
Gene- and Gender-Related Decrease in Serum BDNF Levels in Alzheimer’s Disease
 , ,
, ,
Abstract
:1. Introduction
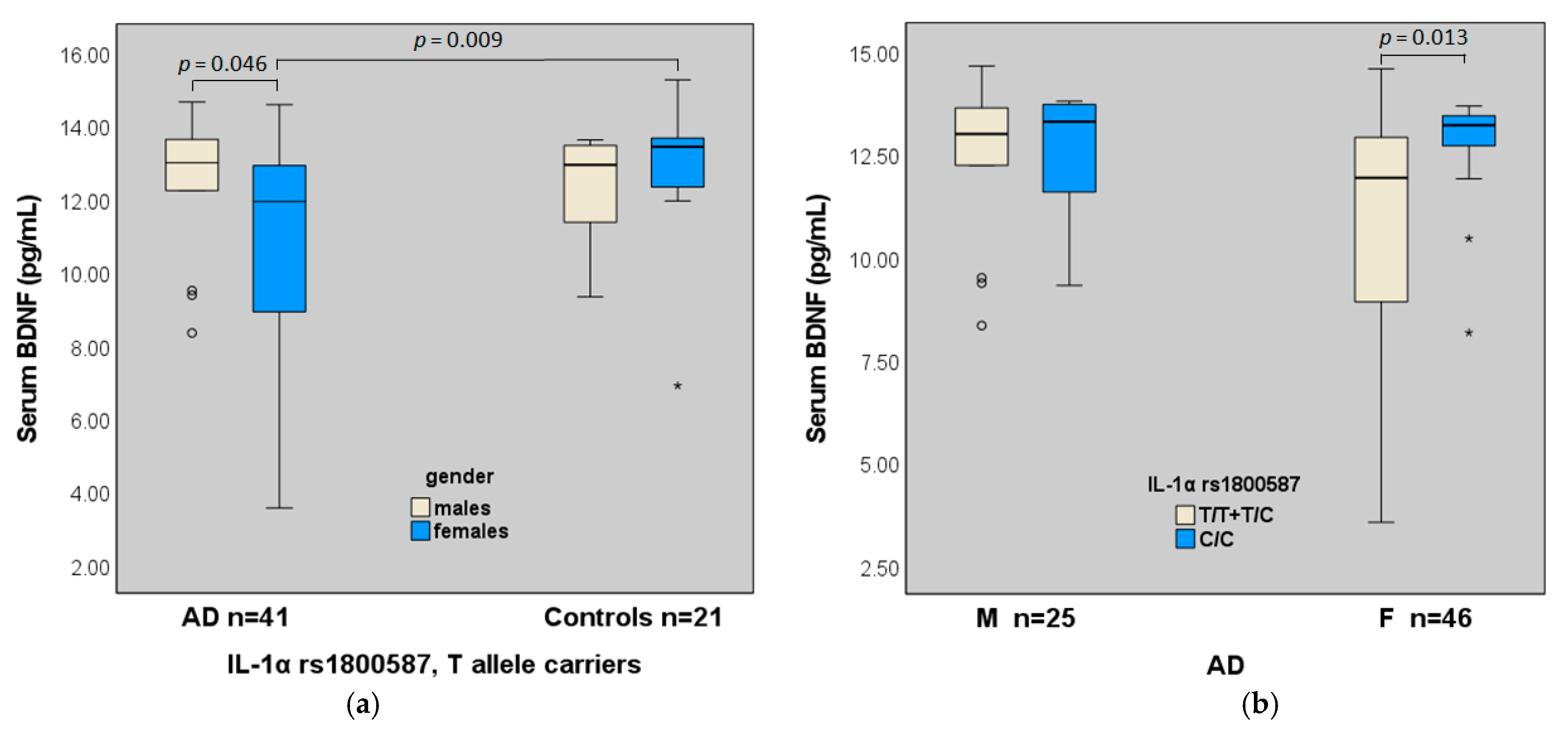
2. Results
2.1. Patients
2.2. Serum BDNF
2.3. Alleles, Genotypes and Haplotypes
2.4. BDNF (Val66Met, C270T), IL-1α Gene Polymorphisms and Serum Levels of BDNF
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Serum Collection, DNA Extraction and SNP Genotyping
4.3. Enzyme-Linked Immunosorbent Assay for Detection of BDNF
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in Neuronal Development and Function. Annu. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, T.; You, Y.; Joseph, C.; Mirzaei, M.; Klistorner, A.; Graham, S.L.; Gupta, V. BDNF Polymorphism: A Review of Its Diagnostic and Clinical Relevance in Neurodegenerative Disorders. Aging Dis. 2018, 9, 523. [Google Scholar] [CrossRef] [Green Version]
- Matyi, J.; Tschanz, J.T.; Rattinger, G.B.; Sanders, C.; Vernon, E.K.; Corcoran, C.; Kauwe, J.S.K.; Buhusi, M. Sex Differences in Risk for Alzheimer’s Disease Related to Neurotrophin Gene Polymorphisms: The Cache County Memory Study. J. Gerontol. Ser. A 2017, 72, 1607–1613. [Google Scholar] [CrossRef] [Green Version]
- Ng, T.; Ho, C.; Tam, W.; Kua, E.; Ho, R. Decreased Serum Brain-Derived Neurotrophic Factor (BDNF) Levels in Patients with Alzheimer’s Disease (AD): A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2019, 20, 257. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.Y.; Lee, S.H.; Graham, P.L.; Angelucci, F.; Lucia, A.; Pareja-Galeano, H.; Leyhe, T.; Turana, Y.; Lee, I.R.; Yoon, J.H.; et al. Peripheral Brain-Derived Neurotrophic Factor Levels in Alzheimer’s Disease and Mild Cognitive Impairment: A Comprehensive Systematic Review and Meta-Analysis. Mol. Neurobiol. 2017, 54, 7297–7311. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, F.; Spalletta, G.; Iulio, F.; Ciaramella, A.; Salani, F.; Varsi, A.; Gianni, W.; Sancesario, G.; Caltagirone, C.; Bossu, P. Alzheimers Disease (AD) and Mild Cognitive Impairment (MCI) Patients Are Characterized by Increased BDNF Serum Levels. Curr. Alzheimer Res. 2010, 7, 15–20. [Google Scholar] [CrossRef]
- O’Bryant, S.E.; Hobson, V.; Hall, J.R.; Waring, S.C.; Chan, W.; Massman, P.; Lacritz, L.; Cullum, C.M.; Diaz-Arrastia, R. Brain-Derived Neurotrophic Factor Levels in Alzheimer’s Disease. J. Alzheimers Dis. 2009, 17, 337–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, D.T.; Vickers, J.C.; Stuart, K.E.; Cechova, K.; Ward, D.D. The BDNF Val66Met Polymorphism Modulates Resilience of Neurological Functioning to Brain Ageing and Dementia: A Narrative Review. Brain Sci. 2020, 10, 195. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Zhang, Y.; Sterling, K.; Song, W. Brain-Derived Neurotrophic Factor in Alzheimer’s Disease and Its Pharmaceutical Potential. Transl. Neurodegener. 2022, 11, 4. [Google Scholar] [CrossRef]
