
NKT and NKT-like Cells in Autoimmune Neuroinflammatory Diseases—Multiple Sclerosis, Myasthenia Gravis and Guillain-Barre Syndrome

 , , ,
, , ,
Abstract
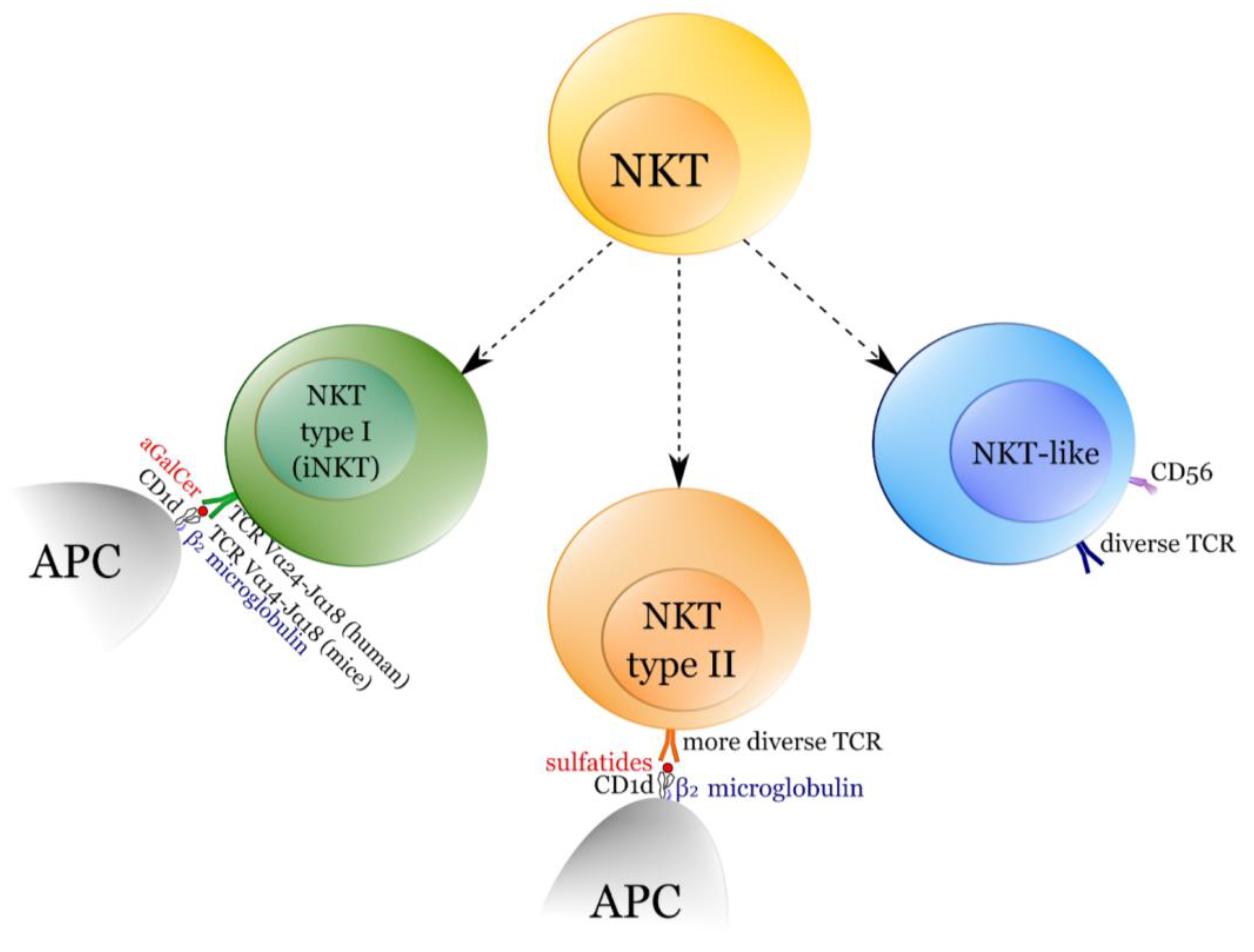
:1. NKT Cells
2. Multiple Sclerosis
2.1. iNKT in MS
2.2. NKT-like
2.3. Effect of Treatment on NKT-like Cells
3. Experimental Autoimmune Encephalomyelitis (EAE)
3.1. iNKT-Mediated Changes in Cytokine Milieu
3.2. Gut Microbiota and iNKT in EAE
3.3. Vitamin D and iNKT Cells in EAE
3.4. NKT Type II in EAE
4. Myasthenia Gravis (MG)
iNKT in MG
5. Guillain-Barré Syndrome
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bojarska-Junak, A.; Tabarkiewicz, J.; Roliński, J. {NKT cells: Their development, mechanisms and effects of action}. Postepy Hig. Med. Dosw. 2013, 67, 65–78. [Google Scholar] [CrossRef]
- Godfrey, D.I.; MacDonald, H.R.; Kronenberg, M.; Smyth, M.J.; Van Kaer, L. NKT cells: What’s in a name? Nat. Rev. Immunol. 2004, 4, 231–237. [Google Scholar] [CrossRef]
- Bendelac, A.; Savage, P.B.; Teyton, L. The Biology of NKT Cells. Annu. Rev. Immunol. 2007, 25, 297–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hapil, F.Z.; Wingender, G. The interaction between invariant Natural Killer T cells and the mucosal microbiota. Immunology 2018, 155, 164–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahng, A.; Maricic, I.; Aguilera, C.; Cardell, S.; Halder, R.C.; Kumar, V. Prevention of Autoimmunity by Targeting a Distinct, Noninvariant CD1d-reactive T Cell Population Reactive to Sulfatide. J. Exp. Med. 2004, 199, 947–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torina, A.; Guggino, G.; La Manna, M.P.; Sireci, G. The Janus Face of NKT Cell Function in Autoimmunity and Infectious Diseases. Int. J. Mol. Sci. 2018, 19, 440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godfrey, D.I.; Stankovic, S.; Baxter, A. Raising the NKT cell family. Nat. Immunol. 2010, 11, 197–206. [Google Scholar] [CrossRef]
- Bendelac, A. Positive selection of mouse NK1+ T cells by CD1-expressing cortical thymocytes. J. Exp. Med. 1995, 182, 2091–2096. [Google Scholar] [CrossRef] [Green Version]
- Hogquist, K.; Georgiev, H. Recent advances in iNKT cell development. F1000Research 2020, 9, 127. [Google Scholar] [CrossRef] [Green Version]
- Lynch, L.; Michelet, X.; Zhang, S.; Brennan, P.J.; Moseman, A.; Lester, C.; Besra, G.; Vomhof-Dekrey, E.E.; Tighe, M.; Koay, H.-F.; et al. Regulatory iNKT cells lack expression of the transcription factor PLZF and control the homeostasis of Treg cells and macrophages in adipose tissue. Nat. Immunol. 2015, 16, 85–95. [Google Scholar] [CrossRef]
- Leadbetter, E.A.; Karlsson, M.C. Reading the room: iNKT cells influence B cell responses. Mol. Immunol. 2021, 130, 49–54. [Google Scholar] [CrossRef]
- Díaz-Basabe, A.; Strati, F.; Facciotti, F. License to Kill: When iNKT Cells Are Granted the Use of Lethal Cytotoxicity. Int. J. Mol. Sci. 2020, 21, 3909. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Zhang, H.; Bai, L. NKT cells in liver diseases. Front. Med. 2018, 12, 249–261. [Google Scholar] [CrossRef]
- Singh, A.K.; Wilson, M.T.; Hong, S.; Olivares-Villagómez, D.; Du, C.; Stanic-Kostic, A.; Joyce, S.; Sriram, S.; Koezuka, Y.; Van Kaer, L. Natural Killer T Cell Activation Protects Mice Against Experimental Autoimmune Encephalomyelitis. J. Exp. Med. 2001, 194, 1801–1811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mars, L.T.; Laloux, V.; Goude, K.; Desbois, S.; Saoudi, A.; Van Kaer, L.; Lassmann, H.; Herbelin, A.; Lehuen, A.; Liblau, R. Cutting Edge: Vα14−Jα281 NKT Cells Naturally Regulate Experimental Autoimmune Encephalomyelitis in Nonobese Diabetic Mice. J. Immunol. 2002, 168, 6007–6011. [Google Scholar] [CrossRef] [Green Version]
- Nowak, M.; Schmidt-Wolf, I.G. Natural Killer T Cells Subsets in Cancer, Functional Defects in Prostate Cancer and Implications for Immunotherapy. Cancers 2011, 3, 3661. [Google Scholar] [CrossRef] [Green Version]
- Lai, L.J.; Shen, J.; Ran, Z.H. Natural killer T cells and ulcerative colitis. Cell. Immunol. 2019, 335, 1–5. [Google Scholar] [CrossRef]
- Zdrazilova-Dubska, L.; Valik, D.; Budinska, E.; Frgala, T.; Bacikova, L.; Demlova, R. NKT-like cells are expanded in solid tumour patients. Klin. Onkol. 2012, 25, 2S21–2S25. [Google Scholar]
