

Transient Receptor Potential (TRP) Channels in Tumor Vascularization
 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Vasculogenesis, Angiogenesis, and Arteriogenesis in Neovessel Formation
3. Neovessel Formation in the Tumor
4. ECFCs in Tumor Vascularization
5. Pro-Angiogenic Ca2+ Signals in Vascular Endothelial Cells and ECFCs
6. TRP Channels
6.1. The Role of TRP Channels in Endothelial Cells
6.2. The Role of Endothelial TRP Channels in Physiological Angiogenesis
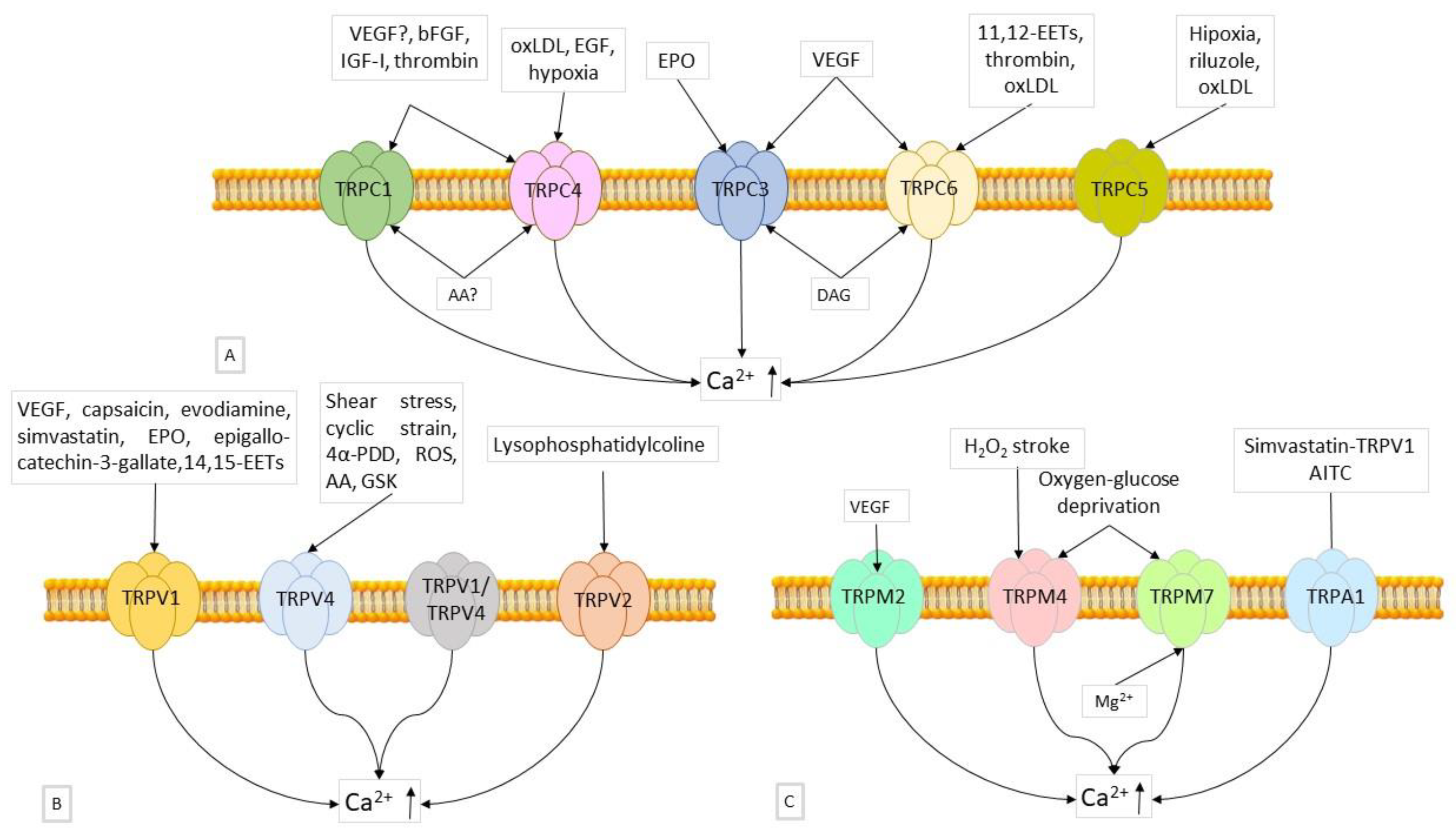
6.2.1. Role of TRPCs
6.2.2. Role of TRPVs
6.2.3. Role of TRPMs
7. The Role of Endothelial TRP Channels in Tumor Vascularization
7.1. Breast Cancer
7.2. Prostate Cancer
7.3. Renal Cellular Carcinoma
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, C.G.; Barbacena, P.; Franco, C.A. Endothelial cells on the move: Dynamics in vascular morphogenesis and disease. Vasc Biol. 2020, 2, H29–H43. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Nolan, D.J.; Mellick, A.S.; Bambino, K.; McDonnell, K.; Mittal, V. Endothelial progenitor cells control the angiogenic switch in mouse lung metastasis. Science 2008, 319, 195–198. [Google Scholar] [CrossRef]
- García-Caballero, M.; Sokol, L.; Cuypers, A.; Carmeliet, P. Metabolic Reprogramming in Tumor Endothelial Cells. Int. J. Mol. Sci. 2022, 23, 11052. [Google Scholar] [CrossRef]
- Saravanan, S.; Vimalraj, S.; Pavani, K.; Nikarika, R.; Sumantran, V.N. Intussusceptive angiogenesis as a key therapeutic target for cancer therapy. Life Sci. 2020, 252, 117670. [Google Scholar] [CrossRef] [PubMed]
- Kuczynski, E.A.; Vermeulen, P.B.; Pezzella, F.; Kerbel, R.S.; Reynolds, A.R. Vessel co-option in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 469–493. [Google Scholar] [CrossRef] [PubMed]
- Negri, S.; Faris, P.; Berra-Romani, R.; Guerra, G.; Moccia, F. Endothelial Transient Receptor Potential Channels and Vascular Remodeling: Extracellular Ca(2+) Entry for Angiogenesis, Arteriogenesis and Vasculogenesis. Front. Physiol. 2019, 10, 1618. [Google Scholar] [CrossRef] [Green Version]
- Yokota, Y.; Nakajima, H.; Wakayama, Y.; Muto, A.; Kawakami, K.; Fukuhara, S.; Mochizuki, N. Endothelial Ca 2+ oscillations reflect VEGFR signaling-regulated angiogenic capacity in vivo. elife 2015, 4, e08817. [Google Scholar] [CrossRef]
- Noren, D.P.; Chou, W.H.; Lee, S.H.; Qutub, A.A.; Warmflash, A.; Wagner, D.S.; Popel, A.S.; Levchenko, A. Endothelial cells decode VEGF-mediated Ca2+ signaling patterns to produce distinct functional responses. Sci. Signal. 2016, 9, ra20. [Google Scholar] [CrossRef] [Green Version]
- Zuccolo, E.; Kheder, D.A.; Lim, D.; Perna, A.; Di Nezza, F.; Botta, L.; Scarpellino, G.; Negri, S.; Martinotti, S.; Soda, T.; et al. Glutamate triggers intracellular Ca. J. Cell Physiol. 2018, 234, 3538–3554. [Google Scholar] [CrossRef]
- Savage, A.M.; Kurusamy, S.; Chen, Y.; Jiang, Z.; Chhabria, K.; Macdonald, R.B.; Kim, H.R.; Wilson, H.L.; Van Eeden, F.J.M.; Armesilla, A.L.; et al. tmem33 is essential for VEGF-mediated endothelial calcium oscillations and angiogenesis. Nat. Commun. 2019, 10, 732. [Google Scholar] [CrossRef] [Green Version]
- Moccia, F.; Negri, S.; Shekha, M.; Faris, P.; Guerra, G. Endothelial Ca(2+) Signaling, Angiogenesis and Vasculogenesis: Just What It Takes to Make a Blood Vessel. Int. J. Mol. Sci. 2019, 20, 3962. [Google Scholar] [CrossRef] [Green Version]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and Therapeutic Aspects of Angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mentzer, S.J.; Konerding, M.A. Intussusceptive angiogenesis: Expansion and remodeling of microvascular networks. Angiogenesis 2014, 17, 499–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moccia, F. Calcium Signaling in Endothelial Colony Forming Cells in Health and Disease. Adv. Exp. Med. Biol. 2020, 1131, 1013–1030. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Zuccolo, E.; Poletto, V.; Cinelli, M.; Bonetti, E.; Guerra, G.; Rosti, V. Endothelial progenitor cells support tumour growth and metastatisation: Implications for the resistance to anti-angiogenic therapy. Tumour Biol. 2015, 36, 6603–6614. [Google Scholar] [CrossRef]
- Hida, K.; Maishi, N.; Torii, C.; Hida, Y. Tumor angiogenesis--characteristics of tumor endothelial cells. Int. J. Clin. Oncol. 2016, 21, 206–212. [Google Scholar] [CrossRef]
- Dimova, I.; Hlushchuk, R.; Makanya, A.; Styp-Rekowska, B.; Ceausu, A.; Flueckiger, S.; Lang, S.; Semela, D.; Le Noble, F.; Chatterjee, S.; et al. Inhibition of Notch signaling induces extensive intussusceptive neo-angiogenesis by recruitment of mononuclear cells. Angiogenesis 2013, 16, 921–937. [Google Scholar] [CrossRef] [Green Version]
