
Learning from TCR Signaling and Immunological Synapse Assembly to Build New Chimeric Antigen Receptors (CARs)



Abstract
:1. Introduction
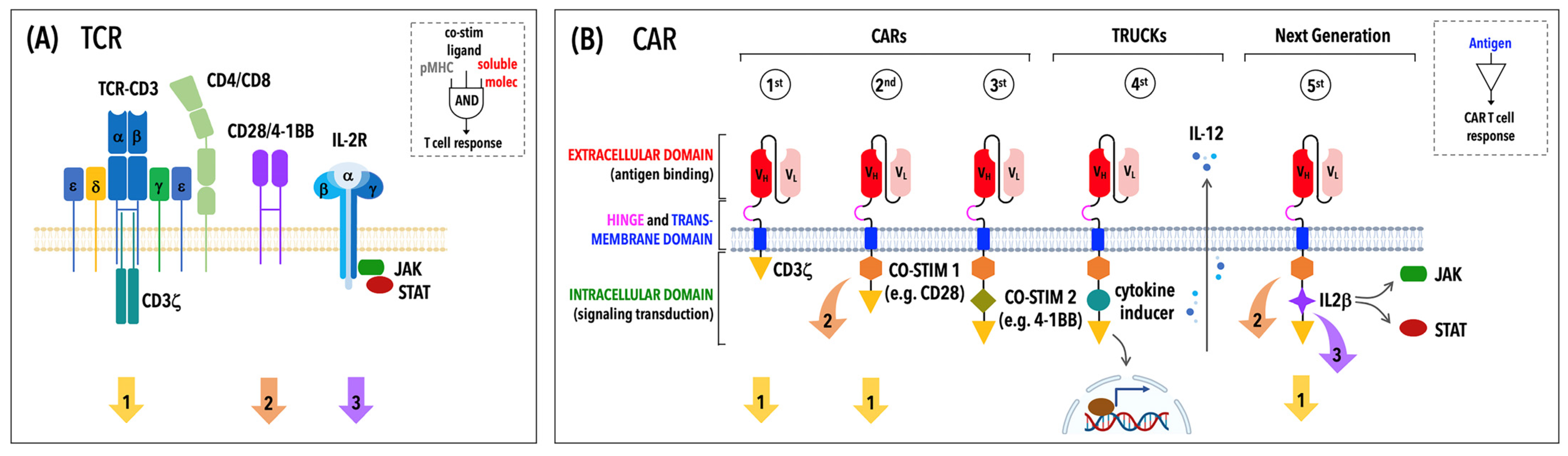
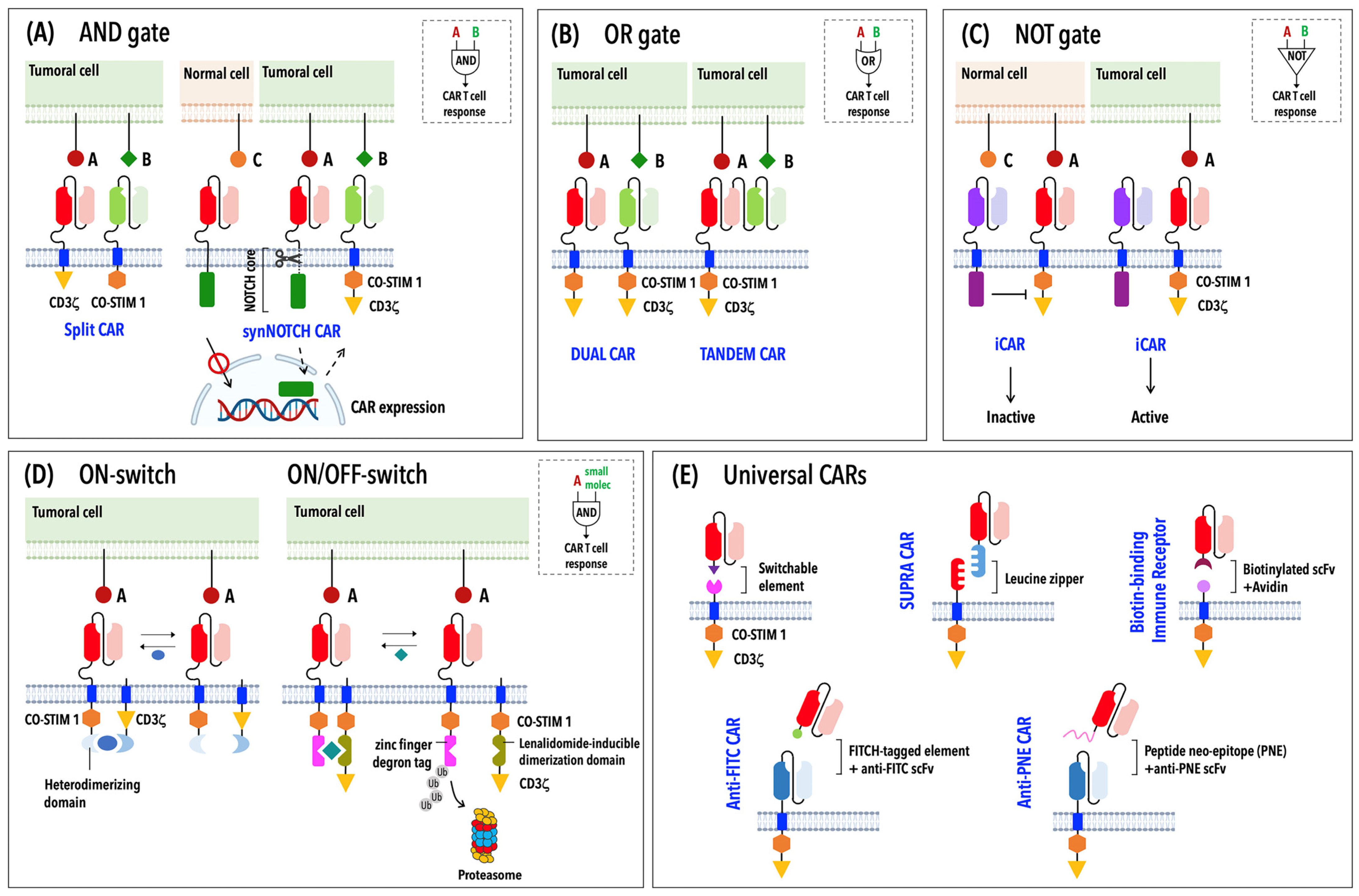
2. Building Optimized CARs
2.1. The Modular Structure of CARs
2.2. The Ongoing Evolution of CARs: From First- to Fifth-Generation CARs and Beyond
3. An Overview of CAR Signaling
4. CAR T Cells Assemble Unconventional but Productive Immunological Synapses
4.1. Structural and Functional Features of the CAR T Cell IS
4.2. CAR T Cell Activation Requires IS Formation and Synaptic Mechanotransduction
5. CAR T Cell-Based Therapy in CLL
5.1. Molecular Targets of CAR T Cell Therapy in CLL
5.2. Determinants of Insufficient CAR T Cell Responses in CLL
6. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A Guide to Cancer Immunotherapy: From T Cell Basic Science to Clinical Practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.M.; Boniface, J.J.; Reich, Z.; Lyons, D.; Hampl, J.; Arden, B.; Chien, Y. Ligand Recognition by Alpha Beta T Cell Receptors. Annu. Rev. Immunol. 1998, 16, 523–544. [Google Scholar] [CrossRef] [PubMed]
- Marrack, P.; Scott-Browne, J.P.; Dai, S.; Gapin, L.; Kappler, J.W. Evolutionarily Conserved Amino Acids That Control TCR-MHC Interaction. Annu. Rev. Immunol. 2008, 26, 171–203. [Google Scholar] [CrossRef] [Green Version]
- Rossjohn, J.; Gras, S.; Miles, J.J.; Turner, S.J.; Godfrey, D.I.; McCluskey, J. T Cell Antigen Receptor Recognition of Antigen-Presenting Molecules. Annu. Rev. Immunol. 2015, 33, 169–200. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A. Use of Tumor-Infiltrating Lymphocytes and Interleukin-2 in the Immunotherapy of Patients with Metastatic Melanoma. A Preliminary Report. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.J.; Mittermüller, J.; Clemm, C.; Holler, E.; Ledderose, G.; Brehm, G.; Heim, M.; Wilmanns, W. Donor Leukocyte Transfusions for Treatment of Recurrent Chronic Myelogenous Leukemia in Marrow Transplant Patients. Blood 1990, 76, 2462–2465. [Google Scholar] [CrossRef] [Green Version]
- Kuwana, Y.; Asakura, Y.; Utsunomiya, N.; Nakanishi, M.; Arata, Y.; Itoh, S.; Nagase, F.; Kurosawa, Y. Expression of Chimeric Receptor Composed of Immunoglobulin-Derived V Regions and T-Cell Receptor-Derived C Regions. Biochem. Biophys. Res. Commun. 1987, 149, 960–968. [Google Scholar] [CrossRef]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of Immunoglobulin-T-Cell Receptor Chimeric Molecules as Functional Receptors with Antibody-Type Specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [Green Version]
- Dustin, M.L.; Chakraborty, A.K.; Shaw, A.S. Understanding the Structure and Function of the Immunological Synapse. Cold Spring Harb. Perspect. Biol. 2010, 2, a002311. [Google Scholar] [CrossRef]
- Ortega-Carrion, A.; Vicente-Manzanares, M. Concerning Immune Synapses: A Spatiotemporal Timeline. F1000Research 2016, 5, F1000 Faculty Rev-418. [Google Scholar] [CrossRef]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric Antigen Receptor-Modified T Cells in Chronic Lymphoid Leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallek, M.; Cheson, B.D.; Catovsky, D.; Caligaris-Cappio, F.; Dighiero, G.; Döhner, H.; Hillmen, P.; Keating, M.; Montserrat, E.; Chiorazzi, N.; et al. IwCLL Guidelines for Diagnosis, Indications for Treatment, Response Assessment, and Supportive Management of CLL. Blood 2018, 131, 2745–2760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.-T.; et al. Determinants of Response and Resistance to CD19 Chimeric Antigen Receptor (CAR) T Cell Therapy of Chronic Lymphocytic Leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Gribben, J.G. The Microenvironment in Chronic Lymphocytic Leukemia (CLL) and Other B Cell Malignancies: Insight into Disease Biology and New Targeted Therapies. Semin. Cancer Biol. 2014, 24, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Palma, M.; Gentilcore, G.; Heimersson, K.; Mozaffari, F.; Näsman-Glaser, B.; Young, E.; Rosenquist, R.; Hansson, L.; Österborg, A.; Mellstedt, H. T Cells in Chronic Lymphocytic Leukemia Display Dysregulated Expression of Immune Checkpoints and Activation Markers. Haematologica 2017, 102, 562–572. [Google Scholar] [CrossRef] [Green Version]
- Capitani, N.; Patrussi, L.; Baldari, C.T. Nature vs. Nurture: The Two Opposing Behaviors of Cytotoxic T Lymphocytes in the Tumor Microenvironment. Int. J. Mol. Sci 2021, 22, 11221. [Google Scholar] [CrossRef]
- Ramsay, A.G.; Johnson, A.J.; Lee, A.M.; Gorgün, G.; Le Dieu, R.; Blum, W.; Byrd, J.C.; Gribben, J.G. Chronic Lymphocytic Leukemia T Cells Show Impaired Immunological Synapse Formation That Can Be Reversed with an Immunomodulating Drug. J. Clin. Investig. 2008, 118, 2427–2437. [Google Scholar] [CrossRef]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering Strategies to Overcome the Current Roadblocks in CAR T Cell Therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef]
- Harris, D.T.; Hager, M.V.; Smith, S.N.; Cai, Q.; Stone, J.D.; Kruger, P.; Lever, M.; Dushek, O.; Schmitt, T.M.; Greenberg, P.D.; et al. Comparison of T Cell Activities Mediated by Human TCRs and CARs That Use the Same Recognition Domains. J. Immunol. 2018, 200, 1088–1100. [Google Scholar] [CrossRef] [Green Version]
- Gudipati, V.; Rydzek, J.; Doel-Perez, I.; Gonçalves, V.D.R.; Scharf, L.; Königsberger, S.; Lobner, E.; Kunert, R.; Einsele, H.; Stockinger, H.; et al. Inefficient CAR-Proximal Signaling Blunts Antigen Sensitivity. Nat. Immunol. 2020, 21, 848–856. [Google Scholar] [CrossRef]
- Salter, A.I.; Rajan, A.; Kennedy, J.J.; Ivey, R.G.; Shelby, S.A.; Leung, I.; Templeton, M.L.; Muhunthan, V.; Voillet, V.; Sommermeyer, D.; et al. Comparative Analysis of TCR and CAR Signaling Informs CAR Designs with Superior Antigen Sensitivity and in Vivo Function. Sci. Signal. 2021, 14, eabe2606. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, J.; Mellody, M.P.; Hou, A.J.; Desai, R.P.; Fung, A.W.; Pham, A.H.T.; Chen, Y.Y.; Zhao, W. CAR-T Design: Elements and Their Synergistic Function. EBioMedicine 2020, 58, 102931. [Google Scholar] [CrossRef] [PubMed]
