

Exploiting ELIOT for Scaffold-Repurposing Opportunities: TRIM33 a Possible Novel E3 Ligase to Expand the Toolbox for PROTAC Design
 , , , , and
, , , , and 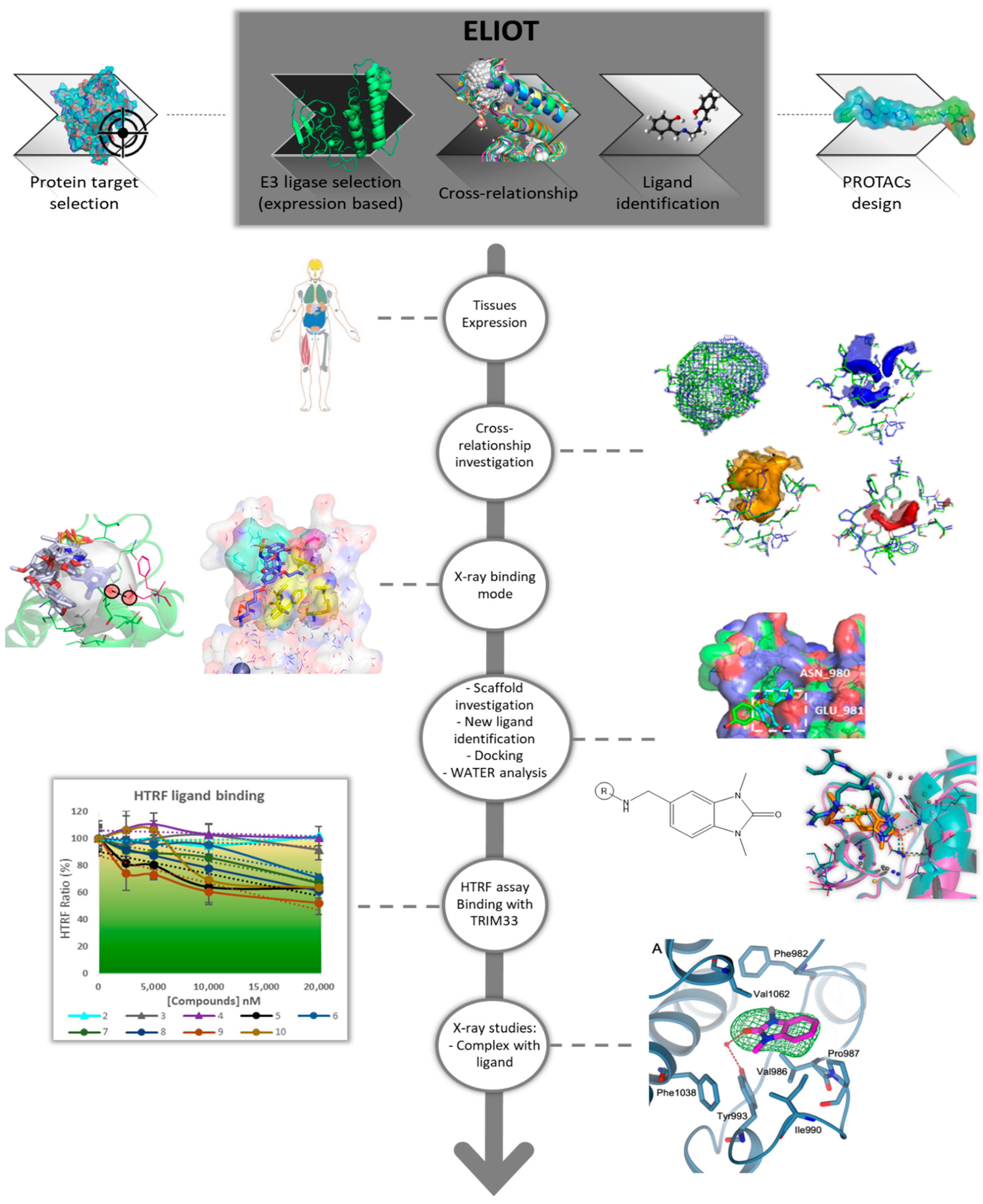{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. ELIOT Application—A Workflow from MIF Comparison to Scaffold-Repurposing Strategy
- TRIM24–TRIM33 Cross-relationship interpretation: in-depth computational analysis to investigate the binding sites’ similarities and differences;
- analysis of the known TRIM24 ligands and their interactions;