- Italiani, P.; Puxeddu, I.; Napoletano, S.; Scala, E.; Melillo, D.; Manocchio, S.; Angiolillo, A.; Migliorini, P.; Boraschi, D.; Vitale, E.; et al. Circulating Levels of IL-1 Family Cytokines and Receptors in Alzheimer’s Disease: New Markers of Disease Progression? J. Neuroinflammation 2018, 15, 342. [Google Scholar] [CrossRef] [Green Version]
- Lai, K.S.P.; Liu, C.S.; Rau, A.; Lanctôt, K.L.; Köhler, C.A.; Pakosh, M.; Carvalho, A.F.; Herrmann, N. Peripheral Inflammatory Markers in Alzheimer’s Disease: A Systematic Review and Meta-Analysis of 175 Studies. J. Neurol. Neurosurg. Psychiatry 2017, 88, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Zhang, L.; Meng, Q.; Gao, Q. Association Between Interleukin-1A, Interleukin-1B, and Bridging Integrator 1 Polymorphisms and Alzheimer’s Disease: A Standard and Cumulative Meta-Analysis. Mol. Neurobiol. 2017, 54, 736–747. [Google Scholar] [CrossRef]
- Kornman, K.S. Interleukin 1 Genetics, Inflammatory Mechanisms, and Nutrigenetic Opportunities to Modulate Diseases of Aging. Am. J. Clin. Nutr. 2006, 83, 475S–483S. [Google Scholar] [CrossRef] [Green Version]
- Gamarra, D.; Elcoroaristizabal, X.; Fernández-Martínez, M.; de Pancorbo, M.M. Association of the C47T Polymorphism in SOD2 with Amnestic Mild Cognitive Impairment and Alzheimer’s Disease in Carriers of the APOEε4 Allele. Dis. Markers 2015, 2015, 746329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, T.; Song, X.; Zhu, C.; Patrick, R.; Skurla, M.; Santangelo, I.; Green, M.; Harper, D.; Ren, B.; Forester, B.P.; et al. Mitochondrial Dysfunction, Oxidative Stress, Neuroinflammation, and Metabolic Alterations in the Progression of Alzheimer’s Disease: A Meta-Analysis of in Vivo Magnetic Resonance Spectroscopy Studies. Ageing Res. Rev. 2021, 72, 101503. [Google Scholar] [CrossRef]
- Saeed, M.; Kausar, M.A.; Singh, R.; Siddiqui, A.J.; Akhter, A. The Role of Glyoxalase in Glycation and Carbonyl Stress Induced Metabolic Disorders. Curr. Protein Pept. Sci. 2020, 21, 846–859. [Google Scholar] [CrossRef]
- Haddad, M.; Perrotte, M.; Ben Khedher, M.R.; Madec, E.; Lepage, A.; Fülöp, T.; Ramassamy, C. Levels of Receptor for Advanced Glycation End Products and Glyoxalase-1 in the Total Circulating Extracellular Vesicles from Mild Cognitive Impairment and Different Stages of Alzheimer’s Disease Patients. J. Alzheimers Dis. 2021, 84, 227–237. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Coelho-Junior, H.; Bucci, C.; Marzetti, E. Circulating Extracellular Vesicles: Friends and Foes in Neurodegeneration. Neural Regen. Res. 2022, 17, 534. [Google Scholar] [CrossRef]