- Moccia, F.; Fotia, V.; Tancredi, R.; Della Porta, M.G.; Rosti, V.; Bonetti, E.; Poletto, V.; Marchini, S.; Beltrame, L.; Gallizzi, G.; et al. Breast and renal cancer-Derived endothelial colony forming cells share a common gene signature. Eur. J. Cancer 2017, 77, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Porro, C.; Soleti, R.; Benameur, T.; Maffione, A.; Andriantsitohaina, R.; Martínez, M. Sonic Hedgehog Pathway as a Target for Therapy in Angiogenesis-Related Diseases. Curr. Signal Transduct. Ther. 2009, 4, 31–45. [Google Scholar] [CrossRef]
- Margheri, G.; Zoppi, A.; Olmi, R.; Trigari, S.; Traversi, R.; Severi, M.; Bani, D.; Bianchini, F.; Torre, E.; Margheri, F.; et al. Tumor-tropic endothelial colony forming cells (ECFCs) loaded with near-infrared sensitive Au nanoparticles: A “cellular stove” approach to the photoablation of melanoma. Oncotarget 2016, 7, 39846–39860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poletto, V.; Rosti, V.; Biggiogera, M.; Guerra, G.; Moccia, F.; Porta, C. The role of endothelial colony forming cells in kidney cancer’s pathogenesis, and in resistance to anti-VEGFR agents and mTOR inhibitors: A speculative review. Crit. Rev. Oncol. Hematol. 2018, 132, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Nolan, D.J.; Ciarrocchi, A.; Mellick, A.S.; Jaggi, J.S.; Bambino, K.; Gupta, S.; Heikamp, E.; McDevitt, M.R.; Scheinberg, D.A.; Benezra, R.; et al. Bone marrow-derived endothelial progenitor cells are a major determinant of nascent tumor neovascularization. Genes Dev. 2007, 21, 1546–1558. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Nolan, D.; McDonnell, K.; Vahdat, L.; Benezra, R.; Altorki, N.; Mittal, V. Bone marrow-derived endothelial progenitor cells contribute to the angiogenic switch in tumor growth and metastatic progression. Biochim. Biophys. Acta Rev. Cancer 2009, 1796, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Moccia, F.; Dragoni, S.; Poletto, V.; Rosti, V.; Tanzi, F.; Ganini, C.; Porta, C. Orai1 and Transient Receptor Potential Channels as Novel Molecular Targets to Impair Tumor Neovascularization in Renal Cell Carcinoma and other Malignancies. Anti-Cancer Agents Med. Chem. 2014, 14, 296–312. [Google Scholar] [CrossRef] [PubMed]
- Dudley, A.C. Tumor endothelial cells. Cold Spring Harb. Perspect Med. 2012, 2, a006536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patenaude, A.; Parker, J.; Karsan, A. Involvement of endothelial progenitor cells in tumor vascularization. Microvasc. Res. 2010, 79, 217–223. [Google Scholar] [CrossRef]
- Moccia, F.; Lodola, F.; Dragoni, S.; Bonetti, E.; Bottino, C.; Guerra, G.; Laforenza, U.; Rosti, V.; Tanzi, F. Ca<sup>2+</sup> Signalling in Endothelial Progenitor Cells: A Novel Means to Improve Cell-Based Therapy and Impair Tumour Vascularisation. Curr. Vasc. Pharmacol. 2014, 12, 87–105. [Google Scholar] [CrossRef] [PubMed]
- Ruzinova, M.B.; Schoer, R.A.; Gerald, W.; Egan, J.E.; Pandolfi, P.P.; Rafii, S.; Manova, K.; Mittal, V.; Benezra, R. Effect of angiogenesis inhibition by Id loss and the contribution of bone-marrow-derived endothelial cells in spontaneous murine tumors. Cancer Cell 2003, 4, 277–289. [Google Scholar] [CrossRef] [Green Version]
- Yoder, M.C. Human endothelial progenitor cells. Cold Spring Harb. Perspect Med. 2012, 2, a006692. [Google Scholar] [CrossRef]
- Basile, D.P.; Yoder, M.C. Circulating and tissue resident endothelial progenitor cells. J. Cell. Physiol. 2014, 229, 10–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bieback, K.; Vinci, M.; Elvers-Hornung, S.; Bartol, A.; Gloe, T.; Czabanka, M.; Klüter, H.; Augustin, H.; Vajkoczy, P. Recruitment of human cord blood-derived endothelial colony-forming cells to sites of tumor angiogenesis. Cytotherapy 2013, 15, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Smadja, D.M.; D’Audigier, C.; Weiswald, L.-B.; Badoual, C.; Dangles-Marie, V.; Mauge, L.; Evrard, S.; Laurendeau, I.; Lallemand, F.; Germain, S.; et al. The Wnt antagonist Dickkopf-1 increases endothelial progenitor cell angiogenic potential. Arterioscler Thromb. Vasc Biol. 2010, 30, 2544–2552. [Google Scholar] [CrossRef]
- Wei, J.; Jarmy, G.; Genuneit, J.; Debatin, K.-M.; Beltinger, C. Human blood late outgrowth endothelial cells for gene therapy of cancer: Determinants of efficacy. Gene Ther. 2007, 14, 344–356. [Google Scholar] [CrossRef]
- Pászty, K.; Caride, A.J.; Bajzer, Z.; Offord, C.P.; Padányi, R.; Hegedűs, L.; Varga, K.; Strehler, E.E.; Enyedi, A. Plasma membrane Ca2+-ATPases can shape the pattern of Ca2+ transients induced by store-operated Ca2+ entry. Sci. Signal. 2015, 8, ra19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuccolo, E.; Lim, D.; Kheder, D.A.; Perna, A.; Catarsi, P.; Botta, L.; Rosti, V.; Riboni, L.; Sancini, G.; Tanzi, F.; et al. Acetylcholine induces intracellular Ca. Cell Calcium 2017, 66, 33–47. [Google Scholar] [CrossRef]