- scaffold-repurposing strategy: the similarities in the binding site provide information to select an interacting and common scaffold from the known TRIM24 ligands, while the differences were exploited to identify specific substituents for TRIM33. Compounds satisfying these features were selected from the literature;
- docking analysis to propose possible binding modes and analysis of the key role of the waters in the binding with TRIM33α;
- HTRF assay: set up of an experimental in house procedure to evaluate the binding in TRIM33;
- Crystallographic studies on TRIM33 to confirm the computational and experimental results.
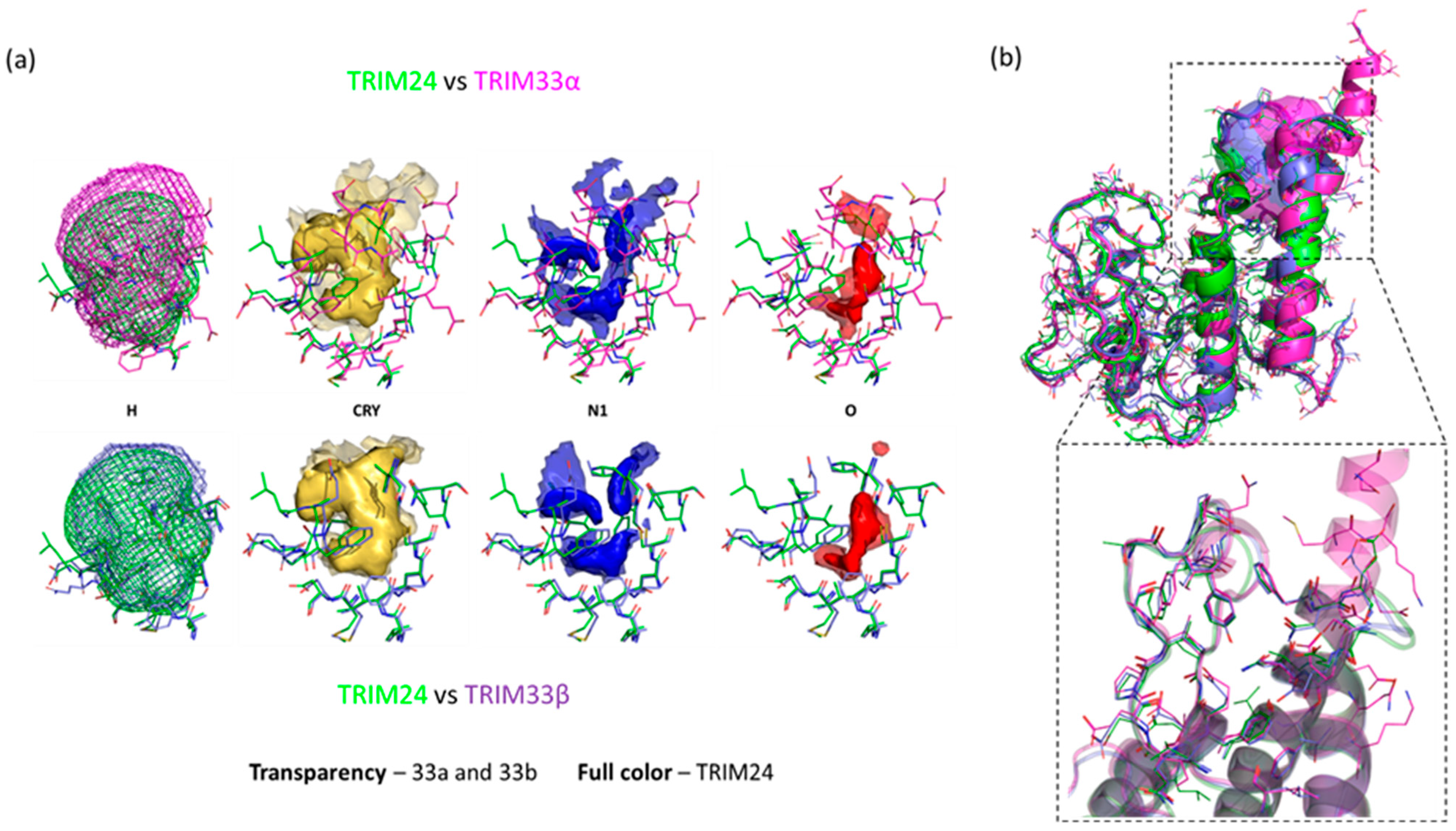
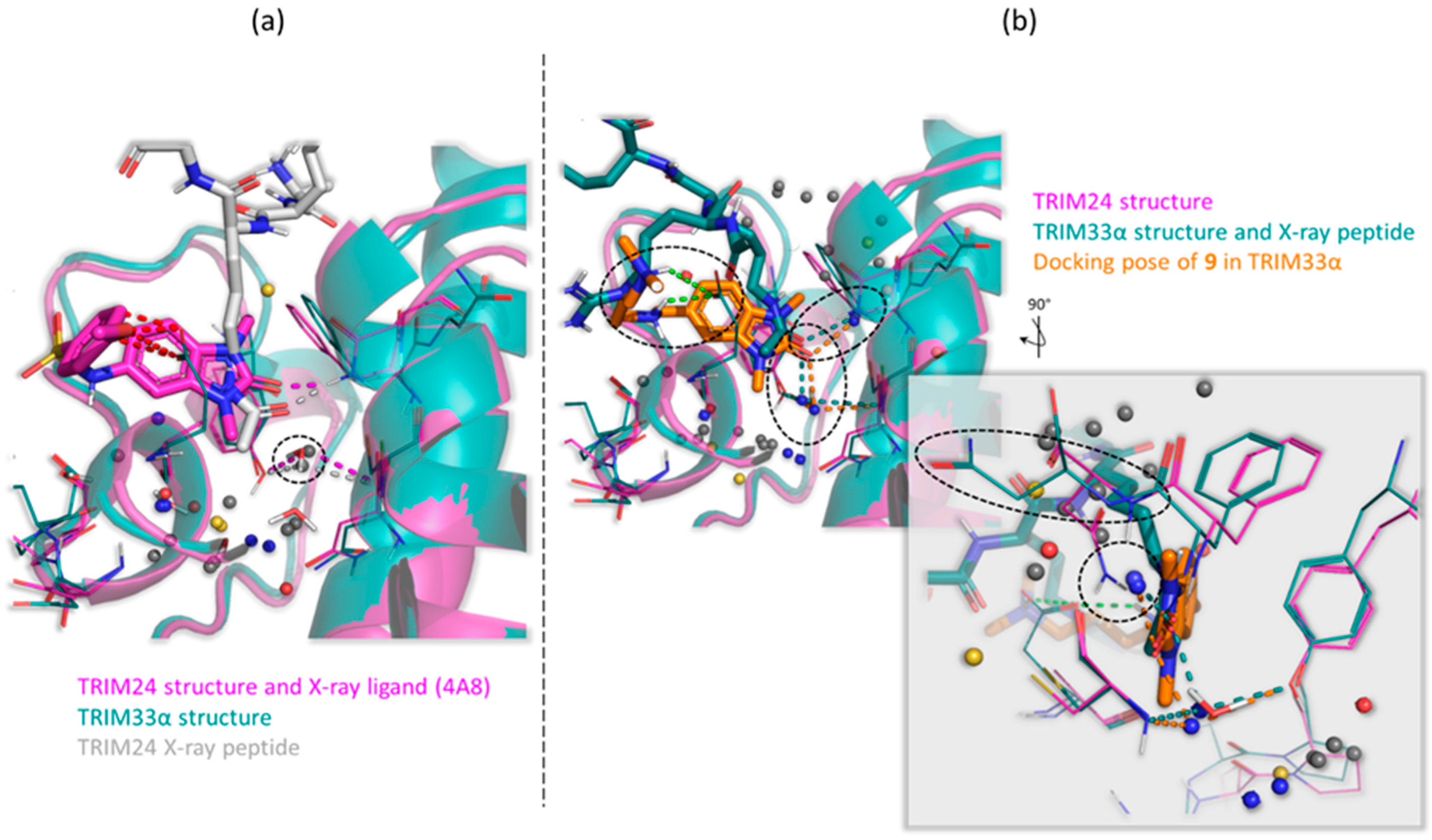
2.2. TRIM24 and TRIM33 Pockets Cross-Relationship
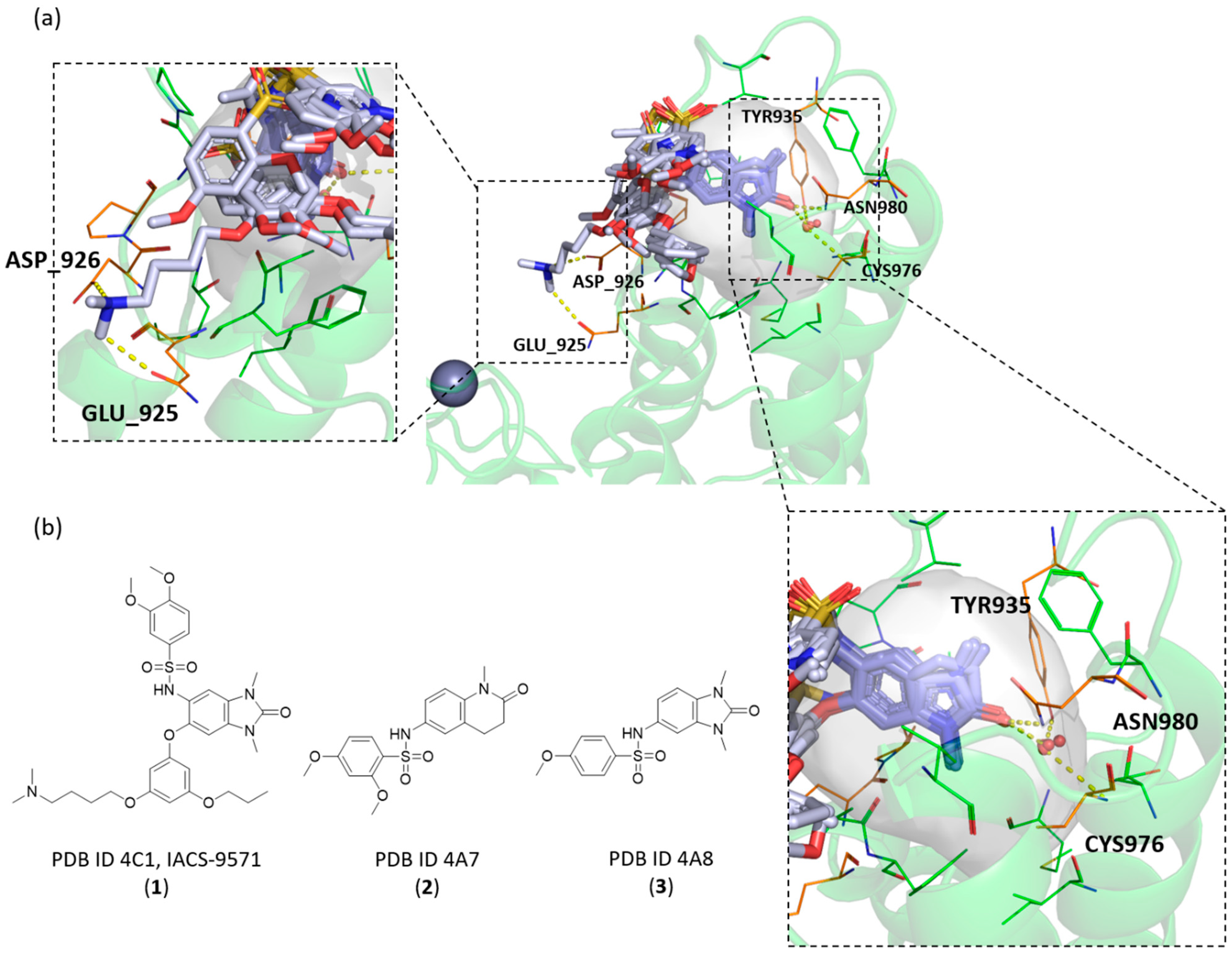
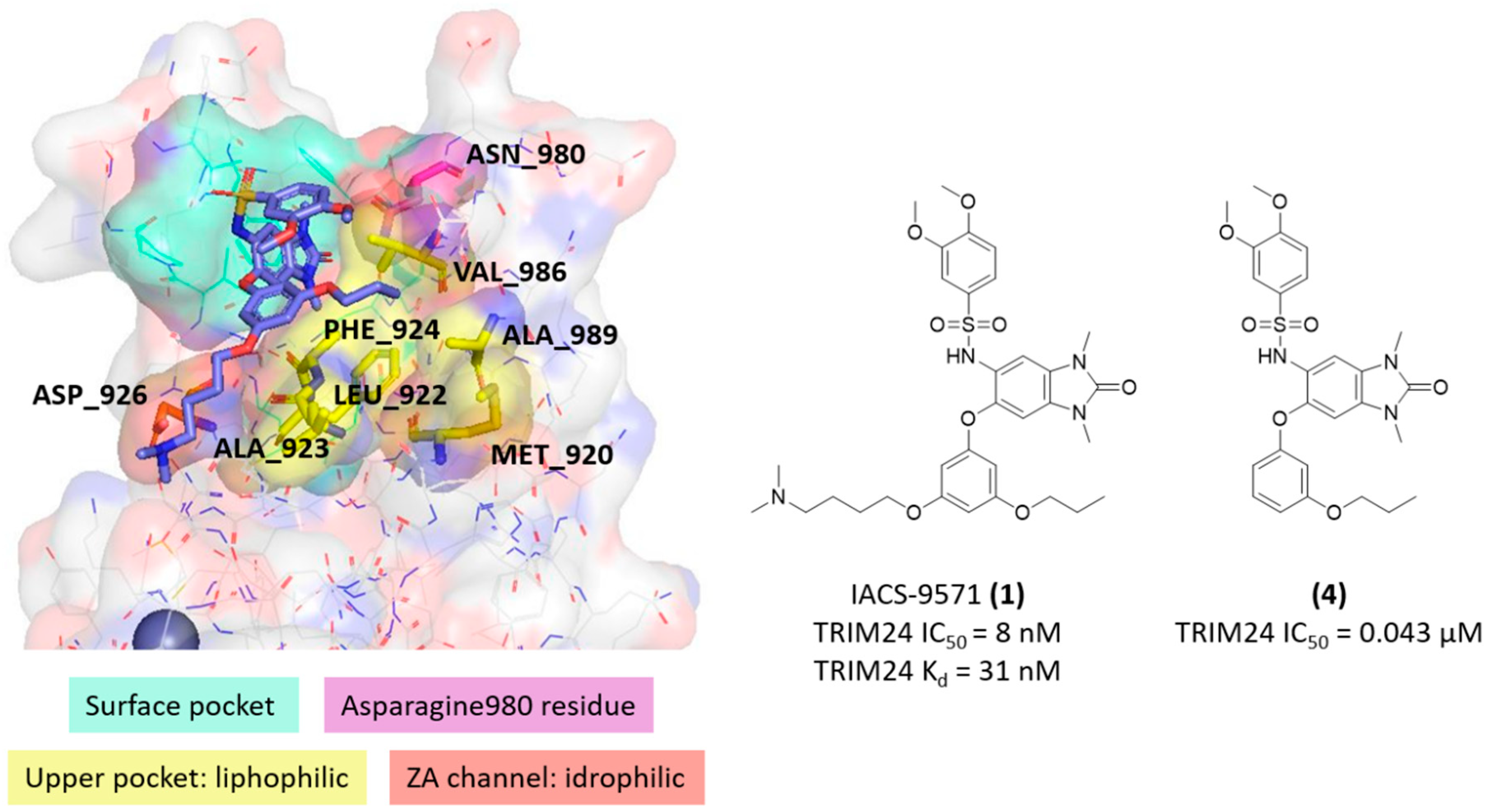
2.3. Known X-ray Crystallographic TRIM24 Ligands
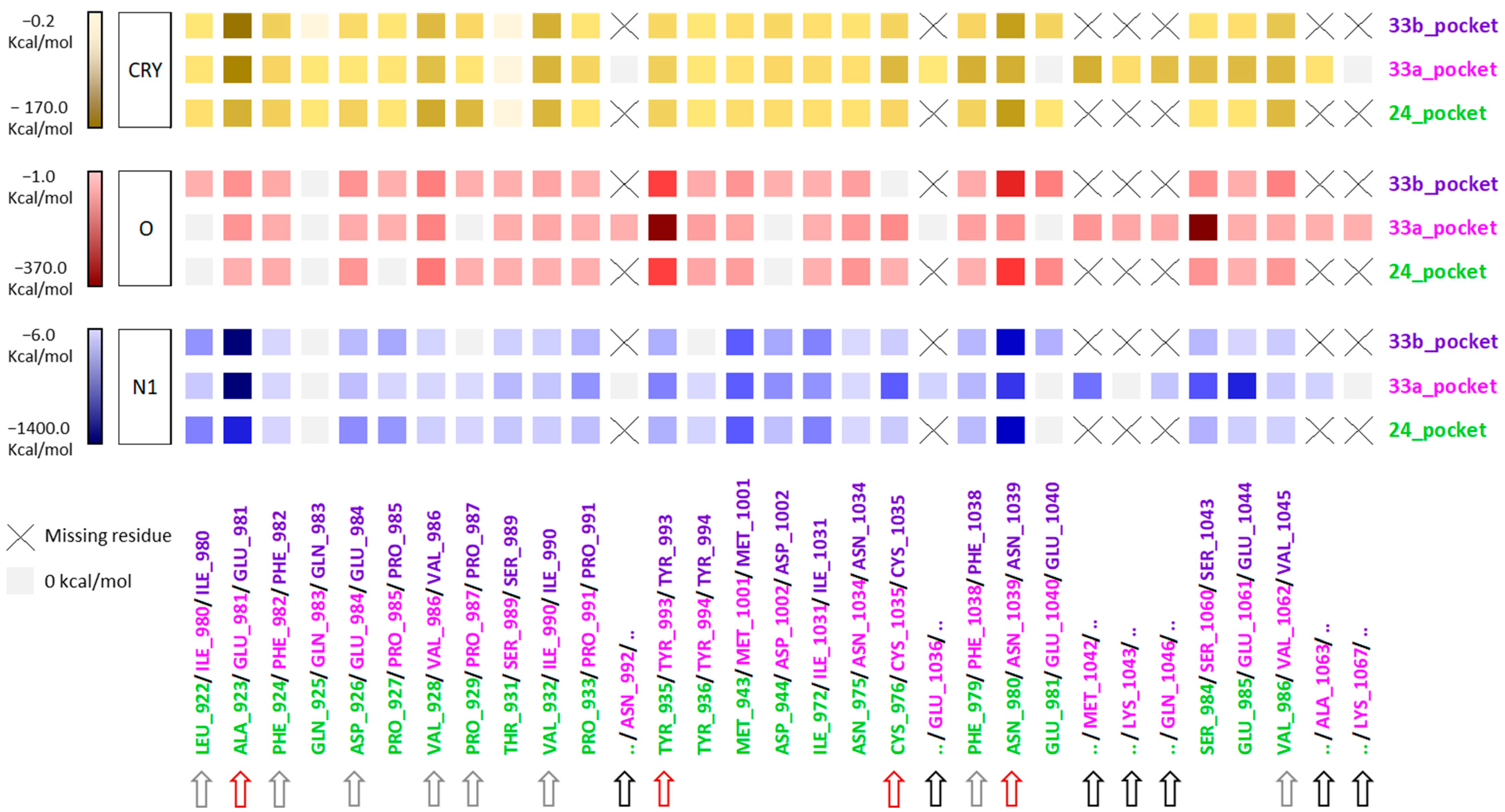
2.4. Similarities and Differences Highlighted Using the MIFs Approach
2.5. Scaffold-Repurposing Strategy: Searching for TRIM33 Ligands
2.6. Docking Analysis for TRIM33α and TRIM33β—The Key Role of a Water Molecule
2.6.1. Water Consideration
2.6.2. Docking Analysis
- -
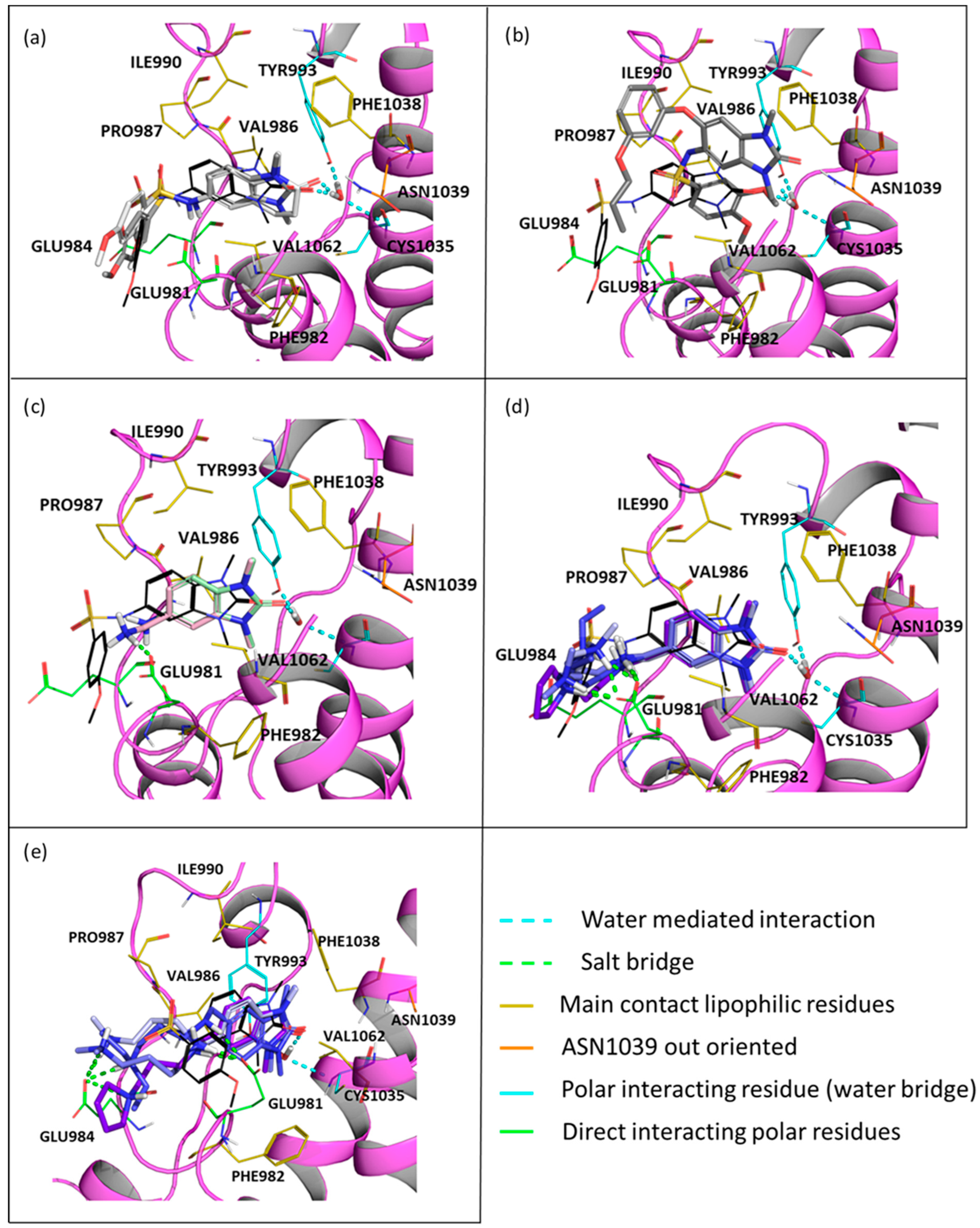
