

Identification of Diosmin and Flavin Adenine Dinucleotide as Repurposing Treatments for Monkeypox Virus: A Computational Study
 , , and
, , and 
Abstract
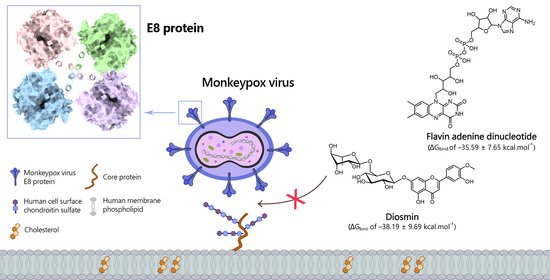
:
1. Introduction
2. Results
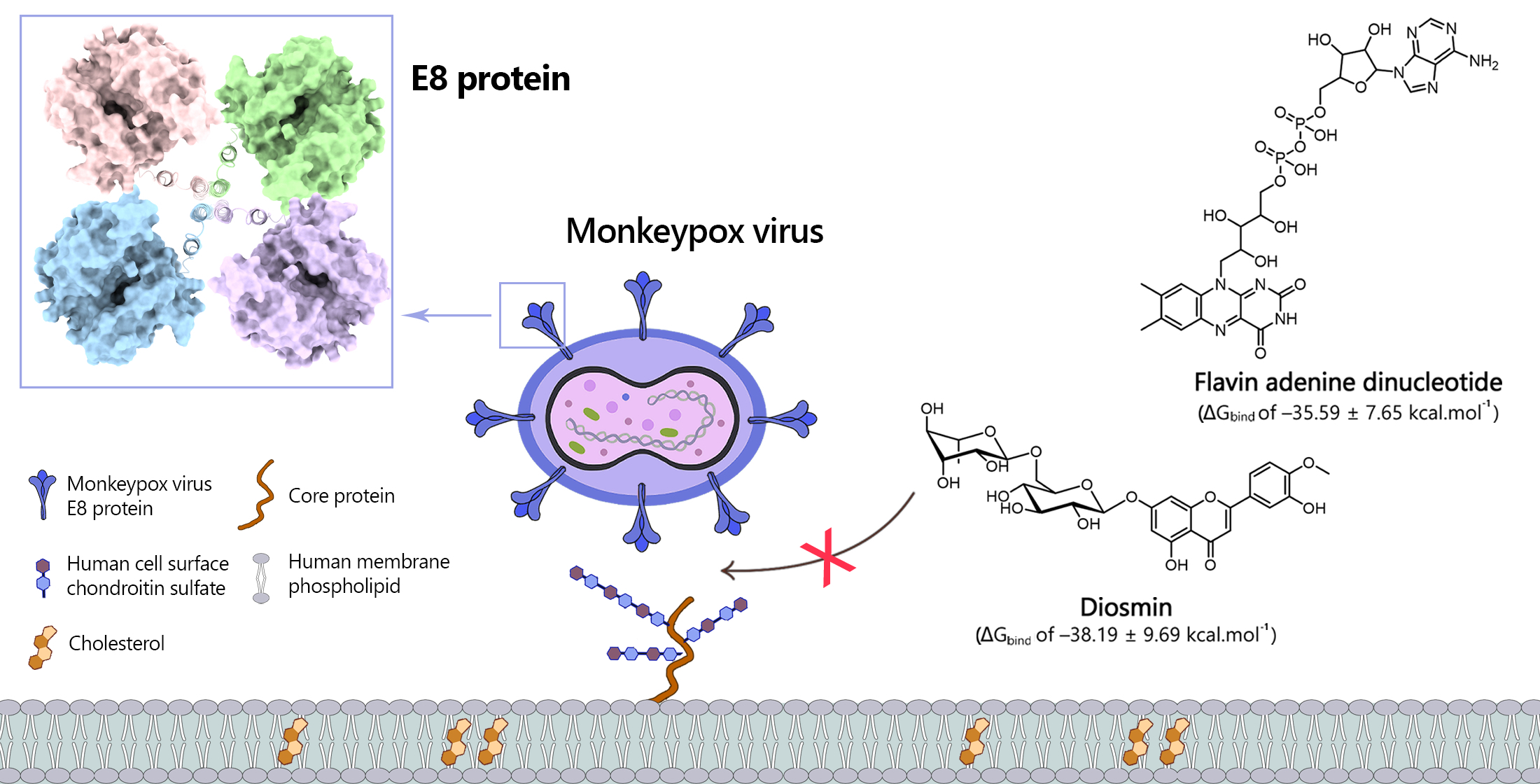
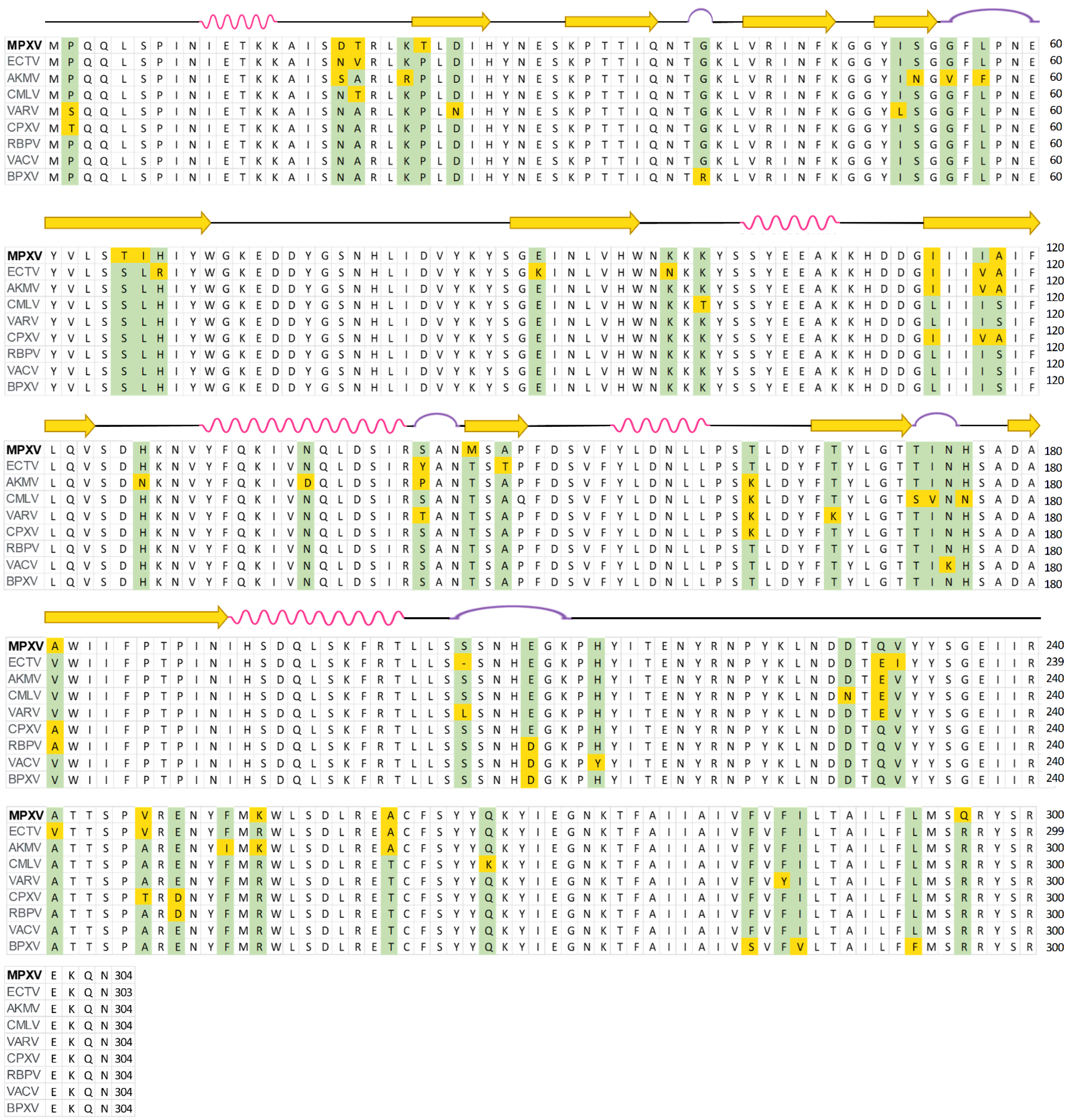