- Hodge, G.; Hodge, S. Steroid Resistant CD8+CD28null NKT-like Pro-inflammatory Cytotoxic Cells in Chronic Obstructive Pulmonary Disease. Front. Immunol. 2016, 7, 617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pugliatti, M.; Rosati, G.; Carton, H.; Riise, T.; Drulovic, J.; Vécsei, L.; Milanov, I. The epidemiology of multiple sclerosis in Europe. Eur. J. Neurol. 2006, 13, 700–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walton, C.; King, R.; Rechtman, L.; Kaye, W.; Leray, E.; Marrie, R.A.; Robertson, N.; La Rocca, N.; Uitdehaag, B.; Van Der Mei, I.; et al. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS, third edition. Mult. Scler. J. 2020, 26, 1816–1821. [Google Scholar] [CrossRef] [PubMed]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2002, 359, 1221–1231. [Google Scholar] [CrossRef]
- Kaskow, B.; Baecher-Allan, C. Effector T Cells in Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8, a029025. [Google Scholar] [CrossRef] [PubMed]
- Gigli, G.; Caielli, S.; Cutuli, D.; Falcone, M. Innate immunity modulates autoimmunity: Type 1 interferon-β treatment in multiple sclerosis promotes growth and function of regulatory invariant natural killer T cells through dendritic cell maturation. Immunology 2007, 122, 409–417. [Google Scholar] [CrossRef] [PubMed]
- O’Keeffe, J.; Gately, C.M.; Counihan, T.; Hennessy, M.; Leahy, T.; Moran, A.P.; Hogan, E.L. T-cells expressing natural killer (NK) receptors are altered in multiple sclerosis and responses to α-galactosylceramide are impaired. J. Neurol. Sci. 2008, 275, 22–28. [Google Scholar] [CrossRef] [Green Version]
- Araki, M.; Kondo, T.; Gumperz, J.E.; Brenner, M.B.; Miyake, S.; Yamamura, T. Th2 bias of CD4+ NKT cells derived from multiple sclerosis in remission. Int. Immunol. 2003, 15, 279–288. [Google Scholar] [CrossRef] [Green Version]
- van der Vliet, H.J.; von Blomberg, B.E.; Nishia, N.; Reijmb, M.; Voskuyl, A.E.; van Bodegraven, A.A.; Polman, C.H.; Rustemeyerf, T.; Lipsg, P.; van den Eertwegh, A.J.; et al. Circulating Vα24+ Vβ11+ NKT Cell Numbers Are Decreased in a Wide Variety of Diseases That Are Characterized by Autoreactive Tissue Damage. Clin. Immunol. 2001, 100, 144–148. [Google Scholar] [CrossRef]
- Démoulins, T.; Gachelin, G.; Bequet, D.; Dormont, D. A biased Vα24+ T-cell repertoire leads to circulating NKT-cell defects in a multiple sclerosis patient at the onset of his disease. Immunol. Lett. 2003, 90, 223–228. [Google Scholar] [CrossRef]
- Illés, Z.; Kondo, T.; Newcombe, J.; Oka, N.; Tabira, T.; Yamamura, T. Differential Expression of NK T Cell Vα24JαQ Invariant TCR Chain in the Lesions of Multiple Sclerosis and Chronic Inflammatory Demyelinating Polyneuropathy. J. Immunol. 2000, 164, 4375–4381. [Google Scholar] [CrossRef] [Green Version]
- Zarobkiewicz, M.K.; Kowalska, W.; Halczuk, P.; Woś, J.; Jodłowska-Jędrych, B.; Rejdak, K.; Roliński, J.; Bojarska-Junak, A.A. RORγT is overexpressed in iNKT and γδ T cells during relapse in relapsing-remitting multiple sclerosis. J. Neuroimmunol. 2019, 337, 577046. [Google Scholar] [CrossRef]
- Gausling, R.; Trollmo, C.; Hafler, D.A. Decreases in Interleukin-4 Secretion by Invariant CD4−CD8−Vα24JαQ T Cells in Peripheral Blood of Patients with Relapsing–Remitting Multiple Sclerosis. Clin. Immunol. 2001, 98, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Misu, T.; Fujihara, K.; Sakoda, S.; Nakatsuji, Y.; Fukaura, H.; Kikuchi, S.; Tashiro, K.; Suzumura, A.; Ishii, N.; et al. Ibudilast, a nonselective phosphodiesterase inhibitor, regulates Th1/Th2 balance and NKT cell subset in multiple sclerosis. Mult. Scler. J. 2004, 10, 494–498. [Google Scholar] [CrossRef]
- Kürtüncü, M.; Yılmaz, V.; Akçay, H.I.; Türkoğlu, R.; Altunrende, B.; Çınar, S.A.; Ulusoy, C.; Gündüz, T.; Içöz, S.; Kasap, M.; et al. Impact of fingolimod on CD4+ T cell subset and cytokine profile of relapsing remitting multiple sclerosis patients. J. Neuroimmunol. 2019, 337, 577065. [Google Scholar] [CrossRef]