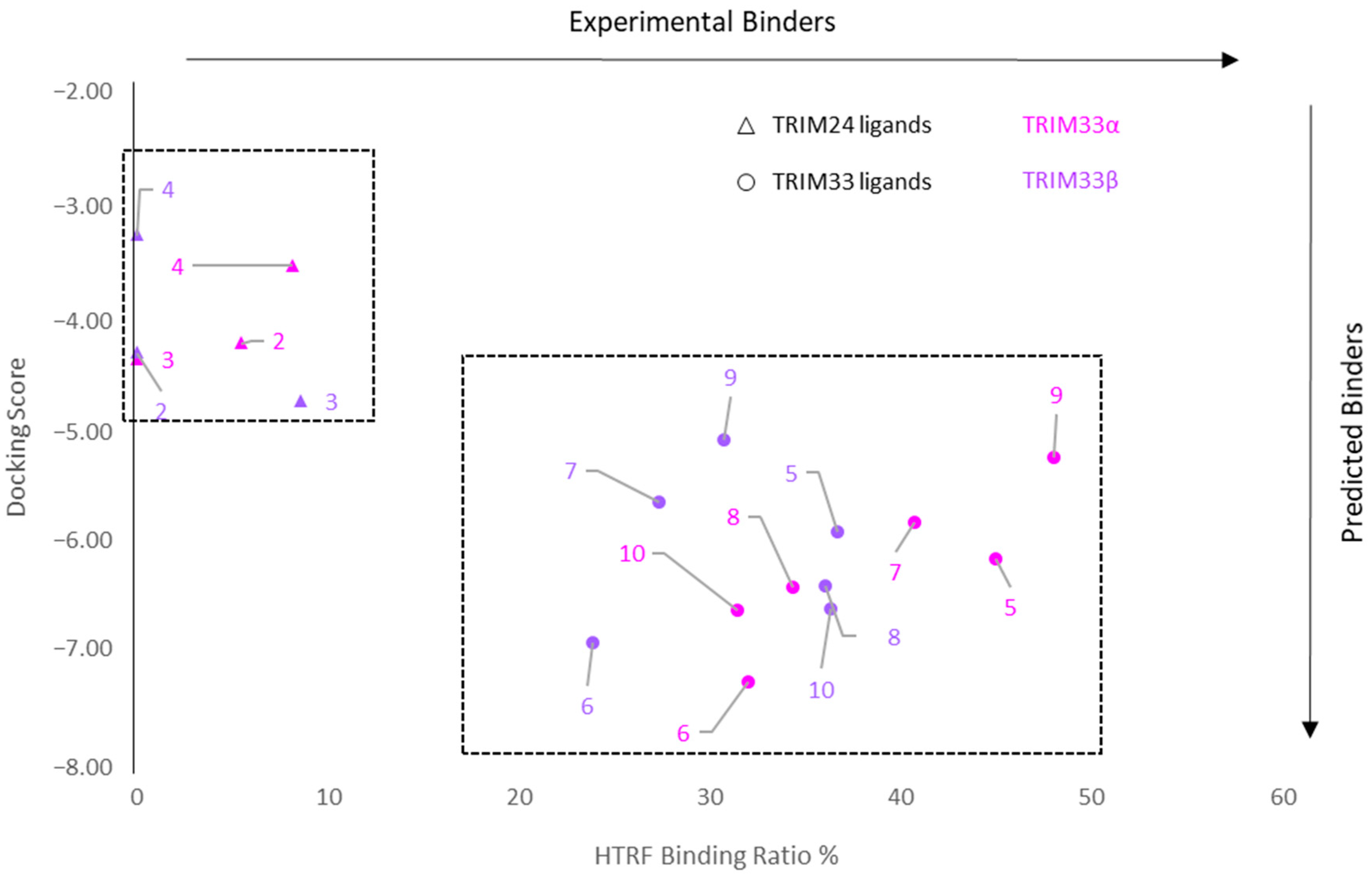
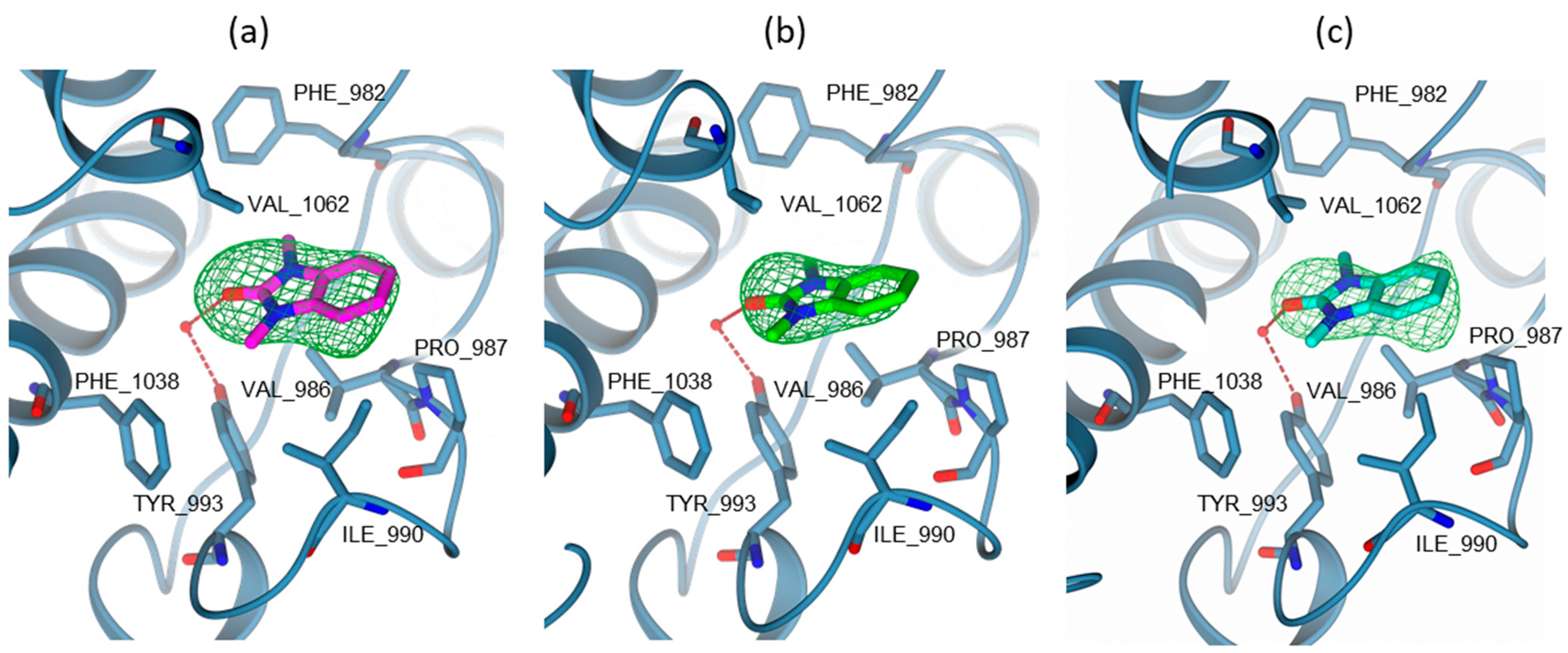
- In TRIM33α, the obtained poses for all the tested compounds replicate the reference binding mode of the X-ray ligand 3 (represented as the black wire in Figure 9).
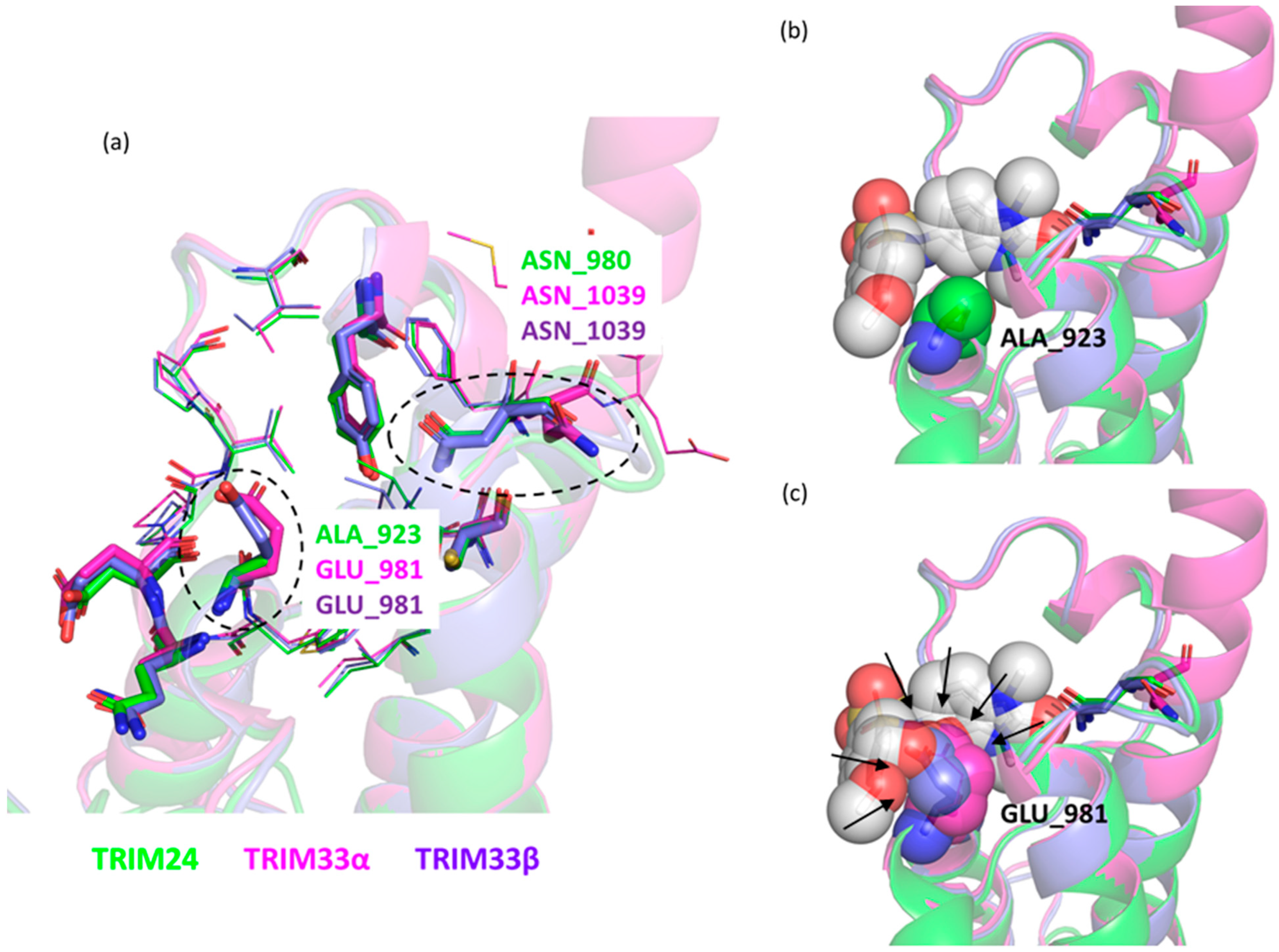
- The presence of the glutamic acid residue (GLU981) should not allow the TRIM24 compounds to fit properly in the pocket. Despite this, for compounds 2 and 3, good poses in TRIM33α were found (Figure 9a) contrary to what was previously assumed, while for compound 4, no plausible poses were obtained (Figure 9b).
