
Antiandrogenic Effects of a Polyphenol in Carex kobomugi through Inhibition of Androgen Synthetic Pathway and Downregulation of Androgen Receptor in Prostate Cancer Cell Lines
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
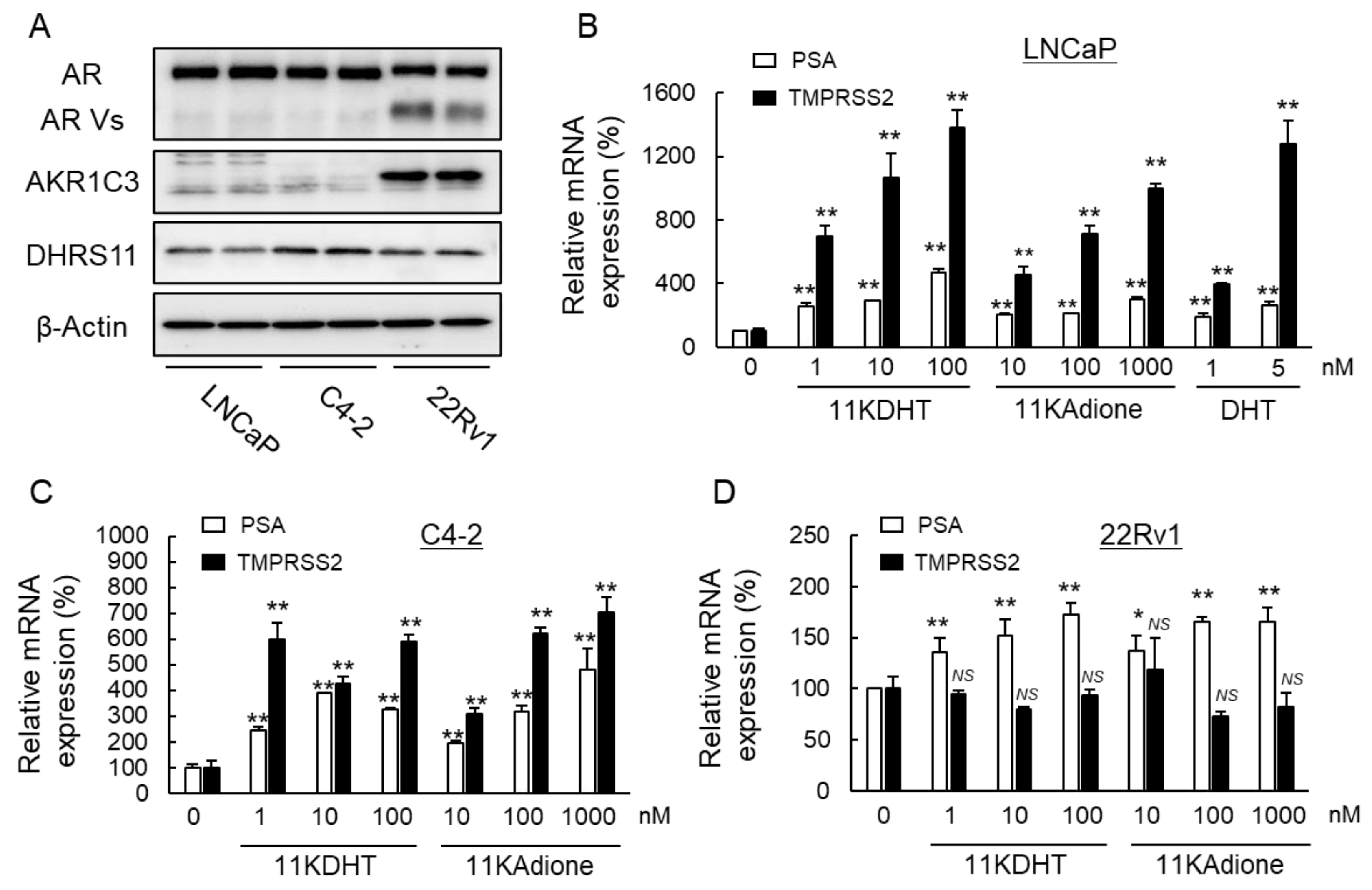
2.1. Activation of Androgen Signaling by 11-Oxygenated Androgens
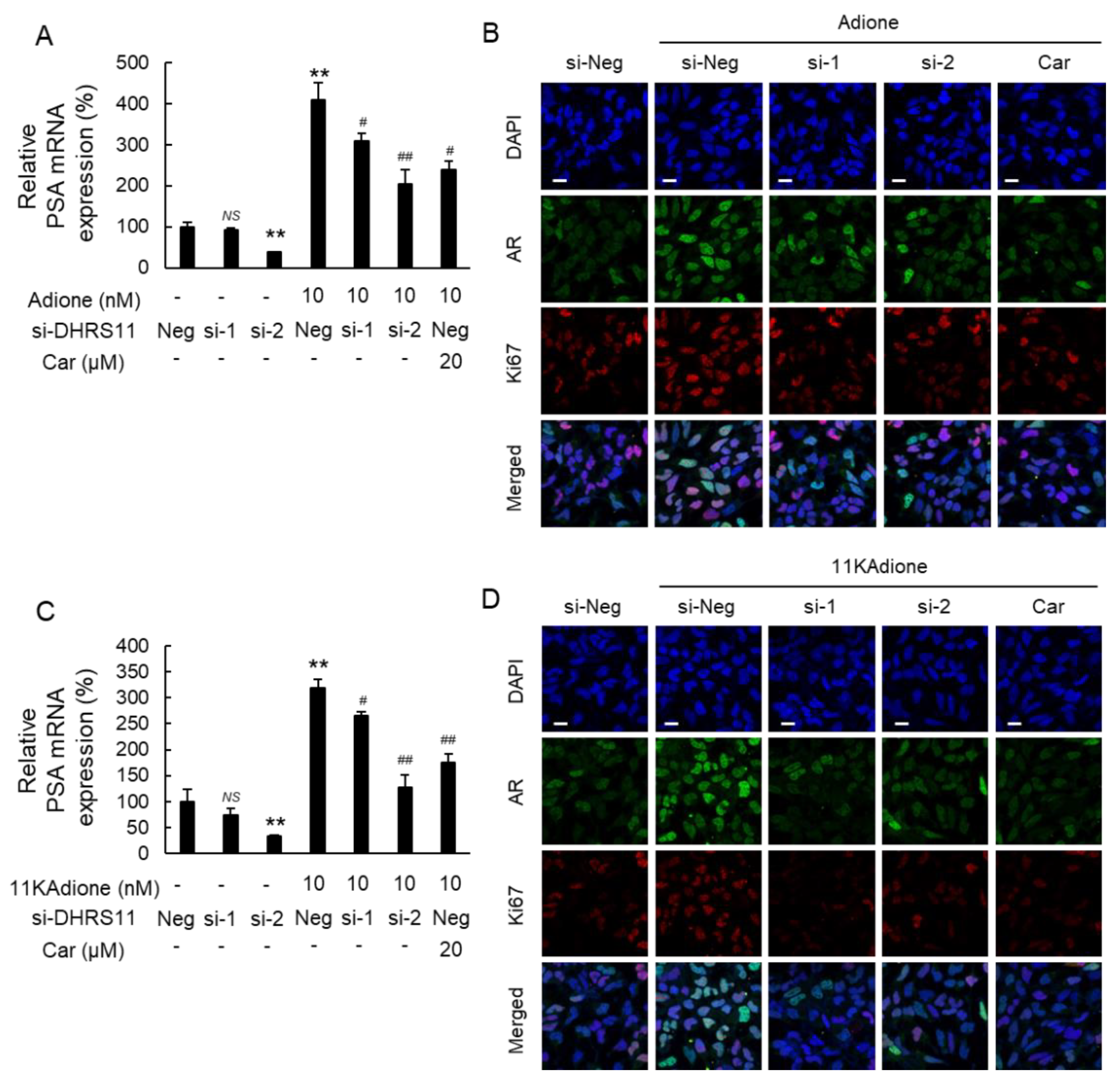
2.2. Effect of DHRS11 Silencing on 11KAdione-Induced Androgen Signaling
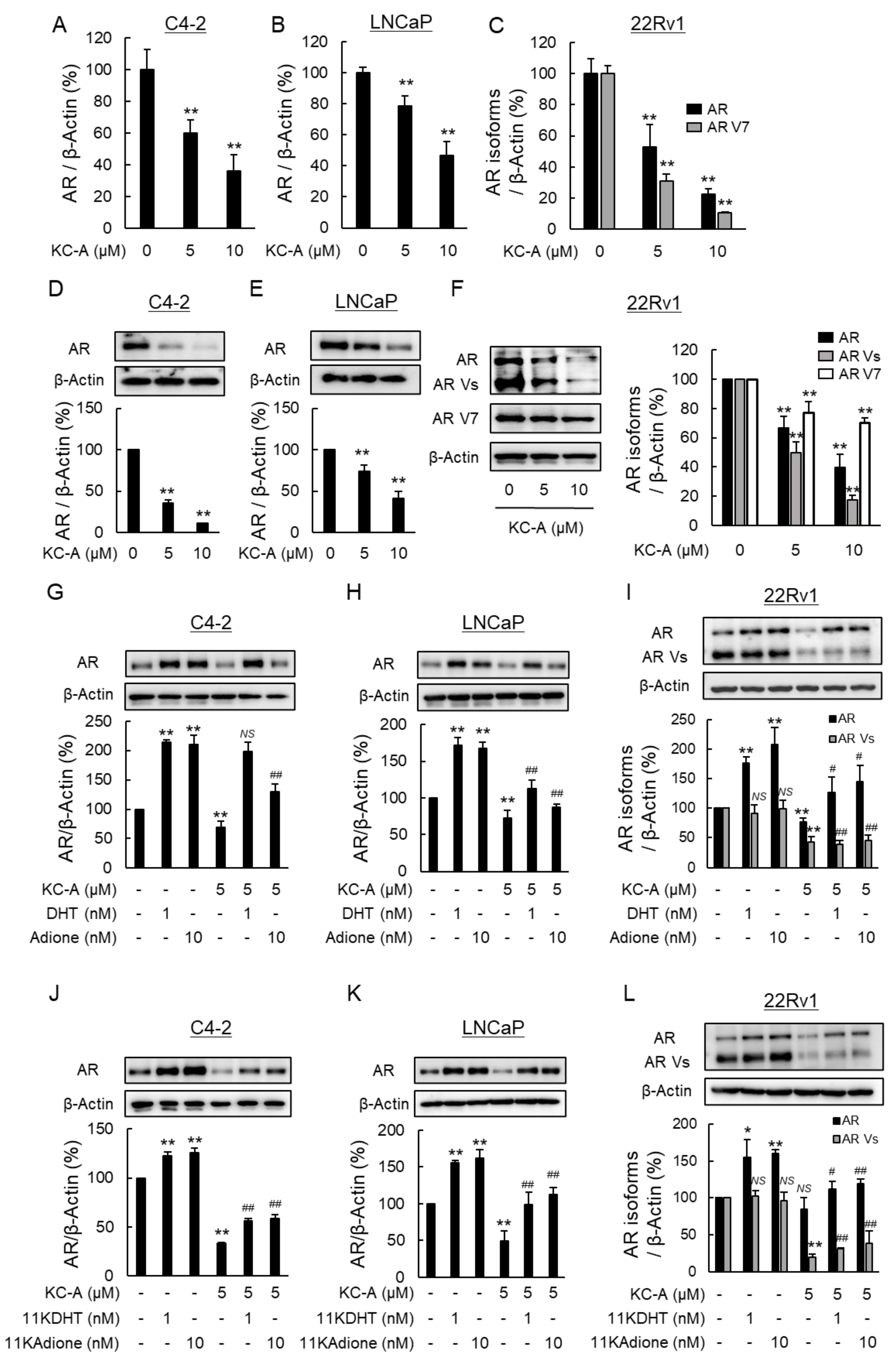
2.3. Suppression of Androgen Signaling Induced by Traditional Androgens and 11-Oxygenated Androgens by Novel Polyphenol KC-A in Carex Kobomugi
2.4. Increased Sensitivity to Abi by KC-A
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. General Experimental Procedures in Chemistry
3.3. Extraction and Isolation of KC-A
3.4. Synthesis of KC-A
3.5. Enzyme Activity Assay
3.6. Cell Culture and Transfection
3.7. Cell Viability
3.8. RNA Extraction and Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)
3.9. Western Blotting
3.10. Immunofluorescence Staining and MT-1 Staining
3.11. AR-EcoScreen Reporter Gene Assay
3.12. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| 11KA4 | 11-keto-4-androstene-3,17-dione |
| 11KAdione | 11-keto-5α-androstane-3,17-dione |
| 11KDHT | 11-keto-5α-dihydrotestosterone |
| 11KT | 11-ketotestosterone |
| 11OHA4 | 11-hydroxy-4-androstene-3,17-dione |
| 17βHSD | 17β-hydroxysteroid dehydrogenase |
| A4 | 4-androstene-3,17-dione |
| Abi | abiraterone acetate |
| Adione | 5α-androstane-3,17-dione |
| AKR | aldo-keto reductase |
| Apa | apalutamide |
| AR | androgen receptor |
| AR Vs | androgen receptor variants |
| CI | combination index |
| CRPC | castration-resistant prostate cancer |
| CS-FBS | charcoal-stripped fetal bovine serum |
| Dar | darolutamide |
| DHEA | dehydroepiandrosterone |
| DHEAS | dehydroepiandrosterone sulfate |
| DHRS11 | dehydrogenase/reductase SDR family member 11 |
| DHT | 5α-dihydrotestosterone |
| DMSO | dimethyl sulfoxide |
| FBS | fetal bovine serum |