- Moccia, F.; Guerra, G. Ca(2+) Signalling in Endothelial Progenitor Cells: Friend or Foe? J. Cell. Physiol. 2016, 231, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Berra-Romani, R.; Tritto, S.; Signorelli, S.; Taglietti, V.; Tanzi, F. Epidermal growth factor induces intracellular Ca2+ oscillations in microvascular endothelial cells. J. Cell. Physiol. 2003, 194, 139–150. [Google Scholar] [CrossRef]
- Andrikopoulos, P.; Kieswich, J.; Harwood, S.M.; Baba, A.; Matsuda, T.; Barbeau, O.; Jones, K.; Eccles, S.A.; Yaqoob, M.M. Endothelial Angiogenesis and Barrier Function in Response to Thrombin Require Ca2+ Influx through the Na+/Ca2+ Exchanger. J. Biol. Chem. 2015, 290, 18412–18428. [Google Scholar] [CrossRef] [Green Version]
- Avanzato, D.; Genova, T.; Fiorio Pla, A.; Bernardini, M.; Bianco, S.; Bussolati, B.; Mancardi, D.; Giraudo, E.; Maione, F.; Cassoni, P.; et al. Activation of P2X7 and P2Y11 purinergic receptors inhibits migration and normalizes tumor-derived endothelial cells via cAMP signaling. Sci. Rep. 2016, 6, 32602. [Google Scholar] [CrossRef]
- Scarpellino, G.; Genova, T.; Avanzato, D.; Bernardini, M.; Bianco, S.; Petrillo, S.; Tolosano, E.; de Almeida Vieira, J.R.; Bussolati, B.; Fiorio Pla, A.; et al. Purinergic Calcium Signals in Tumor-Derived Endothelium. Cancers 2019, 11, 766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyubchenko, T.; Woodward, H.; Veo, K.D.; Burns, N.; Nijmeh, H.; Liubchenko, G.A.; Stenmark, K.R.; Gerasimovskaya, E.V. P2Y1 and P2Y13 purinergic receptors mediate Ca2++ signaling and proliferative responses in pulmonary artery vasa vasorum endothelial cells. Am. J. Physiol. Cell. Physiol. 2011, 300, C266–C275. [Google Scholar] [CrossRef] [Green Version]
- Egleton, R.D.; Brown, K.C.; Dasgupta, P. Angiogenic activity of nicotinic acetylcholine receptors: Implications in tobacco-related vascular diseases. Pharmacol. Ther. 2009, 121, 205–223. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.-B.; Su, K.-H.; Kou, Y.R.; Guo, B.-C.; Lee, K.-I.; Wei, J.; Lee, T.-S. Role of transient receptor potential vanilloid 1 in regulating erythropoietin-induced activation of endothelial nitric oxide synthase. Acta Physiol. (Oxf.) 2017, 219, 465–477. [Google Scholar] [CrossRef]
- Berra-Romani, R.; Raqeeb, A.; Avelino-Cruz, J.E.; Moccia, F.; Oldani, A.; Speroni, F.; Taglietti, V.; Tanzi, F. Ca2+ signaling in injured in situ endothelium of rat aorta. Cell Calcium 2008, 44, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Berra-Romani, R.; Raqeeb, A.; Torres-Jácome, J.; Guzman-Silva, A.; Guerra, G.; Tanzi, F.; Moccia, F. The mechanism of injury-induced intracellular calcium concentration oscillations in the endothelium of excised rat aorta. J. Vasc. Res. 2012, 49, 65–76. [Google Scholar] [CrossRef] [Green Version]
- Balbi, C.; Lodder, K.; Costa, A.; Moimas, S.; Moccia, F.; Van Herwaarden, T.; Rosti, V.; Campagnoli, F.; Palmeri, A.; De Biasio, P.; et al. Reactivating endogenous mechanisms of cardiac regeneration via paracrine boosting using the human amniotic fluid stem cell secretome. Int. J. Cardiol. 2019, 287, 87–95. [Google Scholar] [CrossRef]
- Medina, R.J.; Barber, C.L.; Sabatier, F.; Dignat-George, F.; Melero-Martin, J.M.; Khosrotehrani, K.; Ohneda, O.; Randi, A.M.; Chan, J.K.Y.; Yamaguchi, T.; et al. Endothelial Progenitors: A Consensus Statement on Nomenclature. Stem Cells Transl. Med. 2017, 6, 1316–1320. [Google Scholar] [CrossRef]
- Faris, P.; Negri, S.; Perna, A.; Rosti, V.; Guerra, G.; Moccia, F. Therapeutic Potential of Endothelial Colony-Forming Cells in Ischemic Disease: Strategies to Improve their Regenerative Efficacy. Int. J. Mol. Sci. 2020, 21, 7406. [Google Scholar] [CrossRef]
- Lodola, F.; Laforenza, U.; Cattaneo, F.; Ruffinatti, F.A.; Poletto, V.; Massa, M.; Tancredi, R.; Zuccolo, E.; Khdar, D.A.; Riccardi, A.; et al. VEGF-induced intracellular Ca(2+) oscillations are down-regulated and do not stimulate angiogenesis in breast cancer-derived endothelial colony forming cells. Oncotarget 2017, 8, 95223–95246. [Google Scholar] [CrossRef]
- Zuccolo, E.; Di Buduo, C.; Lodola, F.; Orecchioni, S.; Scarpellino, G.; Kheder, D.A.; Poletto, V.; Guerra, G.; Bertolini, F.; Balduini, A.; et al. Stromal Cell-Derived Factor-1α Promotes Endothelial Colony-Forming Cell Migration Through the Ca(2+)-Dependent Activation of the Extracellular Signal-Regulated Kinase 1/2 and Phosphoinositide 3-Kinase/AKT Pathways. Stem Cells Dev. 2018, 27, 23–34. [Google Scholar] [CrossRef]