- McKay, F.C.; Swain, L.I.; Schibeci, S.D.; Rubio, J.P.; Kilpatrick, T.J.; Heard, R.N.; Stewart, G.J.; Booth, D.R. CD127 immunophenotyping suggests altered CD4+ T cell regulation in primary progressive multiple sclerosis. J. Autoimmun. 2008, 31, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Jons, D.; Kneider, M.; Fogelstrand, L.; Jeppsson, A.; Jacobsson, S.; Andersen, O. Early hematopoiesis in multiple sclerosis patients. J. Neuroimmunol. 2016, 299, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Sellner, J.; Koczi, W.; Harrer, A.; Oppermann, K.; Obregon-Castrillo, E.; Pilz, G.; Wipfler, P.; Afazel, S.; Haschke-Becher, E.; Trinka, E.; et al. Glatiramer acetate attenuates the pro-migratory profile of adhesion molecules on various immune cell subsets in multiple sclerosis. Clin. Exp. Immunol. 2013, 173, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Heming, M.; Schulte-Mecklenbeck, A.; Brix, T.; Wolbert, J.; Ruland, T.; Klotz, L.; Meuth, S.G.; Gross, C.; Wiendl, H.; Zu Hörste, G.M. Immune Cell Profiling of the Cerebrospinal Fluid Provides Pathogenetic Insights Into Inflammatory Neuropathies. Front. Immunol. 2019, 10, 515. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Martín, E.; Picón, C.; Costa-Frossard, L.; Alenda, R.; De La Maza, S.S.; Roldán, E.; Espiño, M.; Villar, L.M.; Álvarez-Cermeño, J.C. Natural killer cell subsets in cerebrospinal fluid of patients with multiple sclerosis. Clin. Exp. Immunol. 2015, 180, 243–249. [Google Scholar] [CrossRef] [Green Version]
- De Andrés, C.; Fernández-Paredes, L.; Tejera-Alhambra, M.; Alonso, B.; Ramos-Medina, R.; Sánchez-Ramón, S. Activation of Blood CD3+CD56+CD8+ T Cells during Pregnancy and Multiple Sclerosis. Front. Immunol. 2017, 8, 196. [Google Scholar] [CrossRef] [Green Version]
- Clerico, M.; Artusi, C.A.; Di Liberto, A.; Rolla, S.; Bardina, V.; Barbero, P.; De Mercanti, S.F.; Durelli, L. Natalizumab in Multiple Sclerosis: Long-Term Management. Int. J. Mol. Sci. 2017, 18, 940. [Google Scholar] [CrossRef] [PubMed]
- Thomas, K.; Proschmann, U.; Ziemssen, T. Fingolimod hydrochloride for the treatment of relapsing remitting multiple sclerosis. Expert Opin. Pharmacother. 2017, 18, 1649–1660. [Google Scholar] [CrossRef]
- Harrer, A.; Pilz, G.; Einhaeupl, M.; Oppermann, K.; Hitzl, W.; Wipfler, P.; Sellner, J.; Golaszewski, S.; Afazel, S.; Haschke-Becher, E.; et al. Lymphocyte Subsets Show Different Response Patterns to In Vivo Bound Natalizumab—A Flow Cytometric Study on Patients with Multiple Sclerosis. PLoS ONE 2012, 7, e31784. [Google Scholar] [CrossRef] [PubMed]
- De La Maza, S.S.; Medina, S.; Villarrubia, N.; Costa-Frossard, L.; Monreal, E.; Tejeda-Velarde, A.; Rodríguez-Martín, E.; Roldán, E.; Álvarez-Cermeño, J.C.; Villar, L.M. Factors associated with dimethyl fumarate-induced lymphopenia. J. Neurol. Sci. 2019, 398, 4–8. [Google Scholar] [CrossRef]
- Moser, T.; Schwenker, K.; Seiberl, M.; Feige, J.; Akgün, K.; Haschke-Becher, E.; Ziemssen, T.; Sellner, J. Long-term peripheral immune cell profiling reveals further targets of oral cladribine in MS. Ann. Clin. Transl. Neurol. 2020, 7, 2199–2212. [Google Scholar] [CrossRef]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; Bradl, M. Multiple sclerosis: Experimental models and reality. Acta Neuropathol. 2017, 133, 223–244. [Google Scholar] [CrossRef] [Green Version]
- Oh, S.J.; Chung, O.H. Invariant NKT Cells Producing IL-4 or IL-10, But Not IFN-γ, Inhibit the Th1 Response in Experimental Autoimmune Encephalomyelitis, Whereas None of These Cells Inhibits the Th17 Response. J. Immunol. 2011, 186, 6815–6821. [Google Scholar] [CrossRef] [Green Version]
- Van de Keere, F.; Tonegawa, S. CD4(+) T Cells Prevent Spontaneous Experimental Autoimmune Encephalomyelitis in Anti-Myelin Basic Protein T Cell Receptor Transgenic Mice. J. Exp. Med. 1998, 188, 1875–1882. [Google Scholar] [CrossRef] [Green Version]
- Denney, L.; Kok, W.L.; Cole, S.L.; Sanderson, S.; McMichael, A.J.; Ho, L.-P. Activation of Invariant NKT Cells in Early Phase of Experimental Autoimmune Encephalomyelitis Results in Differentiation of Ly6ChiInflammatory Monocyte to M2 Macrophages and Improved Outcome. J. Immunol. 2012, 189, 551–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirasinha, R.C.; Vijayan, D.; Smith, N.J.; Parnell, G.; Swarbrick, A.; Brink, R.; King, C.; Stewart, G.; Booth, D.R.; Batten, M. GPR65 inhibits experimental autoimmune encephalomyelitis through CD4+ T cell independent mechanisms that include effects on iNKT cells. Immunol. Cell Biol. 2017, 96, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Mars, L.T.; Gautron, A.-S.; Novak, J.; Beaudoin, L.; Diana, J.; Liblau, R.S.; Lehuen, A. Invariant NKT cells regulate experimental autoimmune encephalomyelitis and infiltrate the central nervous system in a CD1d-independent manner. J. Immunol. 2008, 181, 2321–2329. [Google Scholar] [CrossRef] [Green Version]