| HITCC | 8-hydroxy-2-imino-N-(p-tolyl)-2H-chromene-3-carboxamide |
| PC | prostate cancer |
| PSA | prostate specific antigen |
| RT-qPCR | reverse transcription-quantitative polymerase chain reaction |
| T | testosterone |
| TMPRSS2 | transmembrane protease serine 2 |
References
- Huggins, C.; Hodges, C.V. Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA Cancer J. Clin. 1972, 22, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Shafi, A.A.; Yen, A.E.; Weigel, N.L. Androgen receptors in hormone-dependent and castration-resistant prostate cancer. Pharmacol. Ther. 2013, 140, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, N.; Gulley, J.L.; Dahut, W.L. Androgen deprivation therapy for prostate cancer. JAMA 2005, 294, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Feldman, B.J.; Feldman, D. The development of androgen-independent prostate cancer. Nat. Rev. Cancer 2001, 1, 34–45. [Google Scholar] [CrossRef]
- El-Amm, J.; Aragon-Ching, J.B. The Current Landscape of Treatment in Non-Metastatic Castration-Resistant Prostate Cancer. Clin. Med. Insights Oncol. 2019, 13, 1179554919833927. [Google Scholar] [CrossRef]
- Ross, R.W.; Xie, W.; Regan, M.M.; Pomerantz, M.; Nakabayashi, M.; Daskivich, T.J.; Sartor, O.; Taplin, M.E.; Kantoff, P.W.; Oh, W.K. Efficacy of androgen deprivation therapy (ADT) in patients with advanced prostate cancer: Association between Gleason score, prostate-specific antigen level, and prior ADT exposure with duration of ADT effect. Cancer 2008, 112, 1247–1253. [Google Scholar] [CrossRef]
- Mohler, J.L.; Gregory, C.W.; Ford, O.H., 3rd; Kim, D.; Weaver, C.M.; Petrusz, P.; Wilson, E.M.; French, F.S. The androgen axis in recurrent prostate cancer. Clin. Cancer Res. 2004, 10, 440–448. [Google Scholar] [CrossRef] [Green Version]
- Mizokami, A.; Namiki, M. Reconsideration of progression to CRPC during androgen deprivation therapy. J. Steroid Biochem. Mol. Biol. 2015, 145, 164–171. [Google Scholar] [CrossRef] [Green Version]