- Balducci, V.; Faris, P.; Balbi, C.; Costa, A.; Negri, S.; Rosti, V.; Bollini, S.; Moccia, F. The human amniotic fluid stem cell secretome triggers intracellular Ca(2+) oscillations, NF-κB nuclear translocation and tube formation in human endothelial colony-forming cells. J. Cell Mol. Med. 2021, 25, 8074–8086. [Google Scholar] [CrossRef]
- Galeano-Otero, I.; Del-Toro, R.; Khatib, A.-M.; Rosado, J.A.; Ordóñez-Fernández, A.; Smani, T. Corrigendum: SARAF and Orai1 Contribute to Endothelial Cell Activation and Angiogenesis. Front. Cell Dev. Biol. 2021, 9, 683097. [Google Scholar] [CrossRef] [PubMed]
- Dragoni, S.; Laforenza, U.; Bonetti, E.; Lodola, F.; Bottino, C.; Berra-Romani, R.; Carlo Bongio, G.; Cinelli, M.P.; Guerra, G.; Pedrazzoli, P.; et al. Vascular Endothelial Growth Factor Stimulates Endothelial Colony Forming Cells Proliferation and Tubulogenesis by Inducing Oscillations in Intracellular Ca2+ Concentration. Stem Cells 2011, 29, 1898–1907. [Google Scholar] [CrossRef] [PubMed]
- Faehling, M.; Koch, E.D.; Raithel, J.; Trischler, G.; Waltenberger, J. Vascular endothelial growth factor-A activates Ca2+ -activated K+ channels in human endothelial cells in culture. Int. J. Biochem. Cell Biol. 2001, 33, 337–346. [Google Scholar] [CrossRef]
- Nilius, B.; Droogmans, G. Ion Channels and Their Functional Role in Vascular Endothelium. Physiol. Rev. 2001, 81, 1415–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, Z.; Xie, J.; Yu, A.S.; Stock, J.; Du, J.; Yue, L. Role of TRP channels in the cardiovascular system. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H157–H182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, W.; Yang, F.; Takanishi, C.L.; Zheng, J. Thermosensitive TRPV Channel Subunits Coassemble into Heteromeric Channels with Intermediate Conductance and Gating Properties. J. Gen. Physiol. 2007, 129, 191–207. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Cheng, K.-T.; Wong, C.-O.; O’Neil, R.G.; Birnbaumer, L.; Ambudkar, I.S.; Yao, X. Heteromeric TRPV4-C1 channels contribute to store-operated Ca2++ entry in vascular endothelial cells. Cell Calcium 2011, 50, 502–509. [Google Scholar] [CrossRef] [Green Version]
- Moccia, F. Endothelial Ca(2+) Signaling and the Resistance to Anticancer Treatments: Partners in Crime. Int. J. Mol. Sci. 2018, 19, 217. [Google Scholar] [CrossRef]
- Scarpellino, G.; Munaron, L.; Cantelmo, A.R.; Pla, A.F. Calcium-Permeable Channels in Tumor Vascularization: Peculiar Sensors of Microenvironmental Chemical and Physical Cues. Rev. Physiol. Biochem. Pharmacol. 2022, 182, 111–137. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Monteith, G.R.; Prevarskaya, N.; Roberts-Thomson, S.J. The calcium-cancer signalling nexus. Nat. Rev. Cancer 2017, 17, 367–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiol. Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moccia, F.; Poletto, V. May the remodeling of the Ca2+ toolkit in endothelial progenitor cells derived from cancer patients suggest alternative targets for anti-angiogenic treatment? Biochim. Biophys. Acta 2015, 1853, 1958–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakore, P.; Earley, S. Transient Receptor Potential Channels and Endothelial Cell Calcium Signaling. Compr. Physiol. 2019, 9, 1249–1277. [Google Scholar]
- Gees, M.; Colsoul, B.; Nilius, B. The role of transient receptor potential cation channels in Ca2++ signaling. Cold Spring Harb. Perspect Biol. 2010, 2, a003962. [Google Scholar] [CrossRef] [Green Version]
- Earley, S.; Brayden, J.E. Transient Receptor Potential Channels in the Vasculature. Physiol. Rev. 2015, 95, 645–690. [Google Scholar] [CrossRef] [Green Version]
- Smani, T.; Gómez, L.J.; Regodon, S.; Woodard, G.E.; Siegfried, G.; Khatib, A.-M.; Rosado, J.A. TRP Channels in Angiogenesis and Other Endothelial Functions. Front. Physiol. 2018, 9, 1731. [Google Scholar] [CrossRef] [PubMed]
- Berrout, J.; Jin, M.; O’Neil, R.G. Critical role of TRPP2 and TRPC1 channels in stretch-induced injury of blood–brain barrier endothelial cells. Brain Res. 2012, 1436, 1–12. [Google Scholar] [CrossRef]
- Storch, U.; Forst, A.-L.; Philipp, M.; Gudermann, T.; Schnitzler, M.M.Y. Transient receptor potential channel 1 (TRPC1) reduces calcium permeability in heteromeric channel complexes. J. Biol. Chem. 2012, 287, 3530–3540. [Google Scholar] [CrossRef]
- Goel, M.; Sinkins, W.G.; Schilling, W.P. Selective association of TRPC channel subunits in rat brain synaptosomes. J. Biol. Chem. 2002, 277, 48303–48310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, T.; Schaefer, M.; Schultz, G.; Gudermann, T. Subunit composition of mammalian transient receptor potential channels in living cells. Proc. Natl. Acad. Sci. USA 2002, 99, 7461–7466. [Google Scholar] [CrossRef] [Green Version]