- Wilkie, S.; Picco, G.; Foster, J.; Davies, D.M.; Julien, S.; Cooper, L.; Arif, S.; Mather, S.J.; Taylor-Papadimitriou, J.; Burchell, J.M.; et al. Retargeting of Human T Cells to Tumor-Associated MUC1: The Evolution of a Chimeric Antigen Receptor. J. Immunol. 2008, 180, 4901–4909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Jiang, D.; Yang, H.; He, Z.; Liu, X.; Qin, W.; Li, L.; Wang, C.; Li, Y.; Li, H.; et al. Modified CAR T Cells Targeting Membrane-Proximal Epitope of Mesothelin Enhances the Antitumor Function against Large Solid Tumor. Cell Death Dis. 2019, 10, 476. [Google Scholar] [CrossRef] [Green Version]
- Alabanza, L.; Pegues, M.; Geldres, C.; Shi, V.; Wiltzius, J.J.W.; Sievers, S.A.; Yang, S.; Kochenderfer, J.N. Function of Novel Anti-CD19 Chimeric Antigen Receptors with Human Variable Regions Is Affected by Hinge and Transmembrane Domains. Mol. Ther. 2017, 25, 2452–2465. [Google Scholar] [CrossRef] [Green Version]
- Qin, L.; Lai, Y.; Zhao, R.; Wei, X.; Weng, J.; Lai, P.; Li, B.; Lin, S.; Wang, S.; Wu, Q.; et al. Incorporation of a Hinge Domain Improves the Expansion of Chimeric Antigen Receptor T Cells. J. Hematol. Oncol. 2017, 10, 68. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, K.; Tsunei, A.; Kusabuka, H.; Ogaki, E.; Tachibana, M.; Okada, N. Hinge and Transmembrane Domains of Chimeric Antigen Receptor Regulate Receptor Expression and Signaling Threshold. Cells 2020, 9, 1182. [Google Scholar] [CrossRef]
- Zhang, A.; Sun, Y.; Du, J.; Dong, Y.; Pang, H.; Ma, L.; Si, S.; Zhang, Z.; He, M.; Yue, Y.; et al. Reducing Hinge Flexibility of CAR-T Cells Prolongs Survival In Vivo With Low Cytokines Release. Front. Immunol. 2021, 12, 724211. [Google Scholar] [CrossRef]
- Bridgeman, J.S.; Hawkins, R.E.; Bagley, S.; Blaylock, M.; Holland, M.; Gilham, D.E. The Optimal Antigen Response of Chimeric Antigen Receptors Harboring the CD3zeta Transmembrane Domain Is Dependent upon Incorporation of the Receptor into the Endogenous TCR/CD3 Complex. J. Immunol. 2010, 184, 6938–6949. [Google Scholar] [CrossRef] [Green Version]
- Majzner, R.G.; Rietberg, S.P.; Sotillo, E.; Dong, R.; Vachharajani, V.T.; Labanieh, L.; Myklebust, J.H.; Kadapakkam, M.; Weber, E.W.; Tousley, A.M.; et al. Tuning the Antigen Density Requirement for CAR T-Cell Activity. Cancer Discov. 2020, 10, 702–723. [Google Scholar] [CrossRef]
- Muller, Y.D.; Nguyen, D.P.; Ferreira, L.M.R.; Ho, P.; Raffin, C.; Valencia, R.V.B.; Congrave-Wilson, Z.; Roth, T.L.; Eyquem, J.; Van Gool, F.; et al. The CD28-Transmembrane Domain Mediates Chimeric Antigen Receptor Heterodimerization With CD28. Front. Immunol. 2021, 12, 639818. [Google Scholar] [CrossRef] [PubMed]
- Salter, A.I.; Ivey, R.G.; Kennedy, J.J.; Voillet, V.; Rajan, A.; Alderman, E.J.; Voytovich, U.J.; Lin, C.; Sommermeyer, D.; Liu, L.; et al. Phosphoproteomic Analysis of Chimeric Antigen Receptor Signaling Reveals Kinetic and Quantitative Differences That Affect Cell Function. Sci. Signal. 2018, 11, eaat6753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramello, M.C.; Benzaïd, I.; Kuenzi, B.M.; Lienlaf-Moreno, M.; Kandell, W.M.; Santiago, D.N.; Pabón-Saldaña, M.; Darville, L.; Fang, B.; Rix, U.; et al. An Immunoproteomic Approach to Characterize the CAR Interactome and Signalosome. Sci. Signal. 2019, 12, eaap9777. [Google Scholar] [CrossRef] [PubMed]
- Tokarew, N.; Ogonek, J.; Endres, S.; von Bergwelt-Baildon, M.; Kobold, S. Teaching an Old Dog New Tricks: Next-Generation CAR T Cells. Br, J. Cancer 2019, 120, 26–37. [Google Scholar] [CrossRef] [Green Version]
- Finney, H.M.; Lawson, A.D.; Bebbington, C.R.; Weir, A.N. Chimeric Receptors Providing Both Primary and Costimulatory Signaling in T Cells from a Single Gene Product. J. Immunol. 1998, 161, 2791–2797. [Google Scholar]
- Carpenito, C.; Milone, M.C.; Hassan, R.; Simonet, J.C.; Lakhal, M.; Suhoski, M.M.; Varela-Rohena, A.; Haines, K.M.; Heitjan, D.F.; Albelda, S.M.; et al. Control of Large, Established Tumor Xenografts with Genetically Retargeted Human T Cells Containing CD28 and CD137 Domains. Proc. Natl. Acad. Sci. USA 2009, 106, 3360–3365. [Google Scholar] [CrossRef] [Green Version]
- Chmielewski, M.; Abken, H. TRUCKs: The Fourth Generation of CARs. Expert Opin Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef]
- Bell, M.; Gottschalk, S. CAR T Cell Therapy for Solid Tumors: Fatal Attraction Requires Adhesion. Med 2022, 3, 353–354. [Google Scholar] [CrossRef]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.-H.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A Novel Chimeric Antigen Receptor Containing a JAK-STAT Signaling Domain Mediates Superior Antitumor Effects. Nat. Med. 2018, 24, 352–359. [Google Scholar] [CrossRef]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced with a Chimeric Antigen Receptor Recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Morgan, R.A.; Chinnasamy, N.; Abate-Daga, D.; Gros, A.; Robbins, P.F.; Zheng, Z.; Dudley, M.E.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; et al. Cancer Regression and Neurological Toxicity Following Anti-MAGE-A3 TCR Gene Therapy. J. Immunother. 2013, 36, 133–151. [Google Scholar] [CrossRef] [PubMed]
- Linette, G.P.; Stadtmauer, E.A.; Maus, M.V.; Rapoport, A.P.; Levine, B.L.; Emery, L.; Litzky, L.; Bagg, A.; Carreno, B.M.; Cimino, P.J.; et al. Cardiovascular Toxicity and Titin Cross-Reactivity of Affinity-Enhanced T Cells in Myeloma and Melanoma. Blood 2013, 122, 863–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubert, M.-L.; Schmitt, M.; Wang, L.; Ramos, C.A.; Jordan, K.; Müller-Tidow, C.; Dreger, P. Side-Effect Management of Chimeric Antigen Receptor (CAR) T-Cell Therapy. Ann. Oncol. 2021, 32, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Wilkie, S.; van Schalkwyk, M.C.I.; Hobbs, S.; Davies, D.M.; van der Stegen, S.J.C.; Pereira, A.C.P.; Burbridge, S.E.; Box, C.; Eccles, S.A.; Maher, J. Dual Targeting of ErbB2 and MUC1 in Breast Cancer Using Chimeric Antigen Receptors Engineered to Provide Complementary Signaling. J. Clin. Immunol. 2012, 32, 1059–1070. [Google Scholar] [CrossRef] [PubMed]
- Kloss, C.C.; Condomines, M.; Cartellieri, M.; Bachmann, M.; Sadelain, M. Combinatorial Antigen Recognition with Balanced Signaling Promotes Selective Tumor Eradication by Engineered T Cells. Nat. Biotechnol. 2013, 31, 71–75. [Google Scholar] [CrossRef]