- Kumagai, J.; Hofland, J.; Erkens-Schulze, S.; Dits, N.F.; Steenbergen, J.; Jenster, G.; Homma, Y.; de Jong, F.H.; van Weerden, W.M. Intratumoral conversion of adrenal androgen precursors drives androgen receptor-activated cell growth in prostate cancer more potently than de novo steroidogenesis. Prostate 2013, 73, 1636–1650. [Google Scholar] [CrossRef]
- Narimoto, K.; Mizokami, A.; Izumi, K.; Mihara, S.; Sawada, K.; Sugata, T.; Shimamura, M.; Miyazaki, K.; Nishino, A.; Namiki, M. Adrenal androgen levels as predictors of outcome in castration-resistant prostate cancer patients treated with combined androgen blockade using flutamide as a second-line anti-androgen. Int. J. Urol. 2010, 17, 337–345. [Google Scholar] [CrossRef]
- Coutinho, I.; Day, T.K.; Tilley, W.D.; Selth, L.A. Androgen receptor signaling in castration-resistant prostate cancer: A lesson in persistence. Endocr. Relat. Cancer 2016, 23, T179–T197. [Google Scholar] [CrossRef] [PubMed]
- Ciccarese, C.; Massari, F.; Iacovelli, R.; Fiorentino, M.; Montironi, R.; Di Nunno, V.; Giunchi, F.; Brunelli, M.; Tortora, G. Prostate cancer heterogeneity: Discovering novel molecular targets for therapy. Cancer Treat. Rev. 2017, 54, 68–73. [Google Scholar] [CrossRef]
- Labrie, F. All sex steroids are made intracellularly in peripheral tissues by the mechanisms of intracrinology after menopause. J. Steroid Biochem. Mol. Biol. 2015, 145, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Aka, J.A.; Mazumdar, M.; Lin, S.X. Reductive 17beta-hydroxysteroid dehydrogenases in the sulfatase pathway: Critical in the cell proliferation of breast cancer. Mol. Cell. Endocrinol. 2009, 301, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, E.; Arlt, W.; Storbeck, K.H. A new dawn for androgens: Novel lessons from 11-oxygenated C19 steroids. Mol. Cell. Endocrinol. 2017, 441, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, L.; Arlt, W.; Storbeck, K.H. Intracrine androgen biosynthesis, metabolism and action revisited. Mol. Cell. Endocrinol. 2018, 465, 4–26. [Google Scholar] [CrossRef]