- Jahng, A.W.; Maricic, I.; Pedersen, B.; Burdin, N.; Naidenko, O.; Kronenberg, M.; Koezuka, Y.; Kumar, V. Activation of Natural Killer T Cells Potentiates or Prevents Experimental Autoimmune Encephalomyelitis. J. Exp. Med. 2001, 194, 1789–1799. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, K.; Miyake, S.; Yamamura, T. A synthetic glycolipid prevents autoimmune encephalomyelitis by inducing TH2 bias of natural killer T cells. Nat. Cell Biol. 2001, 413, 531–534. [Google Scholar] [CrossRef]
- Parekh, V.V.; Wu, L.; Olivares-Villagómez, D.; Wilson, K.T.; Van Kaer, L. Activated invariant NKT cells control central nervous system autoimmunity in a mechanism that involves myeloid-derived suppressor cells. J. Immunol. 2013, 190, 1948–1960. [Google Scholar] [CrossRef] [Green Version]
- Furlan, R.; Bergami, A.; Cantarella, D.; Brambilla, E.; Taniguchi, M.; Dellabona, P.; Casorati, G.; Martino, G. Activation of invariant NKT cells by αGalCer administration protects mice from MOG35–55-induced EAE: Critical roles for administration route and IFN-γ. Eur. J. Immunol. 2003, 33, 1830–1838. [Google Scholar] [CrossRef] [PubMed]
- Maricic, I.; Halder, R.; Bischof, F.; Kumar, V. Dendritic cells and anergic type I NKT cells play a crucial role in sulfatide-mediated immune regulation in experimental autoimmune encephalomyelitis. J. Immunol. 2014, 193, 1035–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiethe, C.; Schiemann, M.; Busch, D.; Haeberle, L.; Kopf, M.; Schuler, G.; Lutz, M.B. Interdependency of MHC class II/self-peptide and CD1d/self-glycolipid presentation by TNF-matured dendritic cells for protection from autoimmunity. J. Immunol. 2007, 178, 4908–4916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waddell, A.; Zhao, J.; Cantorna, M.T. NKT cells can help mediate the protective effects of 1,25-dihydroxyvitamin D3 in experimental autoimmune encephalomyelitis in mice. Int. Immunol. 2015, 27, 237–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teige, A.; Teige, I.; Lavasani, S.; Bockermann, R.; Mondoc, E.; Holmdahl, R.; Issazadeh-Navikas, S. CD1-Dependent Regulation of Chronic Central Nervous System Inflammation in Experimental Autoimmune Encephalomyelitis. J. Immunol. 2004, 172, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Pal, E.; Tabira, T.; Kawano, T.; Taniguchi, M.; Miyake, S.; Yamamura, T. Costimulation-Dependent Modulation of Experimental Autoimmune Encephalomyelitis by Ligand Stimulation of Vα14 NK T Cells. J. Immunol. 2001, 166, 662–668. [Google Scholar] [CrossRef] [Green Version]
- Kadowaki, A.; Quintana, F.J. The Gut–CNS Axis in Multiple Sclerosis. Trends Neurosci. 2020, 43, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Yokote, H.; Miyake, S.; Croxford, J.L.; Oki, S.; Mizusawa, H.; Yamamura, T. NKT Cell-Dependent Amelioration of a Mouse Model of Multiple Sclerosis by Altering Gut Flora. Am. J. Pathol. 2008, 173, 1714–1723. [Google Scholar] [CrossRef] [Green Version]
- Dupuis, M.L.; Pagano, M.T.; Pierdominici, M.; Ortona, E. The role of vitamin D in autoimmune diseases: Could sex make the difference? Biol. Sex Differ. 2021, 12, 12. [Google Scholar] [CrossRef] [PubMed]
- Wasnik, S.; Rundle, C.H.; Baylink, D.J.; Yazdi, M.S.; Carreon, E.E.; Xu, Y.; Qin, X.; Lau, K.-H.W.; Tang, X. 1,25-Dihydroxyvitamin D suppresses M1 macrophages and promotes M2 differentiation at bone injury sites. JCI Insight 2018, 3, e98773. [Google Scholar] [CrossRef] [Green Version]
- Dionne, S.; Duchatelier, C.-F.; Seidman, E.G. The influence of vitamin D on M1 and M2 macrophages in patients with Crohn’s disease. Innate Immun. 2017, 23, 557–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tutaj, M.; Szczepanik, M. Epicutaneous (EC) immunization with myelin basic protein (MBP) induces TCRαβ+ CD4+ CD8+ double positive suppressor cells that protect from experimental autoimmune encephalomyelitis (EAE). J. Autoimmun. 2007, 28, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Kanter, J.L.; Narayana, S.; Ho, P.P.; Catz, I.; Warren, K.G.; Sobel, R.A.; Steinman, L.; Robinson, W.H. Lipid microarrays identify key mediators of autoimmune brain inflammation. Nat. Med. 2005, 12, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Brandl, C.; Ortler, S.; Herrmann, T.; Cardell, S.; Lutz, M.B.; Wiendl, H. B7-H1-Deficiency Enhances the Potential of Tolerogenic Dendritic Cells by Activating CD1d Restricted Type II NKT Cells. PLoS ONE 2010, 5, e10800. [Google Scholar] [CrossRef]