- Lanitis, E.; Poussin, M.; Klattenhoff, A.W.; Song, D.; Sandaltzopoulos, R.; June, C.H.; Powell, D.J.J. Chimeric Antigen Receptor T Cells with Dissociated Signaling Domains Exhibit Focused Antitumor Activity with Reduced Potential for Toxicity in Vivo. Cancer Immunol. Res. 2013, 1, 43–53. [Google Scholar] [CrossRef] [Green Version]
- Globerson Levin, A.; Rawet Slobodkin, M.; Waks, T.; Horn, G.; Ninio-Many, L.; Deshet Unger, N.; Ohayon, Y.; Suliman, S.; Cohen, Y.; Tartakovsky, B.; et al. Treatment of Multiple Myeloma Using Chimeric Antigen Receptor T Cells with Dual Specificity. Cancer Immunol. Res. 2020, 8, 1485–1495. [Google Scholar] [CrossRef]
- Morsut, L.; Roybal, K.T.; Xiong, X.; Gordley, R.M.; Coyle, S.M.; Thomson, M.; Lim, W.A. Engineering Customized Cell Sensing and Response Behaviors Using Synthetic Notch Receptors. Cell 2016, 164, 780–791. [Google Scholar] [CrossRef] [Green Version]
- Roybal, K.T.; Williams, J.Z.; Morsut, L.; Rupp, L.J.; Kolinko, I.; Choe, J.H.; Walker, W.J.; McNally, K.A.; Lim, W.A. Engineering T Cells with Customized Therapeutic Response Programs Using Synthetic Notch Receptors. Cell 2016, 167, 419–432.e16. [Google Scholar] [CrossRef] [Green Version]
- Grada, Z.; Hegde, M.; Byrd, T.; Shaffer, D.R.; Ghazi, A.; Brawley, V.S.; Corder, A.; Schönfeld, K.; Koch, J.; Dotti, G.; et al. TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Mol. Ther. Nucleic Acids 2013, 2, e105. [Google Scholar] [CrossRef]
- Hegde, M.; Mukherjee, M.; Grada, Z.; Pignata, A.; Landi, D.; Navai, S.A.; Wakefield, A.; Fousek, K.; Bielamowicz, K.; Chow, K.K.H.; et al. Tandem CAR T Cells Targeting HER2 and IL13Rα2 Mitigate Tumor Antigen Escape. J. Clin. Investig. 2016, 126, 3036–3052. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, V.D.; Themeli, M.; Sadelain, M. PD-1- and CTLA-4-Based Inhibitory Chimeric Antigen Receptors (ICARs) Divert off-Target Immunotherapy Responses. Sci. Transl. Med. 2013, 5, 215ra172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamburger, A.E.; DiAndreth, B.; Cui, J.; Daris, M.E.; Munguia, M.L.; Deshmukh, K.; Mock, J.-Y.; Asuelime, G.E.; Lim, E.D.; Kreke, M.R.; et al. Engineered T Cells Directed at Tumors with Defined Allelic Loss. Mol. Immunol. 2020, 128, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Farooq, M.A.; Gao, Y.; Zhang, L.; Niu, C.; Ajmal, I.; Zhou, Y.; He, C.; Zhao, G.; Yao, J.; et al. CD19-CAR-T Cells Bearing a KIR/PD-1-Based Inhibitory CAR Eradicate CD19(+)HLA-C1(-) Malignant B Cells While Sparing CD19(+)HLA-C1(+) Healthy B Cells. Cancers 2020, 12, 2612. [Google Scholar] [CrossRef] [PubMed]
- Brandt, L.J.B.; Barnkob, M.B.; Michaels, Y.S.; Heiselberg, J.; Barington, T. Emerging Approaches for Regulation and Control of CAR T Cells: A Mini Review. Front. Immunol. 2020, 11, 326. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-Y.; Roybal, K.T.; Puchner, E.M.; Onuffer, J.; Lim, W.A. Remote Control of Therapeutic T Cells through a Small Molecule-Gated Chimeric Receptor. Science 2015, 350, aab4077. [Google Scholar] [CrossRef] [Green Version]
- Jan, M.; Scarfò, I.; Larson, R.C.; Walker, A.; Schmidts, A.; Guirguis, A.A.; Gasser, J.A.; Słabicki, M.; Bouffard, A.A.; Castano, A.P.; et al. Reversible ON- and OFF-Switch Chimeric Antigen Receptors Controlled by Lenalidomide. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef]
- Weber, E.W.; Parker, K.R.; Sotillo, E.; Lynn, R.C.; Anbunathan, H.; Lattin, J.; Good, Z.; Belk, J.A.; Daniel, B.; Klysz, D.; et al. Transient Rest Restores Functionality in Exhausted CAR-T Cells through Epigenetic Remodeling. Science 2021, 372. [Google Scholar] [CrossRef]
- Huang, Z.; Wu, Y.; Allen, M.E.; Pan, Y.; Kyriakakis, P.; Lu, S.; Chang, Y.-J.; Wang, X.; Chien, S.; Wang, Y. Engineering Light-Controllable CAR T Cells for Cancer Immunotherapy. Sci. Adv. 2020, 6, eaay9209. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Liu, Y.; Huang, Z.; Wang, X.; Jin, Z.; Li, J.; Limsakul, P.; Zhu, L.; Allen, M.; Pan, Y.; et al. Control of the Activity of CAR-T Cells within Tumours via Focused Ultrasound. Nat. Biomed. Eng. 2021, 5, 1336–1347. [Google Scholar] [CrossRef]
- Sutherland, A.R.; Owens, M.N.; Geyer, C.R. Modular Chimeric Antigen Receptor Systems for Universal CAR T Cell Retargeting. Int. J. Mol. Sci. 2020, 21, 7222. [Google Scholar] [CrossRef]
- Wu, L.; Wei, Q.; Brzostek, J.; Gascoigne, N.R.J. Signaling from T Cell Receptors (TCRs) and Chimeric Antigen Receptors (CARs) on T Cells. Cell Mol. Immunol. 2020, 17, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Gascoigne, N.R.J.; Casas, J.; Brzostek, J.; Rybakin, V. Initiation of TCR Phosphorylation and Signal Transduction. Front. Immunol. 2011, 2, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casas, J.; Brzostek, J.; Zarnitsyna, V.I.; Hong, J.; Wei, Q.; Hoerter, J.A.H.; Fu, G.; Ampudia, J.; Zamoyska, R.; Zhu, C.; et al. Ligand-Engaged TCR Is Triggered by Lck Not Associated with CD8 Coreceptor. Nat. Commun. 2014, 5, 5624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Q.; Brzostek, J.; Sankaran, S.; Casas, J.; Hew, L.S.-Q.; Yap, J.; Zhao, X.; Wojciech, L.; Gascoigne, N.R.J. Lck Bound to Coreceptor Is Less Active than Free Lck. Proc. Natl. Acad. Sci. USA 2020, 117, 15809–15817. [Google Scholar] [CrossRef]
- Zhang, H.; Cordoba, S.-P.; Dushek, O.; van der Merwe, P.A. Basic Residues in the T-Cell Receptor ζ Cytoplasmic Domain Mediate Membrane Association and Modulate Signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 19323–19328. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Guo, X.; Shi, X.; Li, C.; Wu, W.; Yan, C.; Wang, H.; Li, H.; Xu, C. Ionic CD3-Lck Interaction Regulates the Initiation of T-Cell Receptor Signaling. Proc. Natl. Acad. Sci. USA 2017, 114, E5891–E5899. [Google Scholar] [CrossRef] [Green Version]
- James, J.R. Tuning ITAM Multiplicity on T Cell Receptors Can Control Potency and Selectivity to Ligand Density. Sci. Signal. 2018, 11, eaan1088. [Google Scholar] [CrossRef] [Green Version]