- Morris, B.J.; Willcox, D.C.; Donlon, T.A.; Willcox, B.J. FOXO3: A Major Gene for Human Longevity—A Mini-Review. Gerontology 2015, 61, 515–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bona, D.; Accardi, G.; Virruso, C.; Candore, G.; Caruso, C. Association between Genetic Variations in the Insulin/Insulin-Like Growth Factor (Igf-1) Signaling Pathway and Longevity: A Systematic Review and Meta-Analysis. Curr. Vasc. Pharmacol. 2013, 12, 674–681. [Google Scholar] [CrossRef]
- Anselmi, C.V.; Malovini, A.; Roncarati, R.; Novelli, V.; Villa, F.; Condorelli, G.; Bellazzi, R.; Puca, A.A. Association of the FOXO3A Locus with Extreme Longevity in a Southern Italian Centenarian Study. Rejuvenation Res. 2009, 12, 95–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albani, D.; Ateri, E.; Mazzuco, S.; Ghilardi, A.; Rodilossi, S.; Biella, G.; Ongaro, F.; Antuono, P.; Boldrini, P.; Di Giorgi, E.; et al. Modulation of Human Longevity by SIRT3 Single Nucleotide Polymorphisms in the Prospective Study “Treviso Longeva (TRELONG)”. AGE 2014, 36, 469–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, J.S.; Liu, L.; Yan, L.L.; Zeng, Y. Comparing Effects of FOXO3 and Residing in Urban Areas on Longevity: A Gene–Environment Interaction Study. J. Gerontol. Ser. A 2022, 77, 1549–1556. [Google Scholar] [CrossRef]
- Peculis, R.; Konrade, I.; Skapare, E.; Fridmanis, D.; Nikitina-Zake, L.; Lejnieks, A.; Pirags, V.; Dambrova, M.; Klovins, J. Identification of Glyoxalase 1 Polymorphisms Associated with Enzyme Activity. Gene 2013, 515, 140–143. [Google Scholar] [CrossRef]
- Camporez, D.; Belcavello, L.; Almeida, J.F.F.; Silva-Sena, G.G.; Pimassoni, L.H.S.; Morelato, R.L.; do Carmo Pimentel Batitucci, M.; de Paula, F. Positive Association of a Sirt1 Variant and Parameters of Oxidative Stress on Alzheimer’s Disease. Neurol. Sci. 2021, 42, 1843–1851. [Google Scholar] [CrossRef]
- Erickson, K.I.; Prakash, R.S.; Voss, M.W.; Chaddock, L.; Heo, S.; McLaren, M.; Pence, B.D.; Martin, S.A.; Vieira, V.J.; Woods, J.A.; et al. Brain-Derived Neurotrophic Factor Is Associated with Age-Related Decline in Hippocampal Volume. J. Neurosci. 2010, 30, 5368–5375. [Google Scholar] [CrossRef] [Green Version]
- Mori, Y.; Tsuji, M.; Oguchi, T.; Kasuga, K.; Kimura, A.; Futamura, A.; Sugimoto, A.; Kasai, H.; Kuroda, T.; Yano, S.; et al. Serum BDNF as a Potential Biomarker of Alzheimer’s Disease: Verification Through Assessment of Serum, Cerebrospinal Fluid, and Medial Temporal Lobe Atrophy. Front. Neurol. 2021, 12, 653267. [Google Scholar] [CrossRef]
- Amidfar, M.; de Oliveira, J.; Kucharska, E.; Budni, J.; Kim, Y.-K. The Role of CREB and BDNF in Neurobiology and Treatment of Alzheimer’s Disease. Life Sci. 2020, 257, 118020. [Google Scholar] [CrossRef]