- Cheng, W.; Sun, C.; Zheng, J. Heteromerization of TRP channel subunits: Extending functional diversity. Protein Cell 2010, 1, 802–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindl, R.; Fritsch, R.; Jardin, I.; Frischauf, I.; Kahr, H.; Muik, M.; Riedl, M.C.; Groschner, K.; Romanin, C. Canonical Transient Receptor Potential (TRPC) 1 Acts as a Negative Regulator for Vanilloid TRPV6-mediated Ca2+ Influx *. J. Biol. Chem. 2012, 287, 35612–35620. [Google Scholar] [CrossRef] [Green Version]
- Goldenberg, N.M.; Wang, L.; Ranke, H.; Liedtke, W.; Tabuchi, A.; Kuebler, W.M. TRPV4 Is Required for Hypoxic Pulmonary Vasoconstriction. Anesthesiology 2015, 122, 1338–1348. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Grimm, C.; Becker, L.; Ricci, A.J.; Heller, S. A Novel Ion Channel Formed by Interaction of TRPML3 with TRPV5. PLoS ONE 2013, 8, e58174. [Google Scholar] [CrossRef] [Green Version]
- Venkatachalam, K.; Montell, C. TRP Channels. Annu. Rev. Biochem. 2007, 76, 387–417. [Google Scholar] [CrossRef] [Green Version]
- Guerra, G.; Lucariello, A.; Perna, A.; Botta, L.; De Luca, A.; Moccia, F. The Role of Endothelial Ca. Int. J. Mol. Sci. 2018, 19, 938. [Google Scholar] [CrossRef] [Green Version]
- Moccia, F.; Dragoni, S.; Lodola, F.; Bonetti, E.; Bottino, C.; Guerra, G.; Laforenza, U.; Rosti, V.; Tanzi, F. Store-dependent Ca(2+) entry in endothelial progenitor cells as a perspective tool to enhance cell-based therapy and adverse tumour vascularization. Curr. Med. Chem. 2012, 19, 5802–5818. [Google Scholar] [CrossRef]
- Moccia, F.; Berra-Romani, R.; Tanzi, F. Update on vascular endothelial Ca(2+) signalling: A tale of ion channels, pumps and transporters. World J. Biol. Chem. 2012, 3, 127–158. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.C.; Wu, L.; Hicks, K.; O’Neil, R.G. Regulation of Blood-Brain Barrier Permeability by Transient Receptor Potential Type C and Type V Calcium-Permeable Channels. Microcirculation 2008, 15, 359–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuccolo, E.; Laforenza, U.; Negri, S.; Botta, L.; Berra-Romani, R.; Faris, P.; Scarpellino, G.; Forcaia, G.; Pellavio, G.; Sancini, G.; et al. Muscarinic M5 receptors trigger acetylcholine-induced Ca2++ signals and nitric oxide release in human brain microvascular endothelial cells. J. Cell. Physiol. 2019, 234, 4540–4562. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, H.Z.; Carlton-Carew, S.R.; Khan, D.M.; Zargaran, A.; Jahan, K.S.; Ho, W.; Albert, A. Heteromeric TRPV4/TRPC1 channels mediate calcium-sensing receptor-induced nitric oxide production and vasorelaxation in rabbit mesenteric arteries. Vasc. Pharmacol. 2017, 96–98, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Poteser, M.; Graziani, A.; Rosker, C.; Eder, P.; Derler, I.; Kahr, H.; Zhu, M.X.; Romanin, C.; Groschner, K. TRPC3 and TRPC4 Associate to Form a Redox-sensitive Cation Channel: Evidence for expression of native trpc3-trpc4 heteromeric channels in endothelial cells *. J. Biol. Chem. 2006, 281, 13588–13595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Leary, C.; McGahon, M.K.; Ashraf, S.; McNaughten, J.; Friedel, T.; Cincolà, P.; Barabas, P.; Fernandez, J.A.; Stitt, A.W.; McGeown, J.G.; et al. Involvement of TRPV1 and TRPV4 Channels in Retinal Angiogenesis. Investig. Ophthalmol. Vis. Sci. 2019, 60, 3297–3309. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Ma, X.; Shen, B.; Huang, Y.; Birnbaumer, L.; Yao, X. TRPV4, TRPC1, and TRPP2 assemble to form a flow-sensitive heteromeric channel. FASEB J. 2014, 28, 4677–4685. [Google Scholar] [CrossRef] [Green Version]
- Pires, P.W.; Earley, S. Redox regulation of transient receptor potential channels in the endothelium. Microcirculation 2017, 24, e12329. [Google Scholar] [CrossRef] [Green Version]
- Mergler, S.; Valtink, M.; Coulson-Thomas, V.J.; Lindemann, D.; Reinach, P.S.; Engelmann, K.; Pleyer, U. TRPV channels mediate temperature-sensing in human corneal endothelial cells. Exp. Eye Res. 2010, 90, 758–770. [Google Scholar] [CrossRef]
- Watanabe, H.; Vriens, J.; Suh, S.H.; Benham, C.D.; Droogmans, G.; Nilius, B. Heat-evoked Activation of TRPV4 Channels in a HEK293 Cell Expression System and in Native Mouse Aorta Endothelial Cells*. J. Biol. Chem. 2002, 277, 47044–47051. [Google Scholar] [CrossRef] [Green Version]
- Negri, S.; Faris, P.; Rosti, V.; Antognazza, M.R.; Lodola, F.; Moccia, F. Endothelial TRPV1 as an Emerging Molecular Target to Promote Therapeutic Angiogenesis. Cells 2020, 9, 1341. [Google Scholar] [CrossRef] [PubMed]
- Armani, G.; Pozzi, E.; Pagani, A.; Porta, C.; Rizzo, M.; Cicognini, D.; Rovati, B.; Moccia, F.; Pedrazzoli, P.; Ferraris, E. The heterogeneity of cancer endothelium: The relevance of angiogenesis and endothelial progenitor cells in cancer microenvironment. Microvasc. Res. 2021, 138, 104189. [Google Scholar] [CrossRef] [PubMed]
