H.D.; Devor, E.J.; Warrier, A.; Newtson, A.M.; Mattson, J.; Wagner, V.; Duncan, G.N.; Leslie, K.K.; Gonzalez-Bosquet, J. Differential DNA methylation in high-grade serous ovarian cancer (HGSOC) is associated with tumor behavior. Sci. Rep. 2019, 9, 17996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez Bosquet, J.; Devor, E.J.; Newtson, A.M.; Smith, B.J.; Bender, D.P.; Goodheart, M.J.; McDonald, M.E.; Braun, T.A.; Thiel, K.W.; Leslie, K.K. Creation and validation of models to predict response to primary treatment in serous ovarian cancer. Sci. Rep. 2021, 11, 5957. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.D.; Salinas, E.A.; Newtson, A.M.; Sharma, D.; Keeney, M.E.; Warrier, A.; Smith, B.J.; Bender, D.P.; Goodheart, M.J.; Thiel, K.W.; et al. An integrated prediction model of recurrence in endometrial endometrioid cancers. Cancer Manag. Res. 2019, 11, 5301–5315. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Hunt, S.E.; Moore, B.; Amode, R.M.; Armean, I.M.; Lemos, D.; Mushtaq, A.; Parton, A.; Schuilenburg, H.; Szpak, M.; Thormann, A.; et al. Annotating and prioritizing genomic variants using the Ensembl Variant Effect Predictor-A tutorial. Hum. Mutat. 2022, 43, 986–997. [Google Scholar] [CrossRef]
- Flensburg, C.; Sargeant, T.; Oshlack, A.; Majewski, I.J. SuperFreq: Integrated mutation detection and clonal tracking in cancer. PLoS Comput. Biol. 2020, 16, e1007603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gudmundsson, S.; Singer-Berk, M.; Watts, N.A.; Phu, W.; Goodrich, J.K.; Solomonson, M.; Genome Aggregation Database Consortium; Rehm, H.L.; MacArthur, D.G.; O’Donnell-Luria, A. Variant interpretation using population databases: Lessons from gnomAD. Hum. Mutat. 2021, 43, 1012–1030. [Google Scholar] [CrossRef] [PubMed]
- Simon, R. Roadmap for developing and validating therapeutically relevant genomic classifiers. J. Clin. Oncol. 2005, 23, 7332–7341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Developers, T. TensorFlow. Available online: https://zenodo.org/record/5949169#.Y3DN3PdBxPY (accessed on 2 February 2022).
- Mohammad, N.; Muad, A.M.; Ahmad, R.; Yusof, M. Accuracy of advanced deep learning with tensorflow and keras for classifying teeth developmental stages in digital panoramic imaging. BMC Med. Imaging 2022, 22, 66. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Bosquet, J.; Marchion, D.C.; Chon, H.; Lancaster, J.M.; Chanock, S. Analysis of chemotherapeutic response in ovarian cancers using publically available high-throughput data. Cancer Res. 2014, 74, 3902–3912. [Google Scholar] [CrossRef] [Green Version]
- Olshen, A.B.; Venkatraman, E.S.; Lucito, R.; Wigler, M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 2004, 5, 557–572. [Google Scholar] [CrossRef]
- Cmero, M.; Schmidt, B.; Majewski, I.J.; Ekert, P.G.; Oshlack, A.; Davidson, N.M. MINTIE: Identifying novel structural and splice variants in transcriptomes using RNA-seq data. Genome Biol. 2021, 22, 296. [Google Scholar] [CrossRef]
- Haas, B.J.; Dobin, A.; Li, B.; Stransky, N.; Pochet, N.; Regev, A. Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biol. 2019, 20, 213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzalez-Bosquet, J.; Cardillo, N.D.; Reyes, H.D.; Smith, B.J.; Leslie, K.K.; Bender, D.P.; Goodheart, M.J.; Devor, E.J. Using Genomic Variation to Distinguish Ovarian High-Grade Serous Carcinoma from Benign Fallopian Tubes. Int. J. Mol. Sci. 2022, 23, 14814. https://doi.org/10.3390/ijms232314814
Gonzalez-Bosquet J, Cardillo ND, Reyes HD, Smith BJ, Leslie KK, Bender DP, Goodheart MJ, Devor EJ. Using Genomic Variation to Distinguish Ovarian High-Grade Serous Carcinoma from Benign Fallopian Tubes. International Journal of Molecular Sciences. 2022; 23(23):14814. https://doi.org/10.3390/ijms232314814
Chicago/Turabian StyleGonzalez-Bosquet, Jesus, Nicholas D. Cardillo, Henry D. Reyes, Brian J. Smith, Kimberly K. Leslie, David P. Bender, Michael J. Goodheart, and Eric J. Devor. 2022. "Using Genomic Variation to Distinguish Ovarian High-Grade Serous Carcinoma from Benign Fallopian Tubes" International Journal of Molecular Sciences 23, no. 23: 14814. https://doi.org/10.3390/ijms232314814