- Rasmussen, P.; Brassard, P.; Adser, H.; Pedersen, M.V.; Leick, L.; Hart, E.; Secher, N.H.; Pedersen, B.K.; Pilegaard, H. Evidence for a Release of Brain-Derived Neurotrophic Factor from the Brain during Exercise: Brain-Derived Neurotrophic Factor Release during Exercise. Exp. Physiol. 2009, 94, 1062–1069. [Google Scholar] [CrossRef]
- Klein, A.B.; Williamson, R.; Santini, M.A.; Clemmensen, C.; Ettrup, A.; Rios, M.; Knudsen, G.M.; Aznar, S. Blood BDNF Concentrations Reflect Brain-Tissue BDNF Levels across Species. Int. J. Neuropsychopharmacol. 2011, 14, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Walsh, J.J.; Tschakovsky, M.E. Exercise and Circulating BDNF: Mechanisms of Release and Implications for the Design of Exercise Interventions. Appl. Physiol. Nutr. Metab. 2018, 43, 1095–1104. [Google Scholar] [CrossRef]
- Lee, B.-H.; Kim, H.; Park, S.-H.; Kim, Y.-K. Decreased Plasma BDNF Level in Depressive Patients. J. Affect. Disord. 2007, 101, 239–244. [Google Scholar] [CrossRef]
- Duman, R.S. Neuronal Damage and Protection in the Pathophysiology and Treatment of Psychiatric Illness: Stress and Depression. Dialogues Clin. Neurosci. 2009, 11, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Bus, B.A.A.; Molendijk, M.L.; Tendolkar, I.; Penninx, B.W.J.H.; Prickaerts, J.; Elzinga, B.M.; Voshaar, R.C.O. Chronic Depression Is Associated with a Pronounced Decrease in Serum Brain-Derived Neurotrophic Factor over Time. Mol. Psychiatry 2015, 20, 602–608. [Google Scholar] [CrossRef]
- Aureli, A.; Del Beato, T.; Sebastiani, P.; Marimpietri, A.; Melillo, C.V.; Sechi, E.; Di Loreto, S. Attention-Deficit Hyperactivity Disorder and Intellectual Disability: A Study of Association with Brain-Derived Neurotrophic Factor Gene Polymorphisms. Int. J. Immunopathol. Pharmacol. 2010, 23, 873–880. [Google Scholar] [CrossRef]
- Shinoda, Y.; Sadakata, T.; Nakao, K.; Katoh-Semba, R.; Kinameri, E.; Furuya, A.; Yanagawa, Y.; Hirase, H.; Furuichi, T. Calcium-Dependent Activator Protein for Secretion 2 (CAPS2) Promotes BDNF Secretion and Is Critical for the Development of GABAergic Interneuron Network. Proc. Natl. Acad. Sci. USA 2011, 108, 373–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbina-Varela, R.; Soto-Espinoza, M.I.; Vargas, R.; Quiñones, L.; del Campo, A. Influence of BDNF Genetic Polymorphisms in the Pathophysiology of Aging-Related Diseases. Aging Dis. 2020, 11, 1513. [Google Scholar] [CrossRef]