- Feucht, J.; Sun, J.; Eyquem, J.; Ho, Y.-J.; Zhao, Z.; Leibold, J.; Dobrin, A.; Cabriolu, A.; Hamieh, M.; Sadelain, M. Calibration of CAR Activation Potential Directs Alternative T Cell Fates and Therapeutic Potency. Nat. Med. 2019, 25, 82–88. [Google Scholar] [CrossRef]
- Karlsson, H.; Svensson, E.; Gigg, C.; Jarvius, M.; Olsson-Strömberg, U.; Savoldo, B.; Dotti, G.; Loskog, A. Evaluation of Intracellular Signaling Downstream Chimeric Antigen Receptors. PLoS ONE 2015, 10, e0144787. [Google Scholar] [CrossRef]
- Davenport, A.J.; Cross, R.S.; Watson, K.A.; Liao, Y.; Shi, W.; Prince, H.M.; Beavis, P.A.; Trapani, J.A.; Kershaw, M.H.; Ritchie, D.S.; et al. Chimeric Antigen Receptor T Cells Form Nonclassical and Potent Immune Synapses Driving Rapid Cytotoxicity. Proc. Natl. Acad. Sci. USA 2018, 115, E2068–E2076. [Google Scholar] [CrossRef] [PubMed]
- Kaech, S.M.; Cui, W. Transcriptional Control of Effector and Memory CD8+ T Cell Differentiation. Nat. Rev. Immunol. 2012, 12, 749–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D.J.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garbi, N.; Hämmerling, G.J.; Probst, H.-C.; van den Broek, M. Tonic T Cell Signalling and T Cell Tolerance as Opposite Effects of Self-Recognition on Dendritic Cells. Curr. Opin. Immunol. 2010, 22, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Ajina, A.; Maher, J. Strategies to Address Chimeric Antigen Receptor Tonic Signaling. Mol. Cancer Ther. 2018, 17, 1795–1815. [Google Scholar] [CrossRef] [Green Version]
- Milone, M.C.; Fish, J.D.; Carpenito, C.; Carroll, R.G.; Binder, G.K.; Teachey, D.; Samanta, M.; Lakhal, M.; Gloss, B.; Danet-Desnoyers, G.; et al. Chimeric Receptors Containing CD137 Signal Transduction Domains Mediate Enhanced Survival of T Cells and Increased Antileukemic Efficacy in Vivo. Mol. Ther. 2009, 17, 1453–1464. [Google Scholar] [CrossRef]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB Costimulation Ameliorates T Cell Exhaustion Induced by Tonic Signaling of Chimeric Antigen Receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [Green Version]
- Frigault, M.J.; Lee, J.; Basil, M.C.; Carpenito, C.; Motohashi, S.; Scholler, J.; Kawalekar, O.U.; Guedan, S.; McGettigan, S.E.; Posey, A.D.J.; et al. Identification of Chimeric Antigen Receptors That Mediate Constitutive or Inducible Proliferation of T Cells. Cancer Immunol. Res. 2015, 3, 356–367. [Google Scholar] [CrossRef] [Green Version]
- Monks, C.R.; Freiberg, B.A.; Kupfer, H.; Sciaky, N.; Kupfer, A. Three-Dimensional Segregation of Supramolecular Activation Clusters in T Cells. Nature 1998, 395, 82–86. [Google Scholar] [CrossRef]
- Freiberg, B.A.; Kupfer, H.; Maslanik, W.; Delli, J.; Kappler, J.; Zaller, D.M.; Kupfer, A. Staging and Resetting T Cell Activation in SMACs. Nat. Immunol. 2002, 3, 911–917. [Google Scholar] [CrossRef]
- Sperling, A.I.; Sedy, J.R.; Manjunath, N.; Kupfer, A.; Ardman, B.; Burkhardt, J.K. TCR Signaling Induces Selective Exclusion of CD43 from the T Cell-Antigen-Presenting Cell Contact Site. J. Immunol. 1998, 161, 6459–6462. [Google Scholar] [PubMed]
- Johnson, K.G.; Bromley, S.K.; Dustin, M.L.; Thomas, M.L. A Supramolecular Basis for CD45 Tyrosine Phosphatase Regulation in Sustained T Cell Activation. Proc. Natl. Acad. Sci. USA 2000, 97, 10138–10143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhuri, K.; Llodrá, J.; Roth, E.W.; Tsai, J.; Gordo, S.; Wucherpfennig, K.W.; Kam, L.C.; Stokes, D.L.; Dustin, M.L. Polarized Release of T-Cell-Receptor-Enriched Microvesicles at the Immunological Synapse. Nature 2014, 507, 118–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finetti, F.; Cassioli, C.; Baldari, C.T. Transcellular Communication at the Immunological Synapse: A Vesicular Traffic-Mediated Mutual Exchange. F1000Research 2017, 6, 1880. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Messina, L.; Rodríguez-Galán, A.; de Yébenes, V.G.; Gutiérrez-Vázquez, C.; Tenreiro, S.; Seabra, M.C.; Ramiro, A.R.; Sánchez-Madrid, F. Transfer of Extracellular Vesicle-MicroRNA Controls Germinal Center Reaction and Antibody Production. EMBO Rep. 2020, 21, e48925. [Google Scholar] [CrossRef]
- Céspedes, P.F.; Jainarayanan, A.; Fernández-Messina, L.; Valvo, S.; Saliba, D.G.; Kurz, E.; Kvalvaag, A.; Chen, L.; Ganskow, C.; Colin-York, H.; et al. T-Cell Trans-Synaptic Vesicles Are Distinct and Carry Greater Effector Content than Constitutive Extracellular Vesicles. Nat. Commun. 2022, 13, 3460. [Google Scholar] [CrossRef]
- Stinchcombe, J.C.; Bossi, G.; Booth, S.; Griffiths, G.M. The Immunological Synapse of CTL Contains a Secretory Domain and Membrane Bridges. Immunity 2001, 15, 751–761. [Google Scholar] [CrossRef] [Green Version]
- Bálint, Š.; Müller, S.; Fischer, R.; Kessler, B.M.; Harkiolaki, M.; Valitutti, S.; Dustin, M.L. Supramolecular Attack Particles Are Autonomous Killing Entities Released from Cytotoxic T Cells. Science 2020, 368, 897–901. [Google Scholar] [CrossRef]
- Cassioli, C.; Baldari, C.T. The Expanding Arsenal of Cytotoxic T Cells. Front. Immunol. 2022, 13, 883010. [Google Scholar] [CrossRef]
- McKenzie, B.; Khazen, R.; Valitutti, S. Greek Fire, Poison Arrows, and Scorpion Bombs: How Tumor Cells Defend Against the Siege Weapons of Cytotoxic T Lymphocytes. Front. Immunol. 2022, 13, 894306. [Google Scholar] [CrossRef]
- Dong, R.; Libby, K.A.; Blaeschke, F.; Fuchs, W.; Marson, A.; Vale, R.D.; Su, X. Rewired Signaling Network in T Cells Expressing the Chimeric Antigen Receptor (CAR). EMBO J. 2020, 39, e104730. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, F.; Müller, S.; Roh, K.-H.; Laurent, C.; Dupré, L.; Valitutti, S. An Initial and Rapid Step of Lytic Granule Secretion Precedes Microtubule Organizing Center Polarization at the Cytotoxic T Lymphocyte/Target Cell Synapse. Proc. Natl. Acad. Sci. USA 2013, 110, 6073–6078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davenport, A.J.; Jenkins, M.R.; Cross, R.S.; Yong, C.S.; Prince, H.M.; Ritchie, D.S.; Trapani, J.A.; Kershaw, M.H.; Darcy, P.K.; Neeson, P.J. CAR-T Cells Inflict Sequential Killing of Multiple Tumor Target Cells. Cancer Immunol. Res. 2015, 3, 483–494. [Google Scholar] [CrossRef] [Green Version]