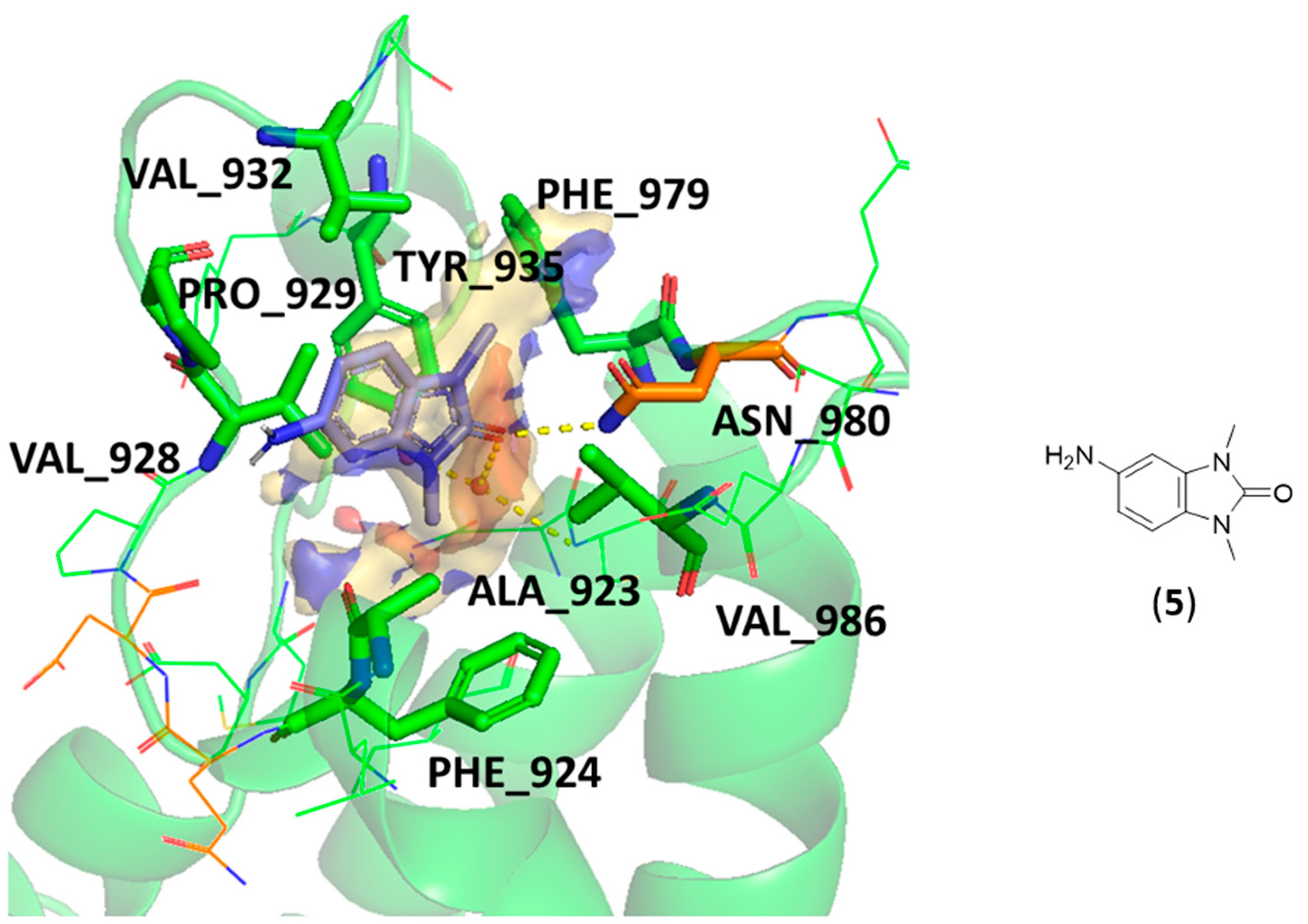
- Both scaffold 5 and 6 fit properly in the pocket, but scaffold 6 can also exploit the salt bridge with GLU981 (Figure 9c).
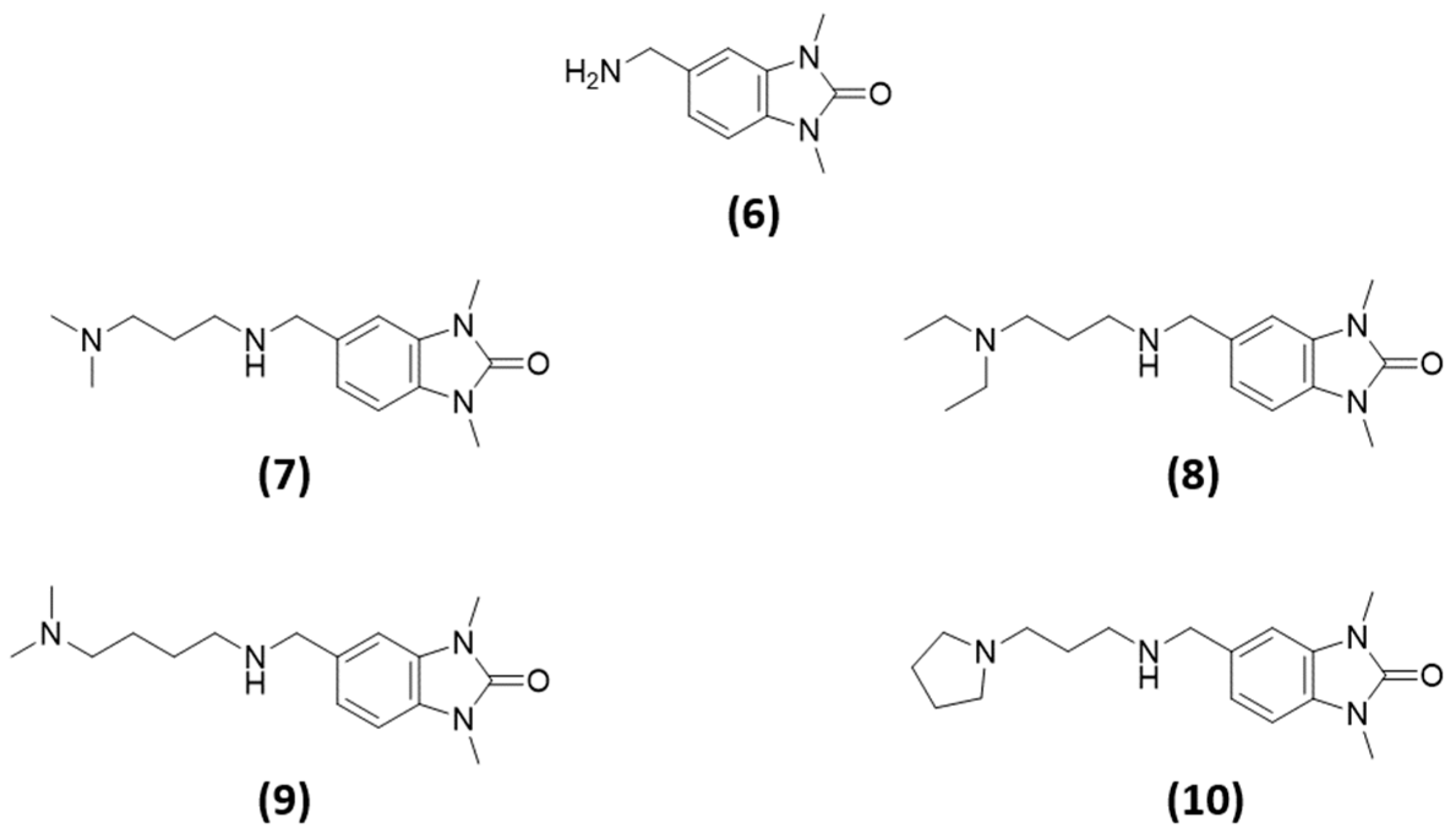
- All the specific TRIM33 ligands (7, 8, 9, and 10) fit very well in the pocket (Figure 9d,e).
- The salt bridge with GLU981 is missing because the amide group can not establish it. Furthermore, even a possible hydrogen bond interaction between the amide and the GLU981 should not occur as at pH 7.4, the amide of the most abundant protomer (66.9%—predicted pKa = 7.19 [56]) of compounds 2 and 3 is deprotonated. Additionally, the reduced mobility due to the rigidity of the sulfonamide does not allow the correct directionality of the amide group towards the glutamic acid.
- As discussed above, after aligning the crystallographic pose of the TRIM24 ligand onto TRIM33α, it is evident how the glutamic acid residue in TRIM33α prevents the access of the ligand to the binding site. The dimethoxybenzenesulfonamide group in its TRIM24 conformation clashes with TRIM33. For this reason, during the docking process, ligands should adapt to the presence of the glutamic acid residue, but their rigidity does not allow them to assume the optimal conformation.
- The absence of an amino terminal aliphatic chain does not allow them to establish additional interactions with the backbone.