- Pretorius, E.; Africander, D.J.; Vlok, M.; Perkins, M.S.; Quanson, J.; Storbeck, K.H. 11-Ketotestosterone and 11-Ketodihydrotestosterone in Castration Resistant Prostate Cancer: Potent Androgens Which Can No Longer Be Ignored. PLoS ONE 2016, 11, e0159867. [Google Scholar] [CrossRef] [Green Version]
- Turcu, A.F.; Rege, J.; Auchus, R.J.; Rainey, W.E. 11-Oxygenated androgens in health and disease. Nat. Rev. Endocrinol. 2020, 16, 284–296. [Google Scholar] [CrossRef]
- Storbeck, K.H.; Bloem, L.M.; Africander, D.; Schloms, L.; Swart, P.; Swart, A.C. 11β-Hydroxydihydrotestosterone and 11-ketodihydrotestosterone, novel C19 steroids with androgenic activity: A putative role in castration resistant prostate cancer? Mol. Cell. Endocrinol. 2013, 377, 135–146. [Google Scholar] [CrossRef]
- Barnard, M.; Quanson, J.L.; Mostaghel, E.; Pretorius, E.; Snoep, J.L.; Storbeck, K.H. 11-Oxygenated androgen precursors are the preferred substrates for aldo-keto reductase 1C3 (AKR1C3): Implications for castration resistant prostate cancer. J. Steroid Biochem. Mol. Biol. 2018, 183, 192–201. [Google Scholar] [CrossRef]
- Snaterse, G.; Visser, J.A.; Arlt, W.; Hofland, J. Circulating steroid hormone variations throughout different stages of prostate cancer. Endocr. Relat. Cancer 2017, 24, R403–R420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnard, M.; Mostaghel, E.A.; Auchus, R.J.; Storbeck, K.H. The role of adrenal derived androgens in castration resistant prostate cancer. J. Steroid Biochem. Mol. Biol. 2020, 197, 105506. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.; O’Day, P.; Alyamani, M.; Sharifi, N.; Auchus, R.J. Abiraterone acetate treatment lowers 11-oxygenated androgens. Eur. J. Endocrinol. 2020, 182, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Endo, S.; Morikawa, Y.; Kudo, Y.; Suenami, K.; Matsunaga, T.; Ikari, A.; Hara, A. Human dehydrogenase/reductase SDR family member 11 (DHRS11) and aldo-keto reductase 1C isoforms in comparison: Substrate and reaction specificity in the reduction of 11-keto-C(19)-steroids. J. Steroid Biochem. Mol. Biol. 2020, 199, 105586. [Google Scholar] [CrossRef]