- Purbhoo, M.A.; Irvine, D.J.; Huppa, J.B.; Davis, M.M. T Cell Killing Does Not Require the Formation of a Stable Mature Immunological Synapse. Nat. Immunol. 2004, 5, 524–530. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, J.P.; Gajewski, T.F. Cutting Edge: Cytotoxic Granule Polarization and Cytolysis Can Occur without Central Supramolecular Activation Cluster Formation in CD8+ Effector T Cells. J. Immunol. 2005, 175, 5581–5585. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Qiu, S.; Chen, J.; Jiang, S.; Chen, W.; Jiang, J.; Wang, F.; Si, W.; Shu, Y.; Wei, P.; et al. Chimeric Antigen Receptor Designed to Prevent Ubiquitination and Downregulation Showed Durable Antitumor Efficacy. Immunity 2020, 53, 456–470.e6. [Google Scholar] [CrossRef]
- Darcy, P.K.; Haynes, N.M.; Snook, M.B.; Trapani, J.A.; Cerruti, L.; Jane, S.M.; Smyth, M.J. Redirected Perforin-Dependent Lysis of Colon Carcinoma by Ex Vivo Genetically Engineered CTL. J. Immunol. 2000, 164, 3705–3712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamonkin, M.; Rouce, R.H.; Tashiro, H.; Brenner, M.K. A T-Cell-Directed Chimeric Antigen Receptor for the Selective Treatment of T-Cell Malignancies. Blood 2015, 126, 983–992. [Google Scholar] [CrossRef] [Green Version]
- Hong, L.K.; Chen, Y.; Smith, C.C.; Montgomery, S.A.; Vincent, B.G.; Dotti, G.; Savoldo, B. CD30-Redirected Chimeric Antigen Receptor T Cells Target CD30(+) and CD30(-) Embryonal Carcinoma via Antigen-Dependent and Fas/FasL Interactions. Cancer Immunol. Res. 2018, 6, 1274–1287. [Google Scholar] [CrossRef] [Green Version]
- Cemerski, S.; Shaw, A. Immune Synapses in T-Cell Activation. Curr. Opin. Immunol. 2006, 18, 298–304. [Google Scholar] [CrossRef]
- Geisler, C. TCR Trafficking in Resting and Stimulated T Cells. Crit. Rev. Immunol. 2004, 24, 67–86. [Google Scholar] [CrossRef] [PubMed]
- Valitutti, S.; Müller, S.; Salio, M.; Lanzavecchia, A. Degradation of T Cell Receptor (TCR)-CD3-Zeta Complexes after Antigenic Stimulation. J. Exp. Med. 1997, 185, 1859–1864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salio, M.; Valitutti, S.; Lanzavecchia, A. Agonist-Induced T Cell Receptor down-Regulation: Molecular Requirements and Dissociation from T Cell Activation. Eur. J. Immunol. 1997, 27, 1769–1773. [Google Scholar] [CrossRef] [PubMed]
- Utzny, C.; Coombs, D.; Müller, S.; Valitutti, S. Analysis of Peptide/MHC-Induced TCR Downregulation: Deciphering the Triggering Kinetics. Cell Biochem. Biophys. 2006, 46, 101–111. [Google Scholar] [CrossRef]
- Naramura, M.; Jang, I.-K.; Kole, H.; Huang, F.; Haines, D.; Gu, H. C-Cbl and Cbl-b Regulate T Cell Responsiveness by Promoting Ligand-Induced TCR down-Modulation. Nat. Immunol. 2002, 3, 1192–1199. [Google Scholar] [CrossRef]
- Vardhana, S.; Choudhuri, K.; Varma, R.; Dustin, M.L. Essential Role of Ubiquitin and TSG101 Protein in Formation and Function of the Central Supramolecular Activation Cluster. Immunity 2010, 32, 531–540. [Google Scholar] [CrossRef] [Green Version]
- Dustin, M.L. The Immunological Synapse. Cancer Immunol. Res. 2014, 2, 1023–1033. [Google Scholar] [CrossRef] [Green Version]
- Lindner, S.E.; Johnson, S.M.; Brown, C.E.; Wang, L.D. Chimeric Antigen Receptor Signaling: Functional Consequences and Design Implications. Sci. Adv. 2020, 6, eaaz3223. [Google Scholar] [CrossRef]
- Kim, S.T.; Takeuchi, K.; Sun, Z.-Y.J.; Touma, M.; Castro, C.E.; Fahmy, A.; Lang, M.J.; Wagner, G.; Reinherz, E.L. The Alphabeta T Cell Receptor Is an Anisotropic Mechanosensor. J. Biol. Chem. 2009, 284, 31028–31037. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Chen, W.; Lou, J.; Rittase, W.; Li, K. Mechanosensing through Immunoreceptors. Nat. Immunol. 2019, 20, 1269–1278. [Google Scholar] [CrossRef]
- Blumenthal, D.; Burkhardt, J.K. Multiple Actin Networks Coordinate Mechanotransduction at the Immunological Synapse. J. Cell Biol. 2020, 219. [Google Scholar] [CrossRef] [PubMed]
- Uray, I.P.; Uray, K. Mechanotransduction at the Plasma Membrane-Cytoskeleton Interface. Int. J. Mol. Sci. 2021, 22, 11566. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Ma, C.; Cai, H.; Chen, W. The CAR T-Cell Mechanoimmunology at a Glance. Adv Sci 2020, 7, 2002628. [Google Scholar] [CrossRef] [PubMed]
- Chang, Z.L.; Lorenzini, M.H.; Chen, X.; Tran, U.; Bangayan, N.J.; Chen, Y.Y. Rewiring T-Cell Responses to Soluble Factors with Chimeric Antigen Receptors. Nat. Chem. Biol. 2018, 14, 317–324. [Google Scholar] [CrossRef]
- Liu, C.S.C.; Ganguly, D. Mechanical Cues for T Cell Activation: Role of Piezo1 Mechanosensors. Crit. Rev. Immunol. 2019, 39, 15–38. [Google Scholar] [CrossRef]
- Pan, Y.; Yoon, S.; Sun, J.; Huang, Z.; Lee, C.; Allen, M.; Wu, Y.; Chang, Y.-J.; Sadelain, M.; Shung, K.K.; et al. Mechanogenetics for the Remote and Noninvasive Control of Cancer Immunotherapy. Proc. Natl. Acad. Sci. USA 2018, 115, 992–997. [Google Scholar] [CrossRef] [Green Version]
- Moia, R.; Boggione, P.; Mahmoud, A.M.; Kodipad, A.A.; Adhinaveni, R.; Sagiraju, S.; Patriarca, A.; Gaidano, G. Targeting P53 in Chronic Lymphocytic Leukemia. Expert Opin. Ther. Targets 2020, 24, 1239–1250. [Google Scholar] [CrossRef]
- Pekarsky, Y.; Balatti, V.; Croce, C.M. BCL2 and MiR-15/16: From Gene Discovery to Treatment. Cell Death Differ. 2018, 25, 21–26. [Google Scholar] [CrossRef]
- Ortíz-Maldonado, V.; Mozas, P.; Delgado, J. The Biology behind B-Cell Lymphoma 2 as a Target in Chronic Lymphocytic Leukemia. Ther. Adv. Hematol. 2016, 7, 321–329. [Google Scholar] [CrossRef]
- Ten Hacken, E.; Burger, J.A. Microenvironment Interactions and B-Cell Receptor Signaling in Chronic Lymphocytic Leukemia: Implications for Disease Pathogenesis and Treatment. Biochim. Biophys. Acta 2016, 1863, 401–413. [Google Scholar] [CrossRef]
- Fürstenau, M.; Eichhorst, B. Novel Agents in Chronic Lymphocytic Leukemia: New Combination Therapies and Strategies to Overcome Resistance. Cancers 2021, 13, 1336. [Google Scholar] [CrossRef] [PubMed]
- Dreger, P.; Schetelig, J.; Andersen, N.; Corradini, P.; van Gelder, M.; Gribben, J.; Kimby, E.; Michallet, M.; Moreno, C.; Stilgenbauer, S.; et al. Managing High-Risk CLL during Transition to a New Treatment Era: Stem Cell Transplantation or Novel Agents? Blood 2014, 124, 3841–3849. [Google Scholar] [CrossRef] [PubMed]