- -
- In TRIM33β, the obtained poses are similar to the ones obtained in TRIM33α, except for the main interaction of the core. In this case, the asparagine residue is well-oriented and it can interact directly with the ligand (as in TRIM24), and the conserved water makes a water mediated interaction with the TYR993 and CYS1035 residues (See Figure S4 in Supporting Information). The observations and differences in the binding mode and the docking score of the compounds are the same as those made previously for TRIM33α.
2.6.3. WaterFLAP Analysis Confirms the Key Role of the Water in TRIM33α Bond
2.7. HTRF Assay
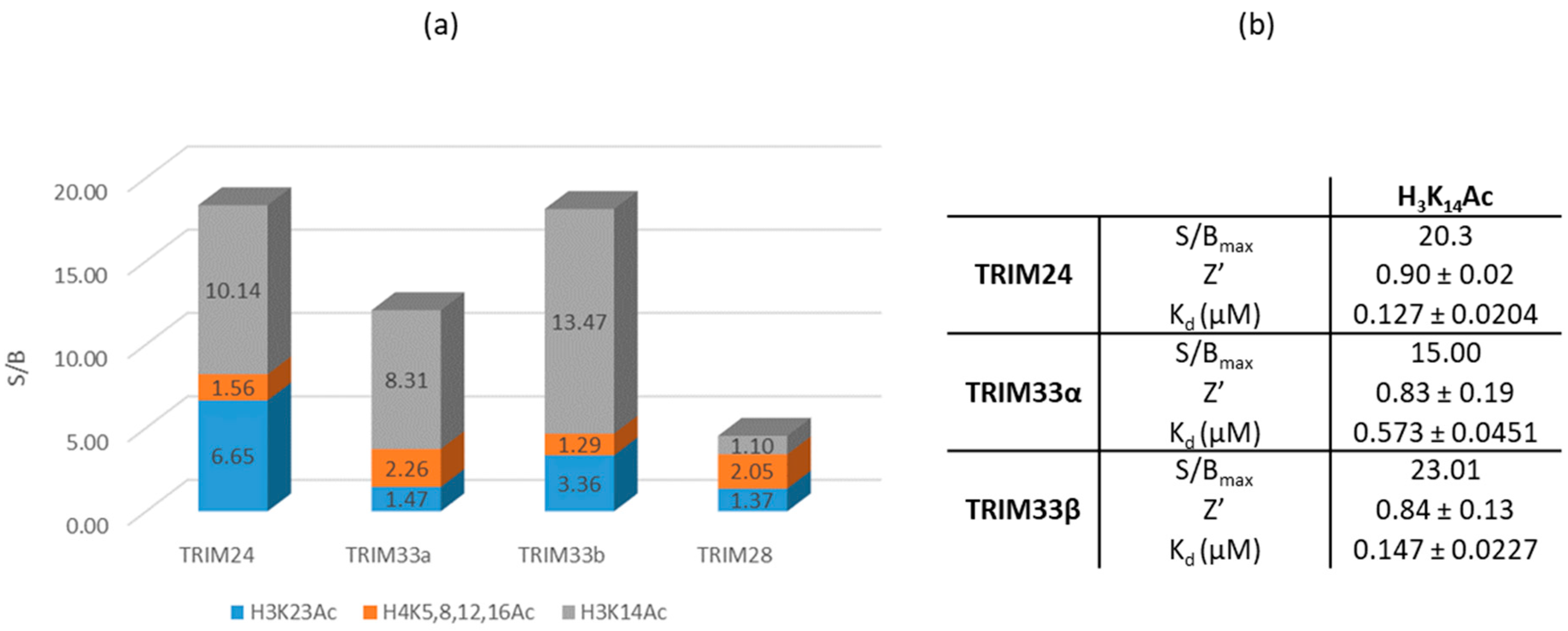
2.7.1. Identification of a Peptide with the Highest Affinity towards the BRD of TRIM24, TRIM33α, and TRIM33β
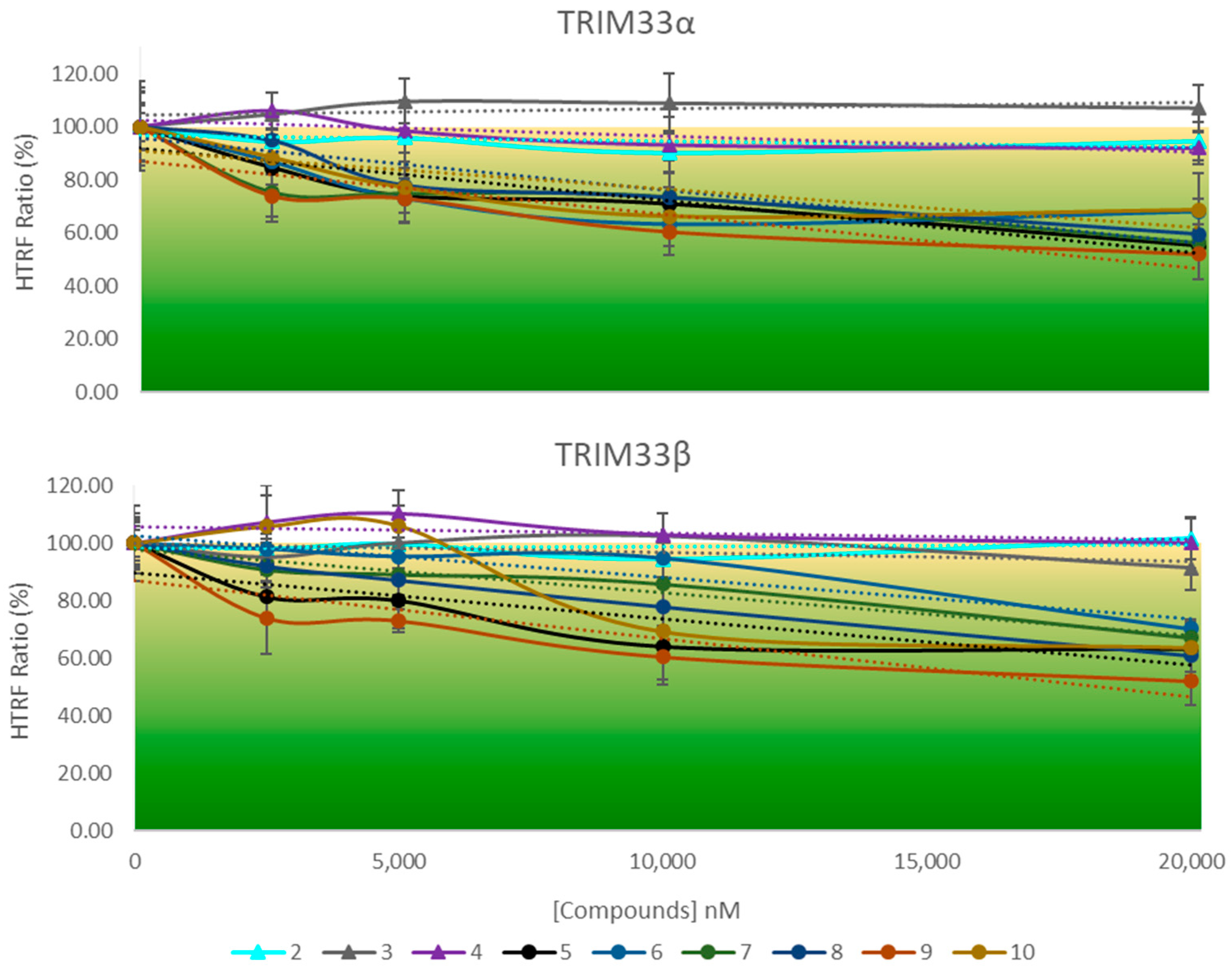
2.7.2. HTRF Competition Assay to Identify TRIM33 Ligands
2.7.3. Correlation between Docking and HTRF Assay
2.8. Crystallography
3. Materials and Methods
3.1. Computational Methods
3.1.1. Data Collection
3.1.2. Pocket Cross-Relationship Analysis
3.1.3. Energetic Contribution of the Residue to the MIFs
3.1.4. Docking Analysis
- Protein files
- Ligand files
- Docking
3.1.5. The WaterFLAP Analysis
- Blue—happy (DG_WAT < −2.0 kcal);
- Grey—bulk-like (−2.0 kcal ≤ DG_WAT < 1.5 kcal);
- Yellow—unhappy (1.5 kcal ≤ DG_WAT < 3.0 kcal);
- Red—very unhappy (DG_WAT ≥ 3.0 kcal).