- Melzer, N.; Ruck, T.; Fuhr, P.; Gold, R.; Hohlfeld, R.; Marx, A.; Melms, A.; Tackenberg, B.; Schalke, B.; Schneider-Gold, C.; et al. Clinical features, pathogenesis, and treatment of myasthenia gravis: A supplement to the Guidelines of the German Neurological Society. J. Neurol. 2016, 263, 1473–1494. [Google Scholar] [CrossRef] [Green Version]
- Zisimopoulou, P.; Brenner, T.; Trakas, N.; Tzartos, S.J. Serological diagnostics in myasthenia gravis based on novel assays and recently identified antigens. Autoimmun. Rev. 2013, 12, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Hemminki, K.; Li, X.; Sundquist, K. Familial Risks for Diseases of Myoneural Junction and Muscle in Siblings Based on Hospitalizations and Deaths in Sweden. Twin Res. Hum. Genet. 2006, 9, 573–579. [Google Scholar] [CrossRef]
- Ramanujam, R.; Pirskanen, R.; Ramanujam, S.; Hammarström, L. Utilizing Twins Concordance Rates to Infer the Predisposition to Myasthenia Gravis. Twin Res. Hum. Genet. 2011, 14, 129–136. [Google Scholar] [CrossRef] [Green Version]
- Lavrnic, D.; Nikolic, A.; Baets, M.D.; Verschuuren, J.; Verduyn, W.; Losen, M.; Stojanovic, V.; Stevic, Z.; Hajdukovic, L.; Apostolski, S. Familial Occurrence of Autoimmune Myasthenia Gravis with Different Antibody Specificity. Neurology 2008, 70, 2011–2013. [Google Scholar] [CrossRef]
- Carr, A.S.; Cardwell, C.; McCarron, P.O.; McConville, J. A systematic review of population based epidemiological studies in Myasthenia Gravis. BMC Neurol. 2010, 10, 46. [Google Scholar] [CrossRef] [Green Version]
- Parr, J.R.; Andrew, M.J.; Finnis, M.; Beeson, D.; Vincent, A.; Jayawant, S. How common is childhood myasthenia? The UK incidence and prevalence of autoimmune and congenital myasthenia. Arch. Dis. Child. 2014, 99, 539–542. [Google Scholar] [CrossRef]
- Cavalcante, P.; Cufi, P.; Mantegazza, R.; Berrih-Aknin, S.; Bernasconi, P.; Le Panse, R. Etiology of myasthenia gravis: Innate immunity signature in pathological thymus. Autoimmun. Rev. 2013, 12, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Patil-Chhablani, P.; Venkatramani, D.V.; Gandhi, R.A.; Nair, A.G. Ocular myasthenia gravis: A review. Indian J. Ophthalmol. 2014, 62, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Christensen, P.B.; Jensen, T.S.; Tsiropoulos, I.; Sorensen, T.; Kjaer, M.; Hojer-Pedersen, E.; Rasmussen, M.J.K.; Lehfeldt, E. Mortality and survival in myasthenia gravis: A Danish population based study. J. Neurol. Neurosurg. Psychiatry 1998, 64, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Le Panse, R.; Bismuth, J.; Cizeron-Clairac, G.; Weiss, J.M.; Cufi, P.; Dartevelle, P.; De Rosbo, N.K.; Berrih-Aknin, S. Thymic remodeling associated with hyperplasia in myasthenia gravis. Autoimmunity 2010, 43, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Verschuuren, J.J.; Huijbers, M.G.; Plomp, J.J.; Niks, E.; Molenaar, P.C.; Martinez, P.M.; Gomez, A.M.; De Baets, M.H.; Losen, M. Pathophysiology of myasthenia gravis with antibodies to the acetylcholine receptor, muscle-specific kinase and low-density lipoprotein receptor-related protein 4. Autoimmun. Rev. 2013, 12, 918–923. [Google Scholar] [CrossRef]
- Maggi, L.; Andreetta, F.; Antozzi, C.; Baggi, F.; Bernasconi, P.; Cavalcante, P.; Cornelio, F.; Muscolino, G.; Novellino, L.; Mantegazza, R. Thymoma-associated myasthenia gravis: Outcome, clinical and pathological correlations in 197 patients on a 20-year experience. J. Neuroimmunol. 2008, 201–202, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Gomez, A.M.; Broeck, J.V.D.; Vrolix, K.; Janssen, S.P.; Lemmens, M.A.M.; Van Der Esch, E.; Duimel, H.; Frederik, P.; Molenaar, P.C.; Martínez-Martíinez, P.; et al. Antibody effector mechanisms in myasthenia gravis—Pathogenesis at the neuromuscular junction. Autoimmunity 2010, 43, 353–370. [Google Scholar] [CrossRef]
- Vandiedonck, C.; Giraud, M.; Garchon, H.-J. Genetics of autoimmune myasthenia gravis: The multifaceted contribution of the HLA complex. J. Autoimmun. 2005, 25, 6–11. [Google Scholar] [CrossRef]