- Smolewski, P.; Robak, T. Current Treatment of Refractory/Relapsed Chronic Lymphocytic Leukemia: A Focus on Novel Drugs. Acta Haematol. 2021, 144, 365–379. [Google Scholar] [CrossRef]
- Mancikova, V.; Smida, M. Current State of CAR T-Cell Therapy in Chronic Lymphocytic Leukemia. Int. J. Mol. Sci. 2021, 22, 5536. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Steeber, D.A.; Jansen, P.J.; Tedder, T.F. CD19 Expression Levels Regulate B Lymphocyte Development: Human CD19 Restores Normal Function in Mice Lacking Endogenous CD19. J. Immunol. 1997, 158, 4662–4669. [Google Scholar] [PubMed]
- Wang, K.; Wei, G.; Liu, D. CD19: A Biomarker for B Cell Development, Lymphoma Diagnosis and Therapy. Exp. Hematol. Oncol. 2012, 1, 36. [Google Scholar] [CrossRef] [Green Version]
- Scheuermann, R.H.; Racila, E. CD19 Antigen in Leukemia and Lymphoma Diagnosis and Immunotherapy. Leuk. Lymphoma 1995, 18, 385–397. [Google Scholar] [CrossRef]
- Gill, S.I.; Vides, V.; Frey, N.V.; Metzger, S.; O’Brien, M.; Hexner, E.; Mato, A.R.; Lacey, S.F.; Melenhorst, J.J.; Pequignot, E.; et al. Prospective Clinical Trial of Anti-CD19 CAR T Cells in Combination with Ibrutinib for the Treatment of Chronic Lymphocytic Leukemia Shows a High Response Rate. Blood 2018, 132, 298. [Google Scholar] [CrossRef]
- Shah, H.R.; Stephens, D.M. Is There a Role for Anti-CD20 Antibodies in CLL? Hematol. Am. Soc. Hematol. Educ. Program 2021, 2021, 68–75. [Google Scholar] [CrossRef]
- Hiraga, J.; Tomita, A.; Sugimoto, T.; Shimada, K.; Ito, M.; Nakamura, S.; Kiyoi, H.; Kinoshita, T.; Naoe, T. Down-Regulation of CD20 Expression in B-Cell Lymphoma Cells after Treatment with Rituximab-Containing Combination Chemotherapies: Its Prevalence and Clinical Significance. Blood 2009, 113, 4885–4893. [Google Scholar] [CrossRef] [Green Version]
- Shah, N.N.; Johnson, B.D.; Schneider, D.; Zhu, F.; Szabo, A.; Keever-Taylor, C.A.; Krueger, W.; Worden, A.A.; Kadan, M.J.; Yim, S.; et al. Bispecific Anti-CD20, Anti-CD19 CAR T Cells for Relapsed B Cell Malignancies: A Phase 1 Dose Escalation and Expansion Trial. Nat. Med. 2020, 26, 1569–1575. [Google Scholar] [CrossRef] [PubMed]
- Fialkow, P.J.; Najfeld, V.; Reddy, A.L.; Singer, J.; Steinmann, L. Chronic Lymphocytic Leukaemia: Clonal Origin in a Committed B-Lymphocyte Progenitor. Lancet 1978, 2, 444–446. [Google Scholar] [CrossRef]
- Ramos, C.A.; Savoldo, B.; Torrano, V.; Ballard, B.; Zhang, H.; Dakhova, O.; Liu, E.; Carrum, G.; Kamble, R.T.; Gee, A.P.; et al. Clinical Responses with T Lymphocytes Targeting Malignancy-Associated κ Light Chains. J. Clin. Investig. 2016, 126, 2588–2596. [Google Scholar] [CrossRef] [Green Version]
- Berger, C.; Sommermeyer, D.; Hudecek, M.; Berger, M.; Balakrishnan, A.; Paszkiewicz, P.J.; Kosasih, P.L.; Rader, C.; Riddell, S.R. Safety of Targeting ROR1 in Primates with Chimeric Antigen Receptor-Modified T Cells. Cancer Immunol. Res. 2015, 3, 206–216. [Google Scholar] [CrossRef] [Green Version]
- Vaisitti, T.; Arruga, F.; Vitale, N.; Lee, T.-T.; Ko, M.; Chadburn, A.; Braggio, E.; Di Napoli, A.; Iannello, A.; Allan, J.N.; et al. ROR1 Targeting with the Antibody-Drug Conjugate VLS-101 Is Effective in Richter Syndrome Patient-Derived Xenograft Mouse Models. Blood 2021, 137, 3365–3377. [Google Scholar] [CrossRef]
- Scarfò, I.; Ormhøj, M.; Frigault, M.J.; Castano, A.P.; Lorrey, S.; Bouffard, A.A.; van Scoyk, A.; Rodig, S.J.; Shay, A.J.; Aster, J.C.; et al. Anti-CD37 Chimeric Antigen Receptor T Cells Are Active against B- and T-Cell Lymphomas. Blood 2018, 132, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-Term Safety and Activity of Axicabtagene Ciloleucel in Refractory Large B-Cell Lymphoma (ZUMA-1): A Single-Arm, Multicentre, Phase 1-2 Trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Porter, D.L.; Hwang, W.-T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric Antigen Receptor T Cells Persist and Induce Sustained Remissions in Relapsed Refractory Chronic Lymphocytic Leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]
- Siddiqi, T.; Soumerai, J.D.; Dorritie, K.A.; Stephens, D.M.; Riedell, P.A.; Arnason, J.; Kipps, T.J.; Gillenwater, H.H.; Gong, L.; Yang, L.; et al. Phase 1 TRANSCEND CLL 004 Study of Lisocabtagene Maraleucel in Patients with Relapsed/Refractory CLL or SLL. Blood 2022, 139, 1794–1806. [Google Scholar] [CrossRef] [PubMed]
- Turtle, C.J.; Hay, K.A.; Hanafi, L.-A.; Li, D.; Cherian, S.; Chen, X.; Wood, B.; Lozanski, A.; Byrd, J.C.; Heimfeld, S.; et al. Durable Molecular Remissions in Chronic Lymphocytic Leukemia Treated With CD19-Specific Chimeric Antigen Receptor-Modified T Cells After Failure of Ibrutinib. J. Clin. Oncol. 2017, 35, 3010–3020. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transpl. 2019, 25, 625–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Locke, F.L.; Lin, Y.; Jain, N.; Daver, N.; Gulbis, A.M.; Adkins, S.; et al. Toxicity Management after Chimeric Antigen Receptor T Cell Therapy: One Size Does Not Fit “ALL”. Nat. Rev. Clin. Oncol. 2018, 15, 218. [Google Scholar] [CrossRef] [PubMed]
- Wang, L. Clinical Determinants of Relapse Following CAR-T Therapy for Hematologic Malignancies: Coupling Active Strategies to Overcome Therapeutic Limitations. Curr. Res. Transl. Med. 2022, 70, 103320. [Google Scholar] [CrossRef]
- Görgün, G.; Holderried, T.A.W.; Zahrieh, D.; Neuberg, D.; Gribben, J.G. Chronic Lymphocytic Leukemia Cells Induce Changes in Gene Expression of CD4 and CD8 T Cells. J. Clin. Investig. 2005, 115, 1797–1805. [Google Scholar] [CrossRef]
- Tinhofer, I.; Weiss, L.; Gassner, F.; Rubenzer, G.; Holler, C.; Greil, R. Difference in the Relative Distribution of CD4+ T-Cell Subsets in B-CLL with Mutated and Unmutated Immunoglobulin (Ig) VH Genes: Implication for the Course of Disease. J. Immunother. 2009, 32, 302–309. [Google Scholar] [CrossRef]
- Forconi, F.; Moss, P. Perturbation of the Normal Immune System in Patients with CLL. Blood 2015, 126, 573–581. [Google Scholar] [CrossRef] [Green Version]
- Annunziato, F.; Romagnani, C.; Romagnani, S. The 3 Major Types of Innate and Adaptive Cell-Mediated Effector Immunity. J. Allergy Clin. Immunol. 2015, 135, 626–635. [Google Scholar] [CrossRef]