3.2. Synthetic Chemistry
3.3. HTRF Assay
3.4. Crystallographic Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nandi, D.; Tahiliani, P.; Kumar, A.; Chandu, D. The Ubiquitin-Proteasome System. J. Biosci. 2006, 31, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A. The Ubiquitin System for Protein Degradation. Annu. Rev. Biochem. 1992, 61, 761–807. [Google Scholar] [CrossRef] [PubMed]
- Behl, T.; Kumar, S.; Althafar, Z.M.; Sehgal, A.; Singh, S.; Sharma, N.; Badavath, V.N.; Yadav, S.; Bhatia, S.; Al-Harrasi, A.; et al. Exploring the Role of Ubiquitin–Proteasome System in Parkinson’s Disease. Mol. Neurobiol. 2022, 59, 4257–4273. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.; Xiao, H.; Wang, H.; Xu, X. Recent Advances in PROTACs for Drug Targeted Protein Research. Int. J. Mol. Sci. 2022, 23, 10328. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric Molecules That Target Proteins to the Skp1-Cullin-F Box Complex for Ubiquitination and Degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Jiang, X.; Feng, F.; Liu, W.; Sun, H. Degradation of Proteins by PROTACs and Other Strategies. Acta Pharm. Sin. B 2020, 10, 207–238. [Google Scholar] [CrossRef]
- Cao, C.; He, M.; Wang, L.; He, Y.; Rao, Y. Chemistries of Bifunctional PROTAC Degraders. Chem. Soc. Rev. 2022, 51, 7066–7114. [Google Scholar] [CrossRef]
- Gao, H.; Sun, X.; Rao, Y. PROTAC Technology: Opportunities and Challenges. ACS Med. Chem. Lett. 2020, 11, 237–240. [Google Scholar] [CrossRef] [Green Version]
- Desantis, J.; Goracci, L. Proteolysis Targeting Chimeras in Antiviral Research. Future Med. Chem. 2022, 14, 459–462. [Google Scholar] [CrossRef]
- Gu, S.; Cui, D.; Chen, X.; Xiong, X.; Zhao, Y. PROTACs: An Emerging Targeting Technique for Protein Degradation in Drug Discovery. BioEssays 2018, 40, 1700247–1700257. [Google Scholar] [CrossRef]
- Haouari, S.; Vourc’h, P.; Jeanne, M.; Marouillat, S.; Veyrat-Durebex, C.; Lanznaster, D.; Laumonnier, F.; Corcia, P.; Blasco, H.; Andres, C.R. The Roles of NEDD4 Subfamily of HECT E3 Ubiquitin Ligases in Neurodevelopment and Neurodegeneration. Int. J. Mol. Sci. 2022, 23, 3882. [Google Scholar] [CrossRef]
- Schneider, M.; Radoux, C.J.; Hercules, A.; Ochoa, D.; Dunham, I.; Zalmas, L.P.; Hessler, G.; Ruf, S.; Shanmugasundaram, V.; Hann, M.M.; et al. The PROTACtable Genome. Nat. Rev. Drug Discov. 2021, 20, 789–797. [Google Scholar] [CrossRef]
- Sharma, A.; Khan, H.; Singh, T.G.; Grewal, A.K.; Najda, A.; Kawecka-Radomska, M.; Kamel, M.; Altyar, A.E.; Abdel-Daim, M.M. Pharmacological Modulation of Ubiquitin-Proteasome Pathways in Oncogenic Signaling. Int. J. Mol. Sci. 2021, 22, 11971. [Google Scholar] [CrossRef] [PubMed]
- Vincente, A.T.S.; Salvador, J.A.R. MDM2-Based Proteolysis-Targeting Chimeras (PROTACs): An Innovative Drug Strategy for Cancer Treatment. Int. J. Mol. Sci. 2022, 23, 11068. [Google Scholar] [CrossRef] [PubMed]
- Dale, B.; Cheng, M.; Park, K.-S.; Kaniskan, H.Ü.; Xiong, Y.; Jin, J. Advancing Targeted Protein Degradation for Cancer Therapy. Nat. Rev. Cancer 2021, 21, 638–654. [Google Scholar] [CrossRef]
- Kannt, A.; Đikić, I. Expanding the Arsenal of E3 Ubiquitin Ligases for Proximity-Induced Protein Degradation. Cell Chem. Biol. 2021, 28, 1014–1031. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Riley-Gillis, B.; Vijay, P.; Shen, Y. Acquired Resistance to BET-ProTACS (Proteolysis-Targeting Chimeras) Caused by Genomic Alterations in Core Components of E3 Ligase Complexes. Mol. Cancer Ther. 2019, 18, 1302–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaidman, D.; Prilusky, J.; London, N. ProsetTac: Rosetta Based Modeling of PROTAC Mediated Ternary Complexes. J. Chem. Inf. Model. 2020, 60, 4894–4903. [Google Scholar] [CrossRef]
- Gadd, M.S.; Testa, A.; Lucas, X.; Chan, K.H.; Chen, W.; Lamont, D.J.; Zengerle, M.; Ciulli, A. Structural Basis of PROTAC Cooperative Recognition for Selective Protein Degradation. Nat. Chem. Biol. 2017, 13, 514–521. [Google Scholar] [CrossRef]
- Guenette, R.G.; Yang, S.W.; Min, J.; Pei, B.; Potts, P.R. Target and Tissue Selectivity of PROTAC Degraders. Chem. Soc. Rev. 2022, 51, 5740–5756. [Google Scholar] [CrossRef]
- Paiva, S.; Crews, C.M. Targeted Protein Degradation: Elements of PROTAC Design. Curr. Opin. Chem. Biol. 2018, 50, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Schapira, M.; Crews, C.M. Targeted Proteind Degradation: Expanding the Toolbox. Nat. Rev. Drug Discov. 2019, 18, 949–963. [Google Scholar] [CrossRef] [PubMed]
- Kramer, L.T.; Zhang, X. Expanding the Landscape of E3 Ligases for Targeted Protein Degradation. Curr. Res. Chem. Biol. 2022, 2, 100020–100024. [Google Scholar] [CrossRef]
- Palomba, T.; Cross, S.; Baroni, M.; Cruciani, G.; Siragusa, L. ELIOT: A Platform to Navigate the E3 Pocketome and Aid the Design of New PROTACs. Chem. Biol. Drug Des. 2022, 1–18. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bica, K.; Gaertner, P. Applications of Chiral Ionic Liquids. Eur. J. Org. Chem. 2008, 19, 3235–3250. [Google Scholar] [CrossRef]
- Jordan, V.C. Antiestrogens and Selective Estrogen Receptor Modulators as Multifunctional Medicines. 2. Clinical Considerations and New Agents. J. Med. Chem. 2003, 46, 1081–1111. [Google Scholar] [CrossRef]
- Hua, Z.; Huang, X.; Bregman, H.; Chakka, N.; Dimauro, E.F.; Doherty, E.M.; Goldstein, J.; Gunaydin, H.; Huang, H.; Mercede, S.; et al. 2-Phenylamino-6-Cyano-1H-Benzimidazole-Based Isoform Selective Casein Kinase 1 Gamma (CK1γ) Inhibitors. Bioorganic Med. Chem. Lett. 2012, 22, 5392–5395. [Google Scholar] [CrossRef]
- Goodfrod, P.J. A Computational Procedure for Determining Energetically Favorable Binding Sites on Biologically Important Macromolecules. J. Med. Chem. 1985, 28, 849–857. [Google Scholar] [CrossRef]
- Siragusa, L.; Cross, S.; Baroni, M.; Goracci, L.; Cruciani, G. BioGPS: Navigating Biological Space to Predict Polypharmacology, off-Targeting, and Selectivity. Proteins Struct. Funct. Bioinforma. 2015, 83, 517–532. [Google Scholar] [CrossRef]
- Siragusa, L.; Luciani, R.; Borsari, C.; Ferrari, S.; Costi, M.P.; Cruciani, G.; Spyrakis, F. Comparing Drug Images and Repurposing Drugs with BioGPS and FLAPdock: The Thymidylate Synthase Case. ChemMedChem 2016, 11, 1653–1666. [Google Scholar] [CrossRef] [PubMed]
- Meroni, G.; Desagher, S. Cellular Function of TRIM E3 Ubiquitin Ligases in Health and Disease. Cells 2022, 11, 250. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Du, Y.; Li, S.; Wu, H. The Role of SUMO E3 Ligases in Signaling Pathway of Cancer Cells. Int. J. Mol. Sci. 2022, 23, 3639. [Google Scholar] [CrossRef]
- Xue, J.; Chen, Y.; Wu, Y.; Wang, Z.; Zhou, A.; Zhang, S.; Lin, K.; Aldape, K.; Majumder, S.; Lu, Z.; et al. Tumour Suppressor TRIM33 Targets Nuclear β-Catenin Degradation. Nat. Commun. 2015, 6, 6156–6171. [Google Scholar] [CrossRef] [Green Version]
- D’Amico, F.; Mukhopadhyay, R.; Ovaa, H.; Mulder, M.P.C. Targeting TRIM Proteins: A Quest towards Drugging an Emerging Protein Class. ChemBioChem 2021, 22, 2011–2031. [Google Scholar] [CrossRef] [PubMed]
- Sekirnik, A.R.; Reynolds, J.K.; See, L.; Bluck, J.P.; Scorah, A.R.; Tallant, C.; Lee, B.; Leszczynska, K.B.; Grimley, R.L.; Storer, R.I.; et al. Identification of Histone Peptide Binding Specificity and Small- Molecule Ligands for the TRIM33α and TRIM33β Bromodomains Published. ACS Chem. Biol. 2022, 17, 2753–2768. [Google Scholar] [CrossRef]
- Hewings, D.S.; Rooney, T.P.C.; Jennings, L.E.; Hay, D.A.; Schofield, C.J.; Brennan, P.E.; Knapp, S.; Conway, S.J. Progress in the Development and Application of Small Molecule Inhibitors of Bromodomain-Acetyl-Lysine Interactions. J. Med. Chem. 2012, 55, 9393–9413. [Google Scholar] [CrossRef]
- Hatakeyama, S. TRIM Family Proteins: Roles in Autophagy, Immunity, and Carcinogenesis. Trends Biochem. Sci. 2017, 42, 297–311. [Google Scholar] [CrossRef]
- Xi, Q.; Wang, Z.; Zaromytidou, A.-I.; Zhang, X.H.-F.; Chow-Tsang, L.-F.; Liu, J.X.; Kim, H.; Barlas, A.; Manova-Todorova, K.; Kaartinen, V. A Poised Chromatin Platform for TGF-β Access to Master Regulators. Cell 2011, 147, 1511–1524. [Google Scholar] [CrossRef] [Green Version]
- Theodoulou, N.H.; Tomkinson, N.C.O.; Prinjha, R.K.; Humphreys, P.G. Clinical Progress and Pharmacology of Small Molecule Bromodomain Inhibitors. Curr. Opin. Chem. Biol. 2016, 33, 58–66. [Google Scholar] [CrossRef]