- Penning, T.M.; Byrns, M.C. Steroid hormone transforming aldo-keto reductases and cancer. Ann. N. Y. Acad. Sci. 2009, 1155, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Endo, S.; Miyagi, N.; Matsunaga, T.; Hara, A.; Ikari, A. Human dehydrogenase/reductase (SDR family) member 11 is a novel type of 17β-hydroxysteroid dehydrogenase. Biochem. Biophys. Res. Commun. 2016, 472, 231–236. [Google Scholar] [CrossRef]
- Liu, L.; Dong, X. Complex impacts of PI3K/AKT inhibitors to androgen receptor gene expression in prostate cancer cells. PLoS ONE 2014, 9, e108780. [Google Scholar] [CrossRef] [Green Version]
- Haendler, B.; Schuttke, I.; Schleuning, W.D. Androgen receptor signalling: Comparative analysis of androgen response elements and implication of heat-shock protein 90 and 14-3-3eta. Mol. Cell. Endocrinol. 2001, 173, 63–73. [Google Scholar] [CrossRef]
- Tararova, N.D.; Narizhneva, N.; Krivokrisenko, V.; Gudkov, A.V.; Gurova, K.V. Prostate cancer cells tolerate a narrow range of androgen receptor expression and activity. Prostate 2007, 67, 1801–1815. [Google Scholar] [CrossRef] [Green Version]
- Endo, S.; Matsunaga, T.; Kanamori, A.; Otsuji, Y.; Nagai, H.; Sundaram, K.; El-Kabbani, O.; Toyooka, N.; Ohta, S.; Hara, A. Selective inhibition of human type-5 17beta-hydroxysteroid dehydrogenase (AKR1C3) by baccharin, a component of Brazilian propolis. J. Nat. Prod. 2012, 75, 716–721. [Google Scholar] [CrossRef]
- Endo, S.; Hu, D.; Matsunaga, T.; Otsuji, Y.; El-Kabbani, O.; Kandeel, M.; Ikari, A.; Hara, A.; Kitade, Y.; Toyooka, N. Synthesis of non-prenyl analogues of baccharin as selective and potent inhibitors for aldo-keto reductase 1C3. Bioorg. Med. Chem. 2014, 22, 5220–5233. [Google Scholar] [CrossRef] [PubMed]
- Endo, S.; Miyagi, N.; Matsunaga, T.; Ikari, A. Rabbit dehydrogenase/reductase SDR family member 11 (DHRS11): Its identity with acetohexamide reductase with broad substrate specificity and inhibitor sensitivity, different from human DHRS11. Chem Biol Interact 2019, 305, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Zwart, N.; Andringa, D.; de Leeuw, W.J.; Kojima, H.; Iida, M.; Houtman, C.J.; de Boer, J.; Kool, J.; Lamoree, M.H.; Hamers, T. Improved androgen specificity of AR-EcoScreen by CRISPR based glucocorticoid receptor knockout. Toxicol. In Vitro 2017, 45, 1–9. [Google Scholar] [CrossRef]