- Miyajima, F.; Ollier, W.; Mayes, A.; Jackson, A.; Thacker, N.; Rabbitt, P.; Pendleton, N.; Horan, M.; Payton, A. Brain-Derived Neurotrophic Factor Polymorphism Val66Met Influences Cognitive Abilities in the Elderly. Genes Brain Behav. 2008, 7, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.E.; Fox, H.; Wright, A.F.; Hayward, C.; Starr, J.M.; Whalley, L.J.; Deary, I.J. The Brain-Derived Neurotrophic Factor Val66Met Polymorphism Is Associated with Age-Related Change in Reasoning Skills. Mol. Psychiatry 2006, 11, 505–513. [Google Scholar] [CrossRef]
- Gajewski, P.D.; Hengstler, J.G.; Golka, K.; Falkenstein, M.; Beste, C. The Met-Allele of the BDNF Val66Met Polymorphism Enhances Task Switching in Elderly. Neurobiol. Aging 2011, 32, 2327.e7–2327.e19. [Google Scholar] [CrossRef]
- Erickson, K.I. Genetic Contributions to Age-Related Decline in Executive Function: A 10-Year Longitudinal Study of COMT and BDNF Polymorphisms. Front. Hum. Neurosci. 2008, 2, 11. [Google Scholar] [CrossRef]
- Wei, Y.-C.; Wang, S.-R.; Xu, X.-H. Sex Differences in Brain-Derived Neurotrophic Factor Signaling: Functions and Implications: Sex Differences in BDNF Signaling. J. Neurosci. Res. 2017, 95, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Jett, S.; Malviya, N.; Schelbaum, E.; Jang, G.; Jahan, E.; Clancy, K.; Hristov, H.; Pahlajani, S.; Niotis, K.; Loeb-Zeitlin, S.; et al. Endogenous and Exogenous Estrogen Exposures: How Women’s Reproductive Health Can Drive Brain Aging and Inform Alzheimer’s Prevention. Front. Aging Neurosci. 2022, 14, 831807. [Google Scholar] [CrossRef] [PubMed]
- Solum, D.T.; Handa, R.J. Estrogen Regulates the Development of Brain-Derived Neurotrophic Factor MRNA and Protein in the Rat Hippocampus. J. Neurosci. 2002, 22, 2650–2659. [Google Scholar] [CrossRef] [Green Version]
- Li, G.-D.; Bi, R.; Zhang, D.-F.; Xu, M.; Luo, R.; Wang, D.; Fang, Y.; Li, T.; Zhang, C.; Yao, Y.-G. Female-Specific Effect of the BDNF Gene on Alzheimer’s Disease. Neurobiol. Aging 2017, 53, 192.e11–192.e19. [Google Scholar] [CrossRef] [Green Version]
- Amantea, D.; Russo, R.; Bagetta, G.; Corasaniti, M.T. From Clinical Evidence to Molecular Mechanisms Underlying Neuroprotection Afforded by Estrogens. Pharmacol. Res. 2005, 52, 119–132. [Google Scholar] [CrossRef]
- Mielke, M.; Vemuri, P.; Rocca, W. Clinical Epidemiology of Alzheimer’s Disease: Assessing Sex and Gender Differences. Clin. Epidemiol. 2014, 6, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Viña, J.; Lloret, A. Why Women Have More Alzheimer’s Disease Than Men: Gender and Mitochondrial Toxicity of Amyloid-β Peptide. J. Alzheimers Dis. 2010, 20, S527–S533. [Google Scholar] [CrossRef] [Green Version]