- Puzzolo, M.C.; Del Giudice, I.; Peragine, N.; Mariglia, P.; De Propris, M.S.; Cappelli, L.V.; Trentin, L.; Reda, G.; Cuneo, A.; Molica, S.; et al. TH2/TH1 Shift Under Ibrutinib Treatment in Chronic Lymphocytic Leukemia. Front. Oncol. 2021, 11, 637186. [Google Scholar] [CrossRef]
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17-Producing CD4+ Effector T Cells Develop via a Lineage Distinct from the T Helper Type 1 and 2 Lineages. Nat. Immunol. 2005, 6, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, N.; Hagner, P.R.; Stokes, M.; Gandhi, A.K.; Apollonio, B.; Fanous, M.; Papazoglou, D.; Sutton, L.-A.; Rosenquist, R.; Amini, R.-M.; et al. Triggering Interferon Signaling in T Cells with Avadomide Sensitizes CLL to Anti-PD-L1/PD-1 Immunotherapy. Blood 2021, 137, 216–231. [Google Scholar] [CrossRef] [PubMed]
- Riches, J.C.; Davies, J.K.; McClanahan, F.; Fatah, R.; Iqbal, S.; Agrawal, S.; Ramsay, A.G.; Gribben, J.G. T Cells from CLL Patients Exhibit Features of T-Cell Exhaustion but Retain Capacity for Cytokine Production. Blood 2013, 121, 1612–1621. [Google Scholar] [CrossRef] [PubMed]
- Roessner, P.M.; Seiffert, M. T-Cells in Chronic Lymphocytic Leukemia: Guardians or Drivers of Disease? Leukemia 2020, 34, 2012–2024. [Google Scholar] [CrossRef] [PubMed]
- Sordo-Bahamonde, C.; Lorenzo-Herrero, S.; González-Rodríguez, A.P.; Payer, Á.R.; González-García, E.; López-Soto, A.; Gonzalez, S. LAG-3 Blockade with Relatlimab (BMS-986016) Restores Anti-Leukemic Responses in Chronic Lymphocytic Leukemia. Cancers 2021, 13, 2112. [Google Scholar] [CrossRef] [PubMed]
- Taghiloo, S.; Allahmoradi, E.; Tehrani, M.; Hossein-Nataj, H.; Shekarriz, R.; Janbabaei, G.; Abediankenari, S.; Asgarian-Omran, H. Frequency and Functional Characterization of Exhausted CD8(+) T Cells in Chronic Lymphocytic Leukemia. Eur. J. Haematol. 2017, 98, 622–631. [Google Scholar] [CrossRef]
- Ramsay, A.G.; Clear, A.J.; Fatah, R.; Gribben, J.G. Multiple Inhibitory Ligands Induce Impaired T-Cell Immunologic Synapse Function in Chronic Lymphocytic Leukemia That Can Be Blocked with Lenalidomide: Establishing a Reversible Immune Evasion Mechanism in Human Cancer. Blood 2012, 120, 1412–1421. [Google Scholar] [CrossRef]
- Bardhan, K.; Patsoukis, N.; Weaver, J.; Freeman, G.; Li, L.; Boussiotis, V.A. PD-1 Inhibits the TCR Signaling Cascade by Sequestering SHP-2 Phosphatase, Preventing Its Translocation to Lipid Rafts and Facilitating Csk-Mediated Inhibitory Phosphorylation of Lck. J. Immunol.. 2016, 196, 128. [Google Scholar]
- Hui, E.; Cheung, J.; Zhu, J.; Su, X.; Taylor, M.J.; Wallweber, H.A.; Sasmal, D.K.; Huang, J.; Kim, J.M.; Mellman, I.; et al. T Cell Costimulatory Receptor CD28 Is a Primary Target for PD-1-Mediated Inhibition. Science 2017, 355, 1428–1433. [Google Scholar] [CrossRef]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T Cells with Cell-Intrinsic PD-1 Checkpoint Blockade Resist Tumor-Mediated Inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef] [Green Version]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted Delivery of a PD-1-Blocking ScFv by CAR-T Cells Enhances Anti-Tumor Efficacy in Vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Zhang, Y.; Yu, L.; Wu, D. Cytotoxic Effect of CLL-1 CAR-T Cell Immunotherapy with PD-1 Silencing on Relapsed/Refractory Acute Myeloid Leukemia. Mol. Med. Rep. 2021, 23, 208. [Google Scholar] [CrossRef] [PubMed]
- Yeku, O.O.; Purdon, T.J.; Koneru, M.; Spriggs, D.; Brentjens, R.J. Armored CAR T Cells Enhance Antitumor Efficacy and Overcome the Tumor Microenvironment. Sci. Rep. 2017, 7, 10541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartl, F.A.; Beck-Garcìa, E.; Woessner, N.M.; Flachsmann, L.J.; Cárdenas, R.M.-H.V.; Brandl, S.M.; Taromi, S.; Fiala, G.J.; Morath, A.; Mishra, P.; et al. Noncanonical Binding of Lck to CD3ε Promotes TCR Signaling and CAR Function. Nat. Immunol. 2020, 21, 902–913. [Google Scholar] [CrossRef]
- Wu, W.; Zhou, Q.; Masubuchi, T.; Shi, X.; Li, H.; Xu, X.; Huang, M.; Meng, L.; He, X.; Zhu, H.; et al. Multiple Signaling Roles of CD3ε and Its Application in CAR-T Cell Therapy. Cell 2020, 182, 855–871.e23. [Google Scholar] [CrossRef]
- Baeuerle, P.A.; Ding, J.; Patel, E.; Thorausch, N.; Horton, H.; Gierut, J.; Scarfo, I.; Choudhary, R.; Kiner, O.; Krishnamurthy, J.; et al. Synthetic TRuC Receptors Engaging the Complete T Cell Receptor for Potent Anti-Tumor Response. Nat. Commun. 2019, 10, 2087. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Liu, G.; Wang, J.; Zheng, Z.-Y.; Jia, L.; Rui, W.; Huang, D.; Zhou, Z.-X.; Zhou, L.; Wu, X.; et al. Chimeric STAR Receptors Using TCR Machinery Mediate Robust Responses against Solid Tumors. Sci. Transl. Med. 2021, 13, eabb5191. [Google Scholar] [CrossRef]
- Trautmann, A.; Valitutti, S. The Diversity of Immunological Synapses. Curr. Opin. Immunol. 2003, 15, 249–254. [Google Scholar] [CrossRef]
- Xiong, W.; Chen, Y.; Kang, X.; Chen, Z.; Zheng, P.; Hsu, Y.-H.; Jang, J.H.; Qin, L.; Liu, H.; Dotti, G.; et al. Immunological Synapse Predicts Effectiveness of Chimeric Antigen Receptor Cells. Mol. Ther. 2018, 26, 963–975. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Badeti, S.; Dotti, G.; Jiang, J.; Wang, H.; Dermody, J.; Soteropoulos, P.; Streck, D.; Birge, R.B.; Liu, C. The Role of Immunological Synapse in Predicting the Efficacy of Chimeric Antigen Receptor (CAR) Immunotherapy. Cell Commun. Signal. 2020, 18, 134. [Google Scholar] [CrossRef]
- Watanabe, K.; Kuramitsu, S.; Posey, A.D.J.; June, C.H. Expanding the Therapeutic Window for CAR T Cell Therapy in Solid Tumors: The Knowns and Unknowns of CAR T Cell Biology. Front. Immunol. 2018, 9, 2486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tötterman, T.H.; Carlsson, M.; Simonsson, B.; Bengtsson, M.; Nilsson, K. T-Cell Activation and Subset Patterns Are Altered in B-CLL and Correlate with the Stage of the Disease. Blood 1989, 74, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Peters, F.S.; Strefford, J.C.; Eldering, E.; Kater, A.P. T-Cell Dysfunction in Chronic Lymphocytic Leukemia from an Epigenetic Perspective. Haematologica 2021, 106, 1234–1243. [Google Scholar] [CrossRef] [PubMed]