- Andrikopoulos, P.; Eccles, S.A.; Yaqoob, M.M. Coupling between the TRPC3 ion channel and the NCX1 transporter contributed to VEGF-induced ERK1/2 activation and angiogenesis in human primary endothelial cells. Cell Signal. 2017, 37, 12–30. [Google Scholar] [CrossRef]
- Dragoni, S.; Laforenza, U.; Bonetti, E.; Lodola, F.; Bottino, C.; Guerra, G.; Borghesi, A.; Stronati, M.; Rosti, V.; Tanzi, F.; et al. Canonical transient receptor potential 3 channel triggers vascular endothelial growth factor-induced intracellular Ca2++ oscillations in endothelial progenitor cells isolated from umbilical cord blood. Stem Cells Dev. 2013, 22, 2561–2580. [Google Scholar] [CrossRef] [PubMed]
- Antigny, F.; Girardin, N.; Frieden, M. Transient receptor potential canonical channels are required for in vitro endothelial tube formation. J. Biol. Chem. 2012, 287, 5917–5927. [Google Scholar] [CrossRef] [Green Version]
- Song, H.B.; Jun, H.-O.; Kim, J.H.; Fruttiger, M.; Kim, J.H. Suppression of transient receptor potential canonical channel 4 inhibits vascular endothelial growth factor-induced retinal neovascularization. Cell Calcium 2015, 57, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Zadeh, M.H.; Glass, C.A.; Magnussen, A.; Hancox, J.C.; Bates, D.O. VEGF-mediated elevated intracellular calcium and angiogenesis in human microvascular endothelial cells in vitro are inhibited by dominant negative TRPC6. Microcirculation 2008, 15, 605–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, R.; Tai, Y.; Sun, Y.; Zhou, K.; Yang, S.; Cheng, T.; Zou, Q.; Shen, F.; Wang, Y. Critical role of TRPC6 channels in VEGF-mediated angiogenesis. Cancer Lett. 2009, 283, 43–51. [Google Scholar] [CrossRef]
- Du, L.-L.; Shen, Z.; Li, Z.; Ye, X.; Wu, M.; Hong, L.; Zhao, Y. TRPC1 Deficiency Impairs the Endothelial Progenitor Cell Function via Inhibition of Calmodulin/eNOS Pathway. J. Cardiovasc. Transl. Res. 2018, 11, 339–345. [Google Scholar] [CrossRef]
- Adapala, R.K.; Thoppil, R.J.; Ghosh, K.; Cappelli, H.C.; Dudley, A.C.; Paruchuri, S.; Keshamouni, V.; Klagsbrun, M.; Meszaros, J.G.; Chilian, W.M.; et al. Activation of mechanosensitive ion channel TRPV4 normalizes tumor vasculature and improves cancer therapy. Oncogene 2016, 35, 314–322. [Google Scholar] [CrossRef] [Green Version]
- Thoppil, R.J.; Cappelli, H.C.; Adapala, R.K.; Kanugula, A.K.; Paruchuri, S.; Thodeti, C.K. TRPV4 channels regulate tumor angiogenesis via modulation of Rho/Rho kinase pathway. Oncotarget 2016, 7, 25849–25861. [Google Scholar] [CrossRef] [PubMed]
- Ching, L.-C.; Kou, Y.R.; Shyue, S.-K.; Su, K.-H.; Wei, J.; Cheng, L.-C.; Yu, Y.-B.; Pan, C.-C.; Lee, T.-S. Molecular mechanisms of activation of endothelial nitric oxide synthase mediated by transient receptor potential vanilloid type 1. Cardiovasc. Res. 2011, 91, 492–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, K.-H.; Lee, K.-I.; Shyue, S.-K.; Chen, H.-Y.; Wei, J.; Lee, T.-S. Implication of transient receptor potential vanilloid type 1 in 14,15-epoxyeicosatrienoic acid-induced angiogenesis. Int. J. Biol. Sci. 2014, 10, 990–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, K.-H.; Lin, S.-J.; Wei, J.; Lee, K.-I.; Zhao, J.-F.; Shyue, S.-K.; Lee, T.-S. The essential role of transient receptor potential vanilloid 1 in simvastatin-induced activation of endothelial nitric oxide synthase and angiogenesis. Acta Physiol. (Oxf.) 2014, 212, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Guo, S.; Xiong, Z.; Liu, M. Oncogenic role and therapeutic target of transient receptor potential melastatin 7 channel in malignancy. Expert Opin. Ther. Targets 2014, 18, 1177–1196. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Urao, N.; Hecquet, C.M.; Zhang, M.; Sudhahar, V.; Gao, X.-P.; Komarova, Y.; Ushio-Fukai, M.; Malik, A.B. Novel role of reactive oxygen species-activated Trp melastatin channel-2 in mediating angiogenesis and postischemic neovascularization. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 877–887. [Google Scholar] [CrossRef] [Green Version]
- Loh, K.; Ng, G.; Yu, C.Y.; Fhu, C.K.; Yu, D.; Vennekens, R.; Nilius, B.; Soong, T.W.; Liao, P. TRPM4 inhibition promotes angiogenesis after ischemic stroke. Pflugers Arch. 2014, 466, 563–576. [Google Scholar] [CrossRef]
- Baldoli, E.; Maier, J.A.M. Silencing TRPM7 mimics the effects of magnesium deficiency in human microvascular endothelial cells. Angiogenesis 2012, 15, 47–57. [Google Scholar] [CrossRef]