- Bus, B.A.A.; Arias-Vasquez, A.; Franke, B.; Prickaerts, J.; de Graaf, J.; Voshaar, R.C.O. Increase in Serum Brain-Derived Neurotrophic Factor in Met Allele Carriers of the BDNF Val66Met Polymorphism Is Specific to Males. Neuropsychobiology 2012, 65, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Egan, M.F.; Kojima, M.; Callicott, J.H.; Goldberg, T.E.; Kolachana, B.S.; Bertolino, A.; Zaitsev, E.; Gold, B.; Goldman, D.; Dean, M.; et al. The BDNF Val66met Polymorphism Affects Activity-Dependent Secretion of BDNF and Human Memory and Hippocampal Function. Cell 2003, 112, 257–269. [Google Scholar] [CrossRef]
- Fukumoto, N.; Fujii, T.; Combarros, O.; Kamboh, M.I.; Tsai, S.-J.; Matsushita, S.; Nacmias, B.; Comings, D.E.; Arboleda, H.; Ingelsson, M.; et al. Sexually Dimorphic Effect of the Val66Met Polymorphism of BDNF on Susceptibility to Alzheimer’s Disease: New Data and Meta-Analysis. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2010, 153B, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liang, X.; Li, B.; Jiang, X.; Xu, Z. Gender-Related Association of Brain-Derived Neurotrophic Factor Gene 196A/G Polymorphism with Alzheimer’s Disease—A Meta-Analysis Including 6854 Cases and 6868 Controls. Int. J. Neurosci. 2014, 124, 724–733. [Google Scholar] [CrossRef]
- Lin, Y.; Cheng, S.; Xie, Z.; Zhang, D. Association of Rs6265 and Rs2030324 Polymorphisms in Brain-Derived Neurotrophic Factor Gene with Alzheimer’s Disease: A Meta-Analysis. PLoS ONE 2014, 9, e94961. [Google Scholar] [CrossRef] [Green Version]
- Mosconi, L.; Berti, V.; Quinn, C.; McHugh, P.; Petrongolo, G.; Varsavsky, I.; Osorio, R.S.; Pupi, A.; Vallabhajosula, S.; Isaacson, R.S.; et al. Sex Differences in Alzheimer Risk: Brain Imaging of Endocrine vs Chronologic Aging. Neurology 2017, 89, 1382–1390. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.W.; Bennett, D.A.; Dong, H. Sexual Dimorphism in Predisposition to Alzheimer’s Disease. Neurobiol. Aging 2018, 70, 308–324. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, J.; Barton, D.; Overall, R.; Marc, J. Neurotrophic Support and Oxidative Stress: Converging Effects in the Normal and Diseased Nervous System. Neuroscientist 2009, 15, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.H.; Mattson, M.P. In Vivo 2-Deoxyglucose Administration Preserves Glucose and Glutamate Transport and Mitochondrial Function in Cortical Synaptic Terminals after Exposure to Amyloid β-Peptide and Iron: Evidence for a Stress Response. Exp. Neurol. 2000, 166, 173–179. [Google Scholar] [CrossRef]
- Falone, S.; D’Alessandro, A.; Mirabilio, A.; Petruccelli, G.; Cacchio, M.; Di Ilio, C.; Di Loreto, S.; Amicarelli, F. Long Term Running Biphasically Improves Methylglyoxal-Related Metabolism, Redox Homeostasis and Neurotrophic Support within Adult Mouse Brain Cortex. PLoS ONE 2012, 7, e31401. [Google Scholar] [CrossRef] [Green Version]
- Pugazhenthi, S.; Phansalkar, K.; Audesirk, G.; West, A.; Cabell, L. Differential Regulation of C-Jun and CREB by Acrolein and 4-Hydroxynonenal. Free Radic. Biol. Med. 2006, 40, 21–34. [Google Scholar] [CrossRef]