- Kanayama, M.; Lu, C.; Luo, J.; Antonarakis, E.S. AR Splicing Variants and Resistance to AR Targeting Agents. Cancers 2021, 13, 2563. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef] [Green Version]
- Krongrad, A.; Wilson, C.M.; Wilson, J.D.; Allman, D.R.; McPhaul, M.J. Androgen increases androgen receptor protein while decreasing receptor mRNA in LNCaP cells. Mol. Cell. Endocrinol. 1991, 76, 79–88. [Google Scholar] [CrossRef]
- Vija, L.; Boukari, K.; Loosfelt, H.; Meduri, G.; Viengchareun, S.; Binart, N.; Young, J.; Lombes, M. Ligand-dependent stabilization of androgen receptor in a novel mouse ST38c Sertoli cell line. Mol. Cell. Endocrinol. 2014, 384, 32–42. [Google Scholar] [CrossRef] [Green Version]
- Saleem, M.; Kweon, M.H.; Yun, J.M.; Adhami, V.M.; Khan, N.; Syed, D.N.; Mukhtar, H. A novel dietary triterpene Lupeol induces fas-mediated apoptotic death of androgen-sensitive prostate cancer cells and inhibits tumor growth in a xenograft model. Cancer Res. 2005, 65, 11203–11213. [Google Scholar] [CrossRef] [Green Version]
- Montazeri-Najafabady, N.; Chatrabnous, N.; Arabnezhad, M.R.; Azarpira, N. Anti-androgenic effect of astaxanthin in LNCaP cells is mediated through the aryl hydrocarbon-androgen receptors cross talk. J. Food Biochem. 2021, 45, e13702. [Google Scholar] [CrossRef]
- Liu, S.; Yamauchi, H. Hinokitiol, a metal chelator derived from natural plants, suppresses cell growth and disrupts androgen receptor signaling in prostate carcinoma cell lines. Biochem. Biophys. Res. Commun. 2006, 351, 26–32. [Google Scholar] [CrossRef]