- Wang, H.-B.; Li, H.; Shi, F.-D.; Chambers, B.J.; Link, H.; Ljunggren, H.-G. Tumor necrosis factor receptor-1 is critically involved in the development of experimental autoimmune myasthenia gravis. Int. Immunol. 2000, 12, 1381–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmaz, V.; Tütüncü, Y.; Hasbal, N.B.; Parman, Y.; Serdaroglu, P.; Deymeer, F.; Saruhan-Direskeneli, G. Polymorphisms of interferon-γ, interleukin-10, and interleukin-12 genes in myasthenia gravis. Hum. Immunol. 2007, 68, 544–549. [Google Scholar] [CrossRef]
- Lettre, G.; Rioux, J.D. Autoimmune diseases: Insights from genome-wide association studies. Hum. Mol. Genet. 2008, 17, R116–R121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nancy, P.; Berrih-Aknin, S. Differential Estrogen Receptor Expression in Autoimmune Myasthenia Gravis. Endocrinology 2005, 146, 2345–2353. [Google Scholar] [CrossRef] [PubMed]
- Bogdanos, D.P.; Smyk, D.S.; Invernizzi, P.; Rigopoulou, E.I.; Blank, M.; Pouria, S.; Shoenfeld, Y. Infectome: A platform to trace infectious triggers of autoimmunity. Autoimmun. Rev. 2013, 12, 726–740. [Google Scholar] [CrossRef] [PubMed]
- Münz, C.; Lünemann, J.D.; Getts, M.T.; Miller, S.D. Antiviral immune responses: Triggers of or triggered by autoimmunity? Nat. Rev. Immunol. 2009, 9, 246–258. [Google Scholar] [CrossRef]
- Schönbeck, S.; Padberg, F.; Hohlfeld, R.; Wekerle, H. Transplantation of thymic autoimmune microenvironment to severe combined immunodeficiency mice. A new model of myasthenia gravis. J. Clin. Investig. 1992, 90, 245–250. [Google Scholar] [CrossRef] [Green Version]
- Theofilopoulos, A.N. The basis of autoimmunity: Part I mechanisms of aberrant self-recognition. Immunol. Today 1995, 16, 90–98. [Google Scholar] [CrossRef]
- Hammond, K. Natural killer T cells: Natural or unnatural regulators of autoimmunity? Curr. Opin. Immunol. 2003, 15, 683–689. [Google Scholar] [CrossRef]
- Viglietta, V.; Baecher-Allan, C.; Weiner, H.L.; Hafler, D.A. Loss of Functional Suppression by CD4+CD25+ Regulatory T Cells in Patients with Multiple Sclerosis. J. Exp. Med. 2004, 199, 971–979. [Google Scholar] [CrossRef] [Green Version]
- Wagner, M.J.D.; Hussain, S.; Mehan, M.; Verdi, J.M.; Delovitch, T.L. A Defect in Lineage Fate Decision during Fetal Thymic Invariant NKT Cell Development May Regulate Susceptibility to Type 1 Diabetes. J. Immunol. 2005, 174, 6764–6771. [Google Scholar] [CrossRef] [Green Version]
- Balandina, A.; Lecart, S.; Dartevelle, P.; Saoudi, A.; Berrih-Aknin, S. Functional defect of regulatory CD4+CD25+ T cells in the thymus of patients with autoimmune myasthenia gravis. Blood 2005, 105, 735–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, Y.; Onodera, H.; Tago, H.; Saito, R.; Ohuchi, M.; Shimizu, M.; Itoyama, Y. Altered populations of natural killer cell and natural killer T cell subclasses in myasthenia gravis. J. Neuroimmunol. 2005, 167, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, C.; Melms, A. Normalization of Elevated CD4−/CD8− (Double-Negative) T Cells after Thymectomy Parallels Clinical Remission in Myasthenia Gravis Associated with Thymic Hyperplasia but Not Thymoma. Ann. Neurol. 2000, 48, 603–608. [Google Scholar] [CrossRef]
- Shi, F.-D.; Flodström, M.; Balasa, B.; Kim, S.H.; Van Gunst, K.; Strominger, J.L.; Wilson, S.B.; Sarvetnick, N. Germ line deletion of the CD1 locus exacerbates diabetes in the NOD mouse. Proc. Natl. Acad. Sci. USA 2001, 98, 6777–6782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; La Cava, A.; Bai, X.-F.; Jee, Y.; Price, M.; Campagnolo, D.I.; Christadoss, P.; Vollmer, T.L.; Van Kaer, L.; Shi, F.-D. Cooperation of Invariant NKT Cells and CD4+CD25+ T Regulatory Cells in the Prevention of Autoimmune Myasthenia. J. Immunol. 2005, 175, 7898–7904. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Shin, T.; Kawano, T.; Sato, H.; Kondo, E.; Toura, I.; Kaneko, Y.; Koseki, H.; Kanno, M.; Taniguchi, M. Requirement for Vα14 NKT Cells in IL-12-Mediated Rejection of Tumors. Science 1997, 278, 1623–1626. [Google Scholar] [CrossRef]
- Wang, Y.H.; Jia, J.C.; Liu, G.; Wang, Y.F. Research on the Influence of α-GalCer Activating Experimental Autoimmune Myasthenia Gravis Mice NKT Cells at Different Times on Myasthenia Gravis. J. Biol. Regul. Homeost. Agents 2015, 29, 195–200. [Google Scholar]
- Brennan, P.J.; Brigl, M.; Brenner, M.B. Invariant natural killer T cells: An innate activation scheme linked to diverse effector functions. Nat. Rev. Immunol. 2013, 13, 101–117. [Google Scholar] [CrossRef] [PubMed]