- Sánchezn-Hernández, Y.; Laforenza, U.; Bonetti, E.; Fontana, J.; Dragoni, S.; Russo, M.; Avelino-Cruz, J.E.; Schinelli, S.; Testa, D.; Guerra, G.; et al. Store-Operated Ca2++ Entry Is Expressed in Human Endothelial Progenitor Cells. Stem Cells Dev. 2010, 19, 1967–1981. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Cubbon, R.M.; Wilson, L.A.; Amer, M.S.; McKeown, L.; Hou, B.; Majeed, Y.; Tumova, S.; Seymour, V.A.; Taylor, H.; et al. Orai1 and CRAC channel dependence of VEGF-activated Ca2++ entry and endothelial tube formation. Circ. Res. 2011, 108, 1190–1198. [Google Scholar] [CrossRef] [Green Version]
- Lodola, F.; Laforenza, U.; Bonetti, E.; Lim, D.; Dragoni, S.; Bottino, C.; Ong, H.L.; Guerra, G.; Ganini, C.; Massa, M.; et al. Store-operated Ca2++ entry is remodelled and controls in vitro angiogenesis in endothelial progenitor cells isolated from tumoral patients. PLoS ONE 2012, 7, e42541. [Google Scholar] [CrossRef] [PubMed]
- Dragoni, S.; Reforgiato, M.; Zuccolo, E.; Poletto, V.; Lodola, F.; Ruffinatti, F.A.; Bonetti, E.; Guerra, G.; Barosi, G.; Rosti, V.; et al. Dysregulation of VEGF-induced proangiogenic Ca2++ oscillations in primary myelofibrosis-derived endothelial colony-forming cells. Exp. Hematol. 2015, 43, 1019–1030.e3. [Google Scholar] [CrossRef]
- Wang, R.; Gurguis, C.I.; Gu, W.; Ko, E.A.; Lim, I.; Bang, H.; Zhou, T.; Ko, J.-H. Ion channel gene expression predicts survival in glioma patients. Sci. Rep. 2015, 5, 11593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moccia, F.; Berra-Romani, R.; Rosti, V. Manipulating Intracellular Ca2++ Signals to Stimulate Therapeutic Angiogenesis in Cardiovascular Disorders. Curr. Pharm. Biotechnol. 2018, 19, 686–699. [Google Scholar] [CrossRef] [PubMed]
- Negri, S.; Faris, P.; Tullii, G.; Vismara, M.; Pellegata, A.F.; Lodola, F.; Guidetti, G.; Rosti, V.; Antognazza, M.R.; Moccia, F. Conjugated polymers mediate intracellular Ca(2+) signals in circulating endothelial colony forming cells through the reactive oxygen species-dependent activation of Transient Receptor Potential Vanilloid 1 (TRPV1). Cell Calcium. 2022, 101, 102502. [Google Scholar] [CrossRef] [PubMed]
- Dragoni, S.; Guerra, G.; Pla, A.F.; Bertoni, G.; Rappa, A.; Poletto, V.; Bottino, C.; Aronica, A.; Lodola, F.; Cinelli, M.P.; et al. A functional transient receptor potential vanilloid 4 (TRPV4) channel is expressed in human endothelial progenitor cells. J. Cell Physiol. 2015, 230, 95–104. [Google Scholar] [CrossRef]
- Zuccolo, E.; Dragoni, S.; Poletto, V.; Catarsi, P.; Guido, D.; Rappa, A.; Reforgiato, M.; Lodola, F.; Lim, D.; Rosti, V.; et al. Arachidonic acid-evoked Ca(2+) signals promote nitric oxide release and proliferation in human endothelial colony forming cells. Vascul. Pharmacol. 2016, 87, 159–171. [Google Scholar] [CrossRef]
- Moccia, F.; Negri, S.; Faris, P.; Ronchi, C.; Lodola, F. Optical excitation of organic semiconductors as a highly selective strategy to induce vascular regeneration and tissue repair. Vascul. Pharmacol. 2022, 144, 106998. [Google Scholar] [CrossRef]
- Hofmann, N.A.; Barth, S.; Waldeck-Weiermair, M.; Klec, C.; Strunk, D.; Malli, R.; Graier, W.F. TRPV1 mediates cellular uptake of anandamide and thus promotes endothelial cell proliferation and network-formation. Biol. Open 2014, 3, 1164–1172. [Google Scholar] [CrossRef] [Green Version]
- Lodola, F.; Rosti, V.; Tullii, G.; Desii, A.; Tapella, L.; Catarsi, P.; Lim, D.; Moccia, F.; Antognazza, M.R. Conjugated polymers optically regulate the fate of endothelial colony-forming cells. Sci. Adv. 2019, 5, eaav4620. [Google Scholar] [CrossRef] [Green Version]
- Pla, A.F.; Ong, H.L.; Cheng, K.T.; Brossa, A.; Bussolati, B.; Lockwich, T.; Paria, B.; Munaron, L.; Ambudkar, I.S. TRPV4 mediates tumor-derived endothelial cell migration via arachidonic acid-activated actin remodeling. Oncogene 2012, 31, 200–212. [Google Scholar] [CrossRef]
- Pla, A.F.; Grange, C.; Antoniotti, S.; Tomatis, C.; Merlino, A.; Bussolati, B.; Munaron, L. Arachidonic acid-induced Ca2++ entry is involved in early steps of tumor angiogenesis. Mol. Cancer Res. 2008, 6, 535–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genova, T.; Grolez, G.P.; Camillo, C.; Bernardini, M.; Bokhobza, A.; Richard, E.; Scianna, M.; Lemonnier, L.; Valdembri, D.; Munaron, L.; et al. TRPM8 inhibits endothelial cell migration via a non-channel function by trapping the small GTPase Rap1. J. Cell Biol. 2017, 216, 2107–2130. [Google Scholar] [CrossRef] [Green Version]
- Bernardini, M.; Brossa, A.; Chinigo, G.; Grolez, G.P.; Trimaglio, G.; Allart, L.; Hulot, A.; Marot, G.; Genova, T.; Joshi, A.; et al. Transient Receptor Potential Channel Expression Signatures in Tumor-Derived Endothelial Cells: Functional Roles in Prostate Cancer Angiogenesis. Cancers 2019, 11, 956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cappelli, H.C.; Kanugula, A.K.; Adapala, R.K.; Amin, V.; Sharma, P.; Midha, P.; Paruchuri, S.; Thodeti, C.K. Mechanosensitive TRPV4 channels stabilize VE-cadherin junctions to regulate tumor vascular integrity and metastasis. Cancer Lett. 2019, 442, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Thoppil, R.J.; Adapala, R.K.; Cappelli, H.C.; Kondeti, V.; Dudley, A.C.; Gary Meszaros, J.; Paruchuri, S.; Thodeti, C.K. TRPV4 channel activation selectively inhibits tumor endothelial cell proliferation. Sci. Rep. 2015, 5, 14257. [Google Scholar] [CrossRef] [Green Version]