- Chiu, F.L.; Lin, J.K. Downregulation of androgen receptor expression by luteolin causes inhibition of cell proliferation and induction of apoptosis in human prostate cancer cells and xenografts. Prostate 2008, 68, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Naiki-Ito, A.; Naiki, T.; Kato, H.; Iida, K.; Etani, T.; Nagayasu, Y.; Suzuki, S.; Yamashita, Y.; Inaguma, S.; Onishi, M.; et al. Recruitment of miR-8080 by luteolin inhibits androgen receptor splice variant 7 expression in castration-resistant prostate cancer. Carcinogenesis 2020, 41, 1145–1157. [Google Scholar] [CrossRef] [Green Version]
- Ideyama, Y.; Kudoh, M.; Tanimoto, K.; Susaki, Y.; Nanya, T.; Nakahara, T.; Ishikawa, H.; Yoden, T.; Okada, M.; Fujikura, T.; et al. Novel nonsteroidal inhibitor of cytochrome P45017α (17α-hydroxylase/C17–20 lyase), YM116, decreased prostatic weights by reducing serum concentrations of testosterone and adrenal androgens in rats. Prostate 1998, 37, 10–18. [Google Scholar] [CrossRef]
- Haidar, S.; Ehmer, P.B.; Barassin, S.; Batzl-Hartmann, C.; Hartmann, R.W. Effects of novel 17α-hydroxylase/C17, 20-lyase (P450 17, CYP 17) inhibitors on androgen biosynthesis in vitro and in vivo. J. Steroid Biochem. Mol. Biol. 2003, 84, 555–562. [Google Scholar] [CrossRef]
- Han, J.; Zhang, J.; Zhang, W.; Zhang, D.; Li, Y.; Zhang, J.; Zhang, Y.; Diao, T.; Cui, L.; Li, W.; et al. Abiraterone and MDV3100 inhibits the proliferation and promotes the apoptosis of prostate cancer cells through mitophagy. Cancer Cell Int. 2019, 19, 332. [Google Scholar] [CrossRef] [PubMed]
- Soifer, H.S.; Souleimanian, N.; Wu, S.; Voskresenskiy, A.M.; Collak, F.K.; Cinar, B.; Stein, C.A. Direct regulation of androgen receptor activity by potent CYP17 inhibitors in prostate cancer cells. J. Biol. Chem. 2012, 287, 3777–3787. [Google Scholar] [CrossRef] [Green Version]
- Ashrafi, G.; Wu, Z.; Farrell, R.J.; Ryan, T.A. GLUT4 Mobilization Supports Energetic Demands of Active Synapses. Neuron 2017, 93, 606–615.e603. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, J.; Wilson, I.; Orton, T.; Pognan, F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 2000, 267, 5421–5426. [Google Scholar] [CrossRef]