- Di Loreto, S.; Falone, S.; D’Alessandro, A.; Santini, S.; Sebastiani, P.; Cacchio, M.; Amicarelli, F. Regular and Moderate Exercise Initiated in Middle Age Prevents Age-Related Amyloidogenesis and Preserves Synaptic and Neuroprotective Signaling in Mouse Brain Cortex. Exp. Gerontol. 2014, 57, 57–65. [Google Scholar] [CrossRef]
- Chico, L.; Simoncini, C.; Lo Gerfo, A.; Rocchi, A.; Petrozzi, L.; Carlesi, C.; Volpi, L.; Tognoni, G.; Siciliano, G.; Bonuccelli, U. Oxidative Stress and APO E Polymorphisms in Alzheimer’s Disease and in Mild Cognitive Impairment. Free Radic. Res. 2013, 47, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Hall, S.K.; Perregaux, D.G.; Gabel, C.A.; Woodworth, T.; Durham, L.K.; Huizinga, T.W.F.; Breedveld, F.C.; Seymour, A.B. Correlation of Polymorphic Variation in the Promoter Region of the Interleukin-1β Gene with Secretion of Interleukin-1β Protein. Arthritis Rheum. 2004, 50, 1976–1983. [Google Scholar] [CrossRef] [PubMed]
- Shirodaria, S.; Smith, J.; McKay, I.J.; Kennett, C.N.; Hughes, F.J. Polymorphisms in the IL-1A Gene Are Correlated with Levels of Interleukin-1α Protein in Gingival Crevicular Fluid of Teeth with Severe Periodontal Disease. J. Dent. Res. 2000, 79, 1864–1869. [Google Scholar] [CrossRef]
- Caldieraro, M.A.; McKee, M.; Leistner-Segal, S.; Vares, E.A.; Kubaski, F.; Spanemberg, L.; Brusius-Facchin, A.C.; Fleck, M.P.; Mischoulon, D. Val66Met Polymorphism Association with Serum BDNF and Inflammatory Biomarkers in Major Depression. World J. Biol. Psychiatry 2018, 19, 402–409. [Google Scholar] [CrossRef]
- Felger, J.C.; Lotrich, F.E. Inflammatory Cytokines in Depression: Neurobiological Mechanisms and Therapeutic Implications. Neuroscience 2013, 246, 199–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berghella, A.M.; Aureli, A.; Canossi, A.; Beato, T.D.; Colanardi, A.; Pellegrini, P. Redox, Immune and Genetic Biomarker System for Personalized Treatments in Colorectal Cancer. World J. Gastrointest. Oncol. 2019, 11, 117–138. [Google Scholar] [CrossRef]
- Szekeres, G.; Juhász, A.; Rimanóczy, Á.; Kéri, S.; Janka, Z. The C270T Polymorphism of the Brain-Derived Neurotrophic Factor Gene Is Associated with Schizophrenia. Schizophr. Res. 2003, 65, 15–18. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin Suite Ver 3.5: A New Series of Programs to Perform Population Genetics Analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Wang, N.; Tian, B. Brain-derived Neurotrophic Factor in Autoimmune Inflammatory Diseases (Review). Exp. Ther. Med. 2021, 22, 1292. [Google Scholar] [CrossRef] [PubMed]
- Weaver, D.F. Alzheimer’s Disease as an Innate Autoimmune Disease (AD2): A New Molecular Paradigm. Alzheimers Dement. 2022, 1–13. [Google Scholar] [CrossRef]