- Van Kaer, L. α-Galactosylceramide therapy for autoimmune diseases: Prospects and obstacles. Nat. Rev. Immunol. 2005, 5, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Christadoss, P.; Poussin, M.; Deng, C. Animal Models of Myasthenia Gravis. Clin. Immunol. 2000, 94, 75–87. [Google Scholar] [CrossRef]
- Balasa, B.; Deng, C.; Lee, J.; Bradley, L.M.; Dalton, D.K.; Christadoss, P.; Sarvetnick, N. Interferon γ (IFN-γ) Is Necessary for the Genesis of Acetylcholine Receptor–induced Clinical Experimental Autoimmune Myasthenia gravis in Mice. J. Exp. Med. 1997, 186, 385–391. [Google Scholar] [CrossRef] [Green Version]
- Yuki, N.; Hartung, H.-P. Guillain–Barré Syndrome. N. Engl. J. Med. 2012, 366, 2294–2304. [Google Scholar] [CrossRef]
- Koga, M.; Yoshino, H.; Morimatsu, M.; Yuki, N. Anti-GT1a IgG in Guillain-Barre syndrome. J. Neurol. Neurosurg. Psychiatry 2002, 72, 767–771. [Google Scholar] [CrossRef]
- Willison, H.J.; Jacobs, B.C.; van Doorn, P.A. Guillain-Barré syndrome. Lancet 2016, 388, 717–727. [Google Scholar] [CrossRef] [Green Version]
- De Libero, G.; Donda, A.; Gober, H.-J.; Manolova, V.; Mazorra, Z.; Shamshiev, A.; Mori, L. A new aspect in glycolipid biology: Glycosphingolipids as antigens recognized by T lymphocytes. Neurochem. Res. 2002, 27, 675–685. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Yuki, N.; Van Kaer, L.; Furukawa, K.; Hirata, K.; Sugita, M. Cutting Edge: Guillain-Barré Syndrome-Associated IgG Responses to Gangliosides Are Generated Independently of CD1 Function in Mice. J. Immunol. 2008, 180, 39–43. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.-X.; Yang, C.-L.; Zhang, M.; Zhang, P.; Liu, R.-T.; Zhang, N.; Yang, B.; Li, X.-L.; Dou, Y.-C.; Duan, R.-S. Sulfatides ameliorate experimental autoimmune neuritis by suppressing Th1/Th17 cells. J. Neuroimmunol. 2019, 326, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Van Kaer, L.; Wu, L. Therapeutic Potential of Invariant Natural Killer T Cells in Autoimmunity. Front. Immunol. 2018, 9, 519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzo, C.; La Barbera, L.; Pizzo, M.L.; Ciccia, F.; Sireci, G.; Guggino, G. Invariant NKT Cells and Rheumatic Disease: Focus on Primary Sjogren Syndrome. Int. J. Mol. Sci. 2019, 20, 5435. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| No | Study Design | Major Results | Citation |
|---|---|---|---|
| 1 | Female B10.PL and C57BL/6 mice + CD1d or IFN-γ knockouts; immunisation with either MBP or MOG35-55; co-immunisation with α-GalCer at different time points | Co-immunisation with MBP and α-GalCer further promotes Th1 phenotype in T cells and significantly exacerbates EAE. Pre-immunisation alleviates EAE symptoms by promoting IL-4 over IFN-γ production. | Jahng et al., 2001 [52] |
| 2 | C57BL/6 mice. Synthetic analogue of α-GalCer administered along with MOG35-55 | Suppression of EAE by promotion of IL-4 production in iNKT cells | Miyamoto et al., 2001 [53] |
| 3 | C57BL/6 mice. MOG35-55 induced EAE and adoptive transfer of MOG-speciifc T cells with co-administration of α-GalCer | α-GalCer activated iNKT cells promote M-MDSC expansion, lowering symptoms of EAE | Parekh et al., 2013 [54] |
| 4 | C57BL/6 mice and knockout, CFA+ α-GalCer stimulation. MOG35-55 induced EAE | iNKTs are not necessary for establishment of EAE. Activation of iNKT diminishes severity and delays onset of EAE probably through IFN-γ increase | Furlan et al., 2003 [55] |
| 5 | NOD mice, transgenic enrichment of iNKT cells and extrathymic CD1d knockout; EAE induced with MOG35-55 | iNKTs diminish the severity of EAE. DN, cytotoxic iNKT cells infiltrate CNS. CD1d seems not necessary for iNKT mediated protection | Mars et al., 2008 [51] |
| 6 | C57BL/6 and knockouts (iNKT, IL-4, IL-10, IFN-γ), MOG35-55 induced EAE, co-administration of α-GalCer (day before and day after); adoptive transfer of iNKT | iNKTs inhibit Th1 and Th17 response; the former is mediated by IL-4 and IL-10. | Oh and Chung, 2011 [47] |
| 7 | SJL/J and C57BL/6 female mice, transgenic iNKT-deficient C57BL/6 mice; EAE induction with either PLP139-151 or MOG35-55; adoptive transfer (2 days prior to immunisation) of liver dendritic cells from sulfatide-pretreated mice | Activation of type II NKT by sulfatides after EAE is established leads to amelioration of symptoms, probably due to induction of anergy in iNKT cells, rendering more regulatory phenotype of iNKT and reducing number of CNS-infiltrating iNKT cells. Moreover, it also leads to decrease in encephalitogenic total Th as well as Th1 and Th17 cells. | Maricic et al., 2014 [56] |