- Cochran, A.G.; Conery, A.R.; Sims, R.J. Bromodomains: A New Target Class for Drug Development. Nat. Rev. Drug Discov. 2019, 18, 609–628. [Google Scholar] [CrossRef] [PubMed]
- Palmer, W.S.; Poncet-montange, G.; Liu, G.; Petrocchi, A.; Reyna, N.; Subramanian, G.; Thero, J.; Yau, A.; Kost-alimova, M.; Bardenhagen, J.P.; et al. Structure-Guided Design of IACS-9571, a Selective High-A Ffi Nity Dual TRIM24-BRPF1 Bromodomain Inhibitor. J. Med. Chem. 2015, 59, 1440–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, J.; Fedorov, O.; Tallant, C.; Monteiro, O.; Meier, J.; Gamble, V.; Savitsky, P.; Nunez-alonso, G.A.; Haendler, B.; Rogers, C.; et al. Discovery of a Chemical Tool Inhibitor Targeting the Bromodomains of TRIM24 and BRPF. J. Med. Chem. 2016, 59, 1642–1647. [Google Scholar] [CrossRef] [Green Version]
- Palmer, W.S. Development of Small Molecule Inhibitors of BRPF1 and TRIM24 Bromodomains. Drug Discov. Today Technol. 2016, 19, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Lin, X.; Chiu, W.T.; Chen, Y.H.; Yu, G.; Liu, M.; Feng, X.H.; Sawaya, R.; Medema, R.H.; Hung, M.C.; et al. Sustained Activation of SMAD3/SMAD4 by FOXM1 Promotes TGF-β-Dependent Cancer Metastasis. J. Clin. Investig. 2014, 124, 564–579. [Google Scholar] [CrossRef]
- Wang, L.; Yang, H.; Lei, Z.; Zhao, J.; Chen, Y.; Chen, P.; Li, C.; Zeng, Y.; Liu, Z.; Liu, X.; et al. Repression of TIF1γ by SOX2 Promotes TGF-β-Induced Epithelial-Mesenchymal Transition in Non-Small-Cell Lung Cancer. Oncogene 2016, 35, 867–877. [Google Scholar] [CrossRef]
- Jingushi, K.; Ueda, Y.; Kitae, K.; Hase, H.; Egawa, H.; Ohshio, I.; Kawakami, R.; Kashiwagi, Y.; Tsukada, Y.; Kobayashi, T.; et al. MiR-629 Targets TRIM33 to Promote TGFβ/Smad Signaling and Metastatic Phenotypes in CcRCC. Mol. Cancer Res. 2015, 13, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Lingadahalli, S.; Narwade, N.; Lei, K.M.K.; Liu, S.; Zhao, Z.; Zheng, Y.; Lu, Q.; Tang, A.H.N.; Poon, T.C.W.; et al. TRIM33 Drives Prostate Tumor Growth by Stabilizing Androgen Receptor from Skp2-mediated Degradation. EMBO Rep. 2022, 23, e53468. [Google Scholar] [CrossRef]
- Stark, C.; Breitkreutz, B.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. BioGRID: A General Repository for Interaction Datasets. Nucleic Acids Res. 2006, 34, 535–539. [Google Scholar] [CrossRef] [Green Version]
- Bortolato, A.; Tehan, B.G.; Smith, R.T.; Mason, J.S. Methodologies for the Examination of Water in GPCRs. Methods Mol. Biol. 2018, 1705, 207–232. [Google Scholar] [CrossRef]
- Mason, J.S.; Bortolato, A.; Weiss, D.R.; Deflorian, F.; Tehan, B.; Marshall, F.H. High End GPCR Design: Crafted Ligand Design and Druggability Analysis Using Protein Structure, Lipophilic Hotspots and Explicit Water Networks. Silico Pharmacol. 2013, 1, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Toye, H.; Zhan, P.; Gopalakrishnan, G.; Kartadikaria, A.R.; Huang, H.; Knio, O.; Hoteit, I. HTRF: A Technology Tailored for Drug Discovery –A Review of Theoreti- Cal Aspects and Recent Applications. Curr. Chem. Genom. 2017, 67, 915–933. [Google Scholar] [CrossRef]
- Newton, P.; Harrison, P.; Clulow, S. A Novel Method for Determination of the Affinity of Protein: Protein Interactions in Homogeneous Assays. J. Biomol. Screen. 2008, 13, 674–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pontén, F.; Jirstrom, K.; Uhlen, M. The Human Protein Atlas—A Tool for Pathology. J. Pathol. 2008, 216, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Scorah, A.; See, L.; Bluck, J.; Reynolds, J.; McCoull, W.; Conway, S.J. Chemical Tools to Probe the Function of TRIM33. Ph.D. Thesis, University of Oxford, Oxford, UK, 2019. [Google Scholar]
- Milletti, F.; Storchi, L.; Sforna, G.; Cruciani, G. New and Original PKa Prediction Method Using Grid Molecular Interaction Fields. J. Chem. Inf. Model. 2007, 47, 2172–2181. [Google Scholar] [CrossRef]
- Tsai, W.-W.; Wang, Z.; Yiu, T.T.; Akdemir, K.C.; Xia, W.; Winter, S.; Tsai, C.-Y.; Shi, X.; Schwarzer, D.; Plunkett, W. TRIM24 Links a Non-Canonical Histone Signature to Breast Cancer. Nature 2010, 468, 927–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baroni, M.; Cruciani, G.; Sciabola, S.; Perruccio, F.; Mason, J.S. A Common Reference Framework for Analyzing/Comparing Proteins and Ligands. Fingerprints for Ligands And Proteins (FLAP): Theory and Application. J. Chem. Inf. Model. 2007, 47, 279–294. [Google Scholar] [CrossRef]
- Cross, S.; Baroni, M.; Carosati, E.; Benedetti, P.; Clementi, S. FLAP: GRID Molecular Interaction Fields in Virtual Screening. Validation Using the DUD Data Set. J. Chem. Inf. Model. 2010, 50, 1442–1450. [Google Scholar] [CrossRef]
- Zeng, L.; Yap, K.L.; Ivanov, A.V.; Wang, X.; Mujtaba, S.; Plotnikova, O.; Rauscher III, F.J.; Zhou, M.-M. Structural Insights into Human KAP1 PHD Finger–Bromodomain and Its Role in Gene Silencing. Nat. Struct. Mol. Biol. 2008, 15, 626–633. [Google Scholar] [CrossRef] [Green Version]
- Burdick, D.J.; Liang, J. Preparation of Pyrimidines as Aurora Kinase. Inhibitors. Patent WO2007120339, 25 October 2007. [Google Scholar]
- Steeneck, C.; Gege, C.; Richter, F.; Hochguertel, M.; Feuerstein, T.; Bluhm, H. Preparation of Amide Containing Heterobicyclic Metalloprotease. Inhibitors. Patent WO2006128184, 8 March 2007. [Google Scholar]
- Stazi, G.; Battistelli, C.; Piano, V.; Mazzone, R.; Marrocco, B.; Marchese, S.; Louie, S.M.; Zwergel, C.; Antonini, L.; Patsilinakos, A.; et al. Development of Alkyl Glycerone Phosphate Synthase Inhibitors: Structure-Activity Relationship and Effects on Ether Lipids and Epithelial-Mesenchymal Transition in Cancer Cells. Eur. J. Med. Chem. 2019, 163, 722–735. [Google Scholar] [CrossRef]
- Kapust, R.B.; Tözsér, J.; Fox, J.D.; Anderson, D.E.; Cherry, S.; Copeland, T.D.; Waugh, D.S. Tobacco Etch Virus Protease: Mechanism of Autolysis and Rational Design of Stable Mutants with Wild-Type Catalytic Proficiency. Protein Eng. 2001, 14, 993–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benvenuti, M.; Mangani, S. Crystallization of Soluble Proteins in Vapor Diffusion for X-Ray Crystallography. Nat. Protoc. 2007, 2, 1633–1651. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. Xds. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, P.R. An Introduction to Data Reduction: Space-Group Determination, Scaling and Intensity Statistics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 282–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, P. Scaling and Assessment of Data Quality. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 72–82. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 Suite and Current Developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Vagin, A.; Teplyakov, A. Molecular Replacement with MOLREP. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 22–25. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the Refinement of Macromolecular Crystal Structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and Development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. IUCr. PROCHECK: A program to check the stereochemical quality of protein structures. J. App. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Potterton, E.; McNicholas, S.; Krissinel, E.; Cowtan, K.; Noble, M. The CCP4 Molecular-Graphics Project. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 1955–1957. [Google Scholar] [CrossRef] [PubMed]














Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palomba, T.; Tassone, G.; Vacca, C.; Bartalucci, M.; Valeri, A.; Pozzi, C.; Cross, S.; Siragusa, L.; Desantis, J. Exploiting ELIOT for Scaffold-Repurposing Opportunities: TRIM33 a Possible Novel E3 Ligase to Expand the Toolbox for PROTAC Design. Int. J. Mol. Sci. 2022, 23, 14218. https://doi.org/10.3390/ijms232214218
Palomba T, Tassone G, Vacca C, Bartalucci M, Valeri A, Pozzi C, Cross S, Siragusa L, Desantis J. Exploiting ELIOT for Scaffold-Repurposing Opportunities: TRIM33 a Possible Novel E3 Ligase to Expand the Toolbox for PROTAC Design. International Journal of Molecular Sciences. 2022; 23(22):14218. https://doi.org/10.3390/ijms232214218
Chicago/Turabian StylePalomba, Tommaso, Giusy Tassone, Carmine Vacca, Matteo Bartalucci, Aurora Valeri, Cecilia Pozzi, Simon Cross, Lydia Siragusa, and Jenny Desantis. 2022. "Exploiting ELIOT for Scaffold-Repurposing Opportunities: TRIM33 a Possible Novel E3 Ligase to Expand the Toolbox for PROTAC Design" International Journal of Molecular Sciences 23, no. 22: 14218. https://doi.org/10.3390/ijms232214218