- Nordvall, G.; Forsell, P.; Sandin, J. Neurotrophin-Targeted Therapeutics: A Gateway to Cognition and More? Drug Discov. Today 2022, 27, 103318. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| AD (n = 79) | MCI (n = 31) | p | |
|---|---|---|---|
| Age at diagnosis, mean ± SD, years | 73.79 ± 8.54, 55–92 | 67.59 ± 9.83, 43–84 | 0.002 |
| Age at study, mean ± SD, Years 1 | 76.41 ± 8.12, 50–89 | 68.71 ± 9.68, 43–84 | <0.001 |
| Mean period from diagnosis, mean ± SD, years | 2.47 ± 2.31, 0–11 | 1.19 ± 1.55, 0–5 | 0.009 |
| Gender 2 n, % | M = 29, 36.7% F = 50, 63.3% | M = 8, 25.8%; F = 23, 74.2% | ns |
| Early onset (<65 years), n, % | n = 8, 10.1% | n = 11, 35.5% | 0.004 |
| Positive family history, n, % | 14, 17.7% | 10, 32.3% | ns |
| Disease duration, years | 0–11 | 0–5 | - |
| MMSE score, (range) | 16.68 ± 6.22 (2–27) | 24.46 ± 5.27 (6–30) | <0.001 |
| APOE4+ genotypes 3 n, % | 37, 46.8% | 9, 29.0% | ns |
| AD n = 71 | MCI n = 31 | Controls n = 32 | p 1 | |
|---|---|---|---|---|
| Serum BDNF (pg/mL) | 11.97 ± 2.24 | 12.63 ± 2.19 | 12.88 ± 1.51 | 0.029 2 |
| Gender | ||||
| M | 12.47 ± 1.68 | 12.45 ± 2.23 | 12.56 ± 1.38 | ns |
| F | 11.70 ± 2.46 | 12.70 ± 2.23 | 13.05 ± 1.58 | 0.005 3 |
| BDNFVal66Met | ||||
| Val/Val | 11.78 ± 2.40 | 12.63 ± 2.43 | 13.21 ± 1.19 | 0.026 4 |
| Met/Met + Val/Met | 12.45 ± 1.88 | 12.53 ± 1.76 | 12.46 ± 1.80 | ns |
| BDNF 270C < T | ||||
| C/C | 12.10 ± 2.23 | 12.28 ± 2.26 | 12.79 ± 1.62 | ns |
| C/T | 12.82 ± 1.11 | 14.74± 0.30 | 13.31 ± 0.62 | 0.035 5 |
| IL-1α rs1800587 | ||||
| T/T + T/C | 11.44 ± 2.60 | 12.53 ± 1.93 | 12.62 ± 1.80 | ns 6 |
| C/C | 12.69 ± 1.35 | 12.77 ± 2.58 | 13.38 ± 0.39 | ns |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piancatelli, D.; Aureli, A.; Sebastiani, P.; Colanardi, A.; Del Beato, T.; Del Cane, L.; Sucapane, P.; Marini, C.; Di Loreto, S. Gene- and Gender-Related Decrease in Serum BDNF Levels in Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 14599. https://doi.org/10.3390/ijms232314599
Piancatelli D, Aureli A, Sebastiani P, Colanardi A, Del Beato T, Del Cane L, Sucapane P, Marini C, Di Loreto S. Gene- and Gender-Related Decrease in Serum BDNF Levels in Alzheimer’s Disease. International Journal of Molecular Sciences. 2022; 23(23):14599. https://doi.org/10.3390/ijms232314599
Chicago/Turabian StylePiancatelli, Daniela, Anna Aureli, Pierluigi Sebastiani, Alessia Colanardi, Tiziana Del Beato, Lorenza Del Cane, Patrizia Sucapane, Carmine Marini, and Silvia Di Loreto. 2022. "Gene- and Gender-Related Decrease in Serum BDNF Levels in Alzheimer’s Disease" International Journal of Molecular Sciences 23, no. 23: 14599. https://doi.org/10.3390/ijms232314599