- Hill, P.A. Recipient origin of vasculature in renal cell carcinoma in a kidney allograft. Pathology 2010, 42, 479–480. [Google Scholar] [CrossRef]
- Matsuda, K.; Ohga, N.; Hida, Y.; Muraki, C.; Tsuchiya, K.; Kurosu, T.; Akino, T.; Shih, S.-C.; Totsuka, Y.; Klagsbrun, M.; et al. Isolated tumor endothelial cells maintain specific character during long-term culture. Biochem. Biophys. Res. Commun. 2010, 394, 947–954. [Google Scholar] [CrossRef]
- Bhatt, R.S.; Zurita, A.J.; O’Neill, A.; Norden-Zfoni, A.; Zhang, L.; Wu, H.K.; Wen, P.Y.; George, D.; Sukhatme, V.P.; Atkins, M.B.; et al. Increased mobilisation of circulating endothelial progenitors in von Hippel-Lindau disease and renal cell carcinoma. Br. J. Cancer 2011, 105, 112–117. [Google Scholar] [CrossRef]
- Hernandez-Yanez, M.; Heymach, J.V.; Zurita, A.J. Circulating Biomarkers in Advanced Renal Cell Carcinoma: Clinical Applications. Curr. Oncol. Rep. 2012, 14, 221–229. [Google Scholar] [CrossRef]
- Yang, L.; Guan, H.; He, J.; Zeng, L.; Yuan, Z.; Xu, M.; Zhang, W.; Wu, X.; Guan, J. VEGF increases the proliferative capacity and eNOS/NO levels of endothelial progenitor cells through the calcineurin/NFAT signalling pathway. Cell Biol. Int. 2012, 36, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Wang, Y.; Li, X.; Shen, Y.; Yin, M.; Guo, Y.; Diao, L.; Liu, Y.; Yue, D. Critical role of TRPC6 channels in the development of human renal cell carcinoma. Mol. Biol. Rep. 2013, 40, 5115–5122. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Lkhagvadorj, S.; Lee, M.-R.; Hwang, K.-H.; Chung, H.C.; Jung, J.H.; Cha, S.-K.; Eom, M. Orai1 and STIM1 are critical for cell migration and proliferation of clear cell renal cell carcinoma. Biochem. Biophys. Res. Commun. 2014, 448, 76–82. [Google Scholar] [CrossRef]
- Veliceasa, D.; Ivanovic, M.; Hoepfner, F.T.-S.; Thumbikat, P.; Volpert, O.V.; Smith, N.D. Transient potential receptor channel 4 controls thrombospondin-1 secretion and angiogenesis in renal cell carcinoma. FEBS J. 2007, 274, 6365–6377. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TRPC Subunit | Angiogenic Processes Regulated |
|---|---|
| TRPC1/TRPC4/TRPC3/TRPC5/TRPC6 | Proliferation, migration, in vitro tubulogenesis. |
| TRPC1 | Filipodia extension, motility in vivo (sprouting angiogenesis); ECFC and MAC proliferation and tube formation. |
| TRPC4 | Retinal neovascularization. |
| TRPC6 | Wound closure in vitro and carotid artery regeneration in vivo. |
| TRPC5 | Neovascularization in hypoxic retina and in mouse hindlimb ischemia. Negative modulation of migration and wound closure in vitro and arterial regeneration in vivo. |
| TRP Subunit | Angiogenic Processes Regulated |
|---|---|
| TRPV1 | Proliferation, migration and tube formation in vitro, angiogenesis in vivo. |
| TRPV4 | Proliferation, migration, tube formation in vitro, angiogenesis and arteriogenesis in vivo, ECFC proliferation. |
| TRP Subunit | Angiogenic Processes Regulated |
|---|---|
| TRPM2 | Migration in vitro, neovascularization in vivo. |
| TRPM4 | Negative regulation of in vitro tubulogenesis and in vivo angiogenesis; supports H2O2-induced migration. |
| TRPM7 | Negative regulation of HUVEC proliferation, adhesion, and migration in vitro and tubulogenesis in vivo; positive regulation of HMEC proliferation and migration. |
| TRPA1 | Tube formation in vitro and neovascularization upon corneal cauterization in vivo. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perna, A.; Sellitto, C.; Komici, K.; Hay, E.; Rocca, A.; De Blasiis, P.; Lucariello, A.; Moccia, F.; Guerra, G. Transient Receptor Potential (TRP) Channels in Tumor Vascularization. Int. J. Mol. Sci. 2022, 23, 14253. https://doi.org/10.3390/ijms232214253
Perna A, Sellitto C, Komici K, Hay E, Rocca A, De Blasiis P, Lucariello A, Moccia F, Guerra G. Transient Receptor Potential (TRP) Channels in Tumor Vascularization. International Journal of Molecular Sciences. 2022; 23(22):14253. https://doi.org/10.3390/ijms232214253
Chicago/Turabian StylePerna, Angelica, Carmine Sellitto, Klara Komici, Eleonora Hay, Aldo Rocca, Paolo De Blasiis, Angela Lucariello, Francesco Moccia, and Germano Guerra. 2022. "Transient Receptor Potential (TRP) Channels in Tumor Vascularization" International Journal of Molecular Sciences 23, no. 22: 14253. https://doi.org/10.3390/ijms232214253