- Kiesgen, S.; Messinger, J.C.; Chintala, N.K.; Tano, Z.; Adusumilli, P.S. Comparative Analysis of Assays to Measure CAR T-Cell-Mediated Cytotoxicity. Nat. Protoc. 2021, 16, 1331–1342. [Google Scholar] [CrossRef]
- Ramírez-Fernández, Á.; Aguilar-Sopeña, Ó.; Díez-Alonso, L.; Segura-Tudela, A.; Domínguez-Alonso, C.; Roda-Navarro, P.; Álvarez-Vallina, L.; Blanco, B. Synapse Topology and Downmodulation Events Determine the Functional Outcome of Anti-CD19 T Cell-Redirecting Strategies. Oncoimmunology 2022, 11, 2054106. [Google Scholar] [CrossRef]
- Robinson, H.R.; Qi, J.; Cook, E.M.; Nichols, C.; Dadashian, E.L.; Underbayev, C.; Herman, S.E.M.; Saba, N.S.; Keyvanfar, K.; Sun, C.; et al. A CD19/CD3 Bispecific Antibody for Effective Immunotherapy of Chronic Lymphocytic Leukemia in the Ibrutinib Era. Blood 2018, 132, 521–532. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Heipertz, E.L.; Zynda, E.R.; Stav-Noraas, T.E.; Hungler, A.D.; Boucher, S.E.; Kaur, N.; Vemuri, M.C. Current Perspectives on “Off-The-Shelf” Allogeneic NK and CAR-NK Cell Therapies. Front. Immunol. 2021, 12, 732135. [Google Scholar] [CrossRef]
- Miller, J.S.; Soignier, Y.; Panoskaltsis-Mortari, A.; McNearney, S.A.; Yun, G.H.; Fautsch, S.K.; McKenna, D.; Le, C.; Defor, T.E.; Burns, L.J.; et al. Successful Adoptive Transfer and in Vivo Expansion of Human Haploidentical NK Cells in Patients with Cancer. Blood 2005, 105, 3051–3057. [Google Scholar] [CrossRef] [Green Version]
- Romee, R.; Rosario, M.; Berrien-Elliott, M.M.; Wagner, J.A.; Jewell, B.A.; Schappe, T.; Leong, J.W.; Abdel-Latif, S.; Schneider, S.E.; Willey, S.; et al. Cytokine-Induced Memory-like Natural Killer Cells Exhibit Enhanced Responses against Myeloid Leukemia. Sci. Transl. Med. 2016, 8, 357ra123. [Google Scholar] [CrossRef] [Green Version]
- Verneris, M.R.; Miller, J.S. The Phenotypic and Functional Characteristics of Umbilical Cord Blood and Peripheral Blood Natural Killer Cells. Br. J. Haematol. 2009, 147, 185–191. [Google Scholar] [CrossRef]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human IPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-Tumor Activity. Cell Stem Cell 2018, 23, 181–192.e5. [Google Scholar] [CrossRef] [PubMed]
- Saetersmoen, M.L.; Hammer, Q.; Valamehr, B.; Kaufman, D.S.; Malmberg, K.-J. Off-the-Shelf Cell Therapy with Induced Pluripotent Stem Cell-Derived Natural Killer Cells. Semin. Immunopathol. 2019, 41, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.R.; Chung, J.W.; Choi, I. Development of Natural Killer Cells from Hematopoietic Stem Cells. Mol. Cells 2007, 24, 1–8. [Google Scholar] [PubMed]
- Luevano, M.; Madrigal, A.; Saudemont, A. Generation of Natural Killer Cells from Hematopoietic Stem Cells in Vitro for Immunotherapy. Cell. Mol. Immunol. 2012, 9, 310–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klingemann, H.; Boissel, L.; Toneguzzo, F. Natural Killer Cells for Immunotherapy—Advantages of the NK-92 Cell Line over Blood NK Cells. Front. Immunol. 2016, 7, 91. [Google Scholar] [CrossRef] [Green Version]
- Ochoa, M.C.; Minute, L.; Rodriguez, I.; Garasa, S.; Perez-Ruiz, E.; Inogés, S.; Melero, I.; Berraondo, P. Antibody-Dependent Cell Cytotoxicity: Immunotherapy Strategies Enhancing Effector NK Cells. Immunol. Cell Biol. 2017, 95, 347–355. [Google Scholar] [CrossRef]
- Sivori, S.; Vacca, P.; Del Zotto, G.; Munari, E.; Mingari, M.C.; Moretta, L. Human NK Cells: Surface Receptors, Inhibitory Checkpoints, and Translational Applications. Cell. Mol. Immunol. 2019, 16, 430–441. [Google Scholar] [CrossRef]
- Sportoletti, P.; De Falco, F.; Del Papa, B.; Baldoni, S.; Guarente, V.; Marra, A.; Dorillo, E.; Rompietti, C.; Adamo, F.M.; Ruggeri, L.; et al. NK Cells in Chronic Lymphocytic Leukemia and Their Therapeutic Implications. Int. J. Mol. Sci. 2021, 22, 6665. [Google Scholar] [CrossRef]
- Fu, W.; Lei, C.; Liu, S.; Cui, Y.; Wang, C.; Qian, K.; Li, T.; Shen, Y.; Fan, X.; Lin, F.; et al. CAR Exosomes Derived from Effector CAR-T Cells Have Potent Antitumour Effects and Low Toxicity. Nat. Commun. 2019, 10, 4355. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Cao, X.; Cai, H.; Feng, P.; Chen, X.; Zhu, Y.; Yang, Y.; An, W.; Yang, Y.; Jie, J. The Exosomes Derived from CAR-T Cell Efficiently Target Mesothelin and Reduce Triple-Negative Breast Cancer Growth. Cell. Immunol. 2021, 360, 104262. [Google Scholar] [CrossRef] [PubMed]
- Staufer, O.; Dietrich, F.; Rimal, R.; Schröter, M.; Fabritz, S.; Boehm, H.; Singh, S.; Möller, M.; Platzman, I.; Spatz, J.P. Bottom-up Assembly of Biomedical Relevant Fully Synthetic Extracellular Vesicles. Sci. Adv. 2021, 7, eabg6666. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Feature | T Cell | CAR T Cell |
|---|---|---|
| Antigen recognition | Peptide:MHC | Surface antigen (MHC-independent) |
| IS structure | Bull’s eye | Disorganized |
| Time required for functional IS assembly | 5–10 min | <2 min |
| Cytoskeleton remodeling | ||
| Yes | Partial |
| Yes | Yes | |
Mechanisms of IS formation
| ||
| Yes | Yes * (CARKR mutant) | |
| Yes | Partial | |
| Yes | Partial * | |
| Mechanosensing and mechanotransduction | Yes | Yes * |
IS functions
| ||
| Yes | Yes | |
| Yes | Yes | |
| Yes | Yes * | |
| Serial killing | Yes | Yes |
Mechanisms of CTL-mediated cytotoxicity
| ||
| Yes | Yes | |
| Yes | Yes * | |
| Yes | Unknown |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cassioli, C.; Patrussi, L.; Valitutti, S.; Baldari, C.T. Learning from TCR Signaling and Immunological Synapse Assembly to Build New Chimeric Antigen Receptors (CARs). Int. J. Mol. Sci. 2022, 23, 14255. https://doi.org/10.3390/ijms232214255
Cassioli C, Patrussi L, Valitutti S, Baldari CT. Learning from TCR Signaling and Immunological Synapse Assembly to Build New Chimeric Antigen Receptors (CARs). International Journal of Molecular Sciences. 2022; 23(22):14255. https://doi.org/10.3390/ijms232214255
Chicago/Turabian StyleCassioli, Chiara, Laura Patrussi, Salvatore Valitutti, and Cosima T. Baldari. 2022. "Learning from TCR Signaling and Immunological Synapse Assembly to Build New Chimeric Antigen Receptors (CARs)" International Journal of Molecular Sciences 23, no. 22: 14255. https://doi.org/10.3390/ijms232214255