- Endo, S.; Kawai, M.; Hoshi, M.; Segawa, J.; Fujita, M.; Matsukawa, T.; Fujimoto, N.; Matsunaga, T.; Ikari, A. Targeting Nrf2-antioxidant signaling reverses acquired cabazitaxel resistance in prostate cancer cells. J. Biochem. 2021, 170, 89–96. [Google Scholar] [CrossRef]
- Endo, S.; Oguri, H.; Segawa, J.; Kawai, M.; Hu, D.; Xia, S.; Okada, T.; Irie, K.; Fujii, S.; Gouda, H.; et al. Development of Novel AKR1C3 Inhibitors as New Potential Treatment for Castration-Resistant Prostate Cancer. J. Med. Chem. 2020, 63, 10396–10411. [Google Scholar] [CrossRef]
- Endo, S.; Matsuoka, T.; Nishiyama, T.; Arai, Y.; Kashiwagi, H.; Abe, N.; Oyama, M.; Matsunaga, T.; Ikari, A. Flavonol glycosides of Rosa multiflora regulates intestinal barrier function through inhibiting claudin expression in differentiated Caco-2 cells. Nutr Res 2019, 72, 92–104. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kudo, Y.; Endo, S.; Tanio, M.; Saka, T.; Himura, R.; Abe, N.; Takeda, M.; Yamaguchi, E.; Yoshino, Y.; Arai, Y.; et al. Antiandrogenic Effects of a Polyphenol in Carex kobomugi through Inhibition of Androgen Synthetic Pathway and Downregulation of Androgen Receptor in Prostate Cancer Cell Lines. Int. J. Mol. Sci. 2022, 23, 14356. https://doi.org/10.3390/ijms232214356
Kudo Y, Endo S, Tanio M, Saka T, Himura R, Abe N, Takeda M, Yamaguchi E, Yoshino Y, Arai Y, et al. Antiandrogenic Effects of a Polyphenol in Carex kobomugi through Inhibition of Androgen Synthetic Pathway and Downregulation of Androgen Receptor in Prostate Cancer Cell Lines. International Journal of Molecular Sciences. 2022; 23(22):14356. https://doi.org/10.3390/ijms232214356
Chicago/Turabian StyleKudo, Yudai, Satoshi Endo, Masatoshi Tanio, Tomofumi Saka, Rin Himura, Naohito Abe, Mitsumi Takeda, Eiji Yamaguchi, Yuta Yoshino, Yuki Arai, and et al. 2022. "Antiandrogenic Effects of a Polyphenol in Carex kobomugi through Inhibition of Androgen Synthetic Pathway and Downregulation of Androgen Receptor in Prostate Cancer Cell Lines" International Journal of Molecular Sciences 23, no. 22: 14356. https://doi.org/10.3390/ijms232214356