| 8 | C57BL/6 mice, various knockout mice and iNKT transgenic mice. EAE was induced with MOG35-55. Adoptive transfer of dendritic cells at various time points before immunisation, adoptive transfer of iNKT cells one day prior to first dendritic cell injection. | Injection of MOG-pulsed TNF-pretreated dendritic cells protects against EAE by activating iNKT cells and promoting Th2-like response thereof. | Wiethe et al., 2007 [57] |
| 9 | Male Va14-Ja281 and Va8 and transgenic NOD mice; various knockout mice; EAE induction with MOG35-55 | Increased iNKT number significantly decreases EAE severity and delays onset in transgenic NOD mice. | Mars et al., 2002 [15] |
| 10 | C57BL/6 mice, both male and female as well as knockout mice (CD1d-, Jα18- or IL-4-deficient); MOG35-55-induced EAE; supplementation with active vitamin D3 (1,25-hydroxy-D3). Co-administration of α-GalCer on day of immunisation. | iNKT cells are important mediators of vitamin D3-mediated EAE protection. This effect is at least partially dependent on IL-4. | Waddell et al., 2015 [58] |
| 11 | C57BL/6 mice, CD1-knockouts (lacking both iNKT and type II NKT cells), MOG35-55-induced EAE, adoptive transfer | CD1-knockout mice had significantly more severe EAE with a tendency towards more chronic course and higher demyelination. Significantly lower TGF-β production in CD1-deficient mice after acute phase was over. | Teige et al., 2004 [59] |
| 12 | C57BL/6 mice, Jα18-knockout, MOG35-55-induced EAE | iNKT-knockout mice develop more severe EAE course. IL-4-produced by iNKT cells seem to be crucial for induction of M2-polarisation of macrophages, thus decreasing EAE severity | Denney et al., 2012 [49] |
| No | Study Design | Major Results | Citation |
|---|---|---|---|
| 1 | SJL/J and C57BL/6 female mice, transgenic iNKT-deficient C57BL/6 mice; EAE induction with either PLP139-151 or MOG35-55; adoptive transfer (2 days prior to immunisation) of liver dendritic cells from sulfatide-pretreated mice | Activation of type II NKT by sulfatides after EAE is established leads to amelioration of symptoms, probably due to induction of anergy in iNKT cells, rendering more regulatory phenotype of iNKT and reducing number of CNS-infiltrating iNKT cells. Moreover, it also leads to decrease in encephalitogenic total Th as well as Th1 and Th17 cells. | Maricic et al., 2014 [56] |
| 2 | C57BL/6 mice, various knockouts; EAE induced with MOG35-55. Administration of sulfatides either at the same time with MOG35-55 or one week before or after. | Type II NKT cells are present in CNS during EAE and are more prevalent than iNKT. Administration of sulfatide 7 days prior, along with or 7 days after MOG35-55 significantly lowers disease burden | Jahng et al., 2004 [5] |
| 3 | C57BL/6 mice and knockout mice (PD-L1-, CD1d or Jα281-deficient); MOG35-55 induced EAE. Adoptive transfer of tolerogenic TNF-pretreated DC either expressing PD-L1 or PD-L1-deficient | Tolerogenic DC, especially those PD-L1 deficient promote production of Th2-type cytokines by type II NKT cells thus decreasing severity of EAE | Brandl et al., 2010 [68] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zarobkiewicz, M.K.; Morawska, I.; Michalski, A.; Roliński, J.; Bojarska-Junak, A. NKT and NKT-like Cells in Autoimmune Neuroinflammatory Diseases—Multiple Sclerosis, Myasthenia Gravis and Guillain-Barre Syndrome. Int. J. Mol. Sci. 2021, 22, 9520. https://doi.org/10.3390/ijms22179520
Zarobkiewicz MK, Morawska I, Michalski A, Roliński J, Bojarska-Junak A. NKT and NKT-like Cells in Autoimmune Neuroinflammatory Diseases—Multiple Sclerosis, Myasthenia Gravis and Guillain-Barre Syndrome. International Journal of Molecular Sciences. 2021; 22(17):9520. https://doi.org/10.3390/ijms22179520
Chicago/Turabian StyleZarobkiewicz, Michał K., Izabela Morawska, Adam Michalski, Jacek Roliński, and Agnieszka Bojarska-Junak. 2021. "NKT and NKT-like Cells in Autoimmune Neuroinflammatory Diseases—Multiple Sclerosis, Myasthenia Gravis and Guillain-Barre Syndrome" International Journal of Molecular Sciences 22, no. 17: 9520. https://doi.org/10.3390/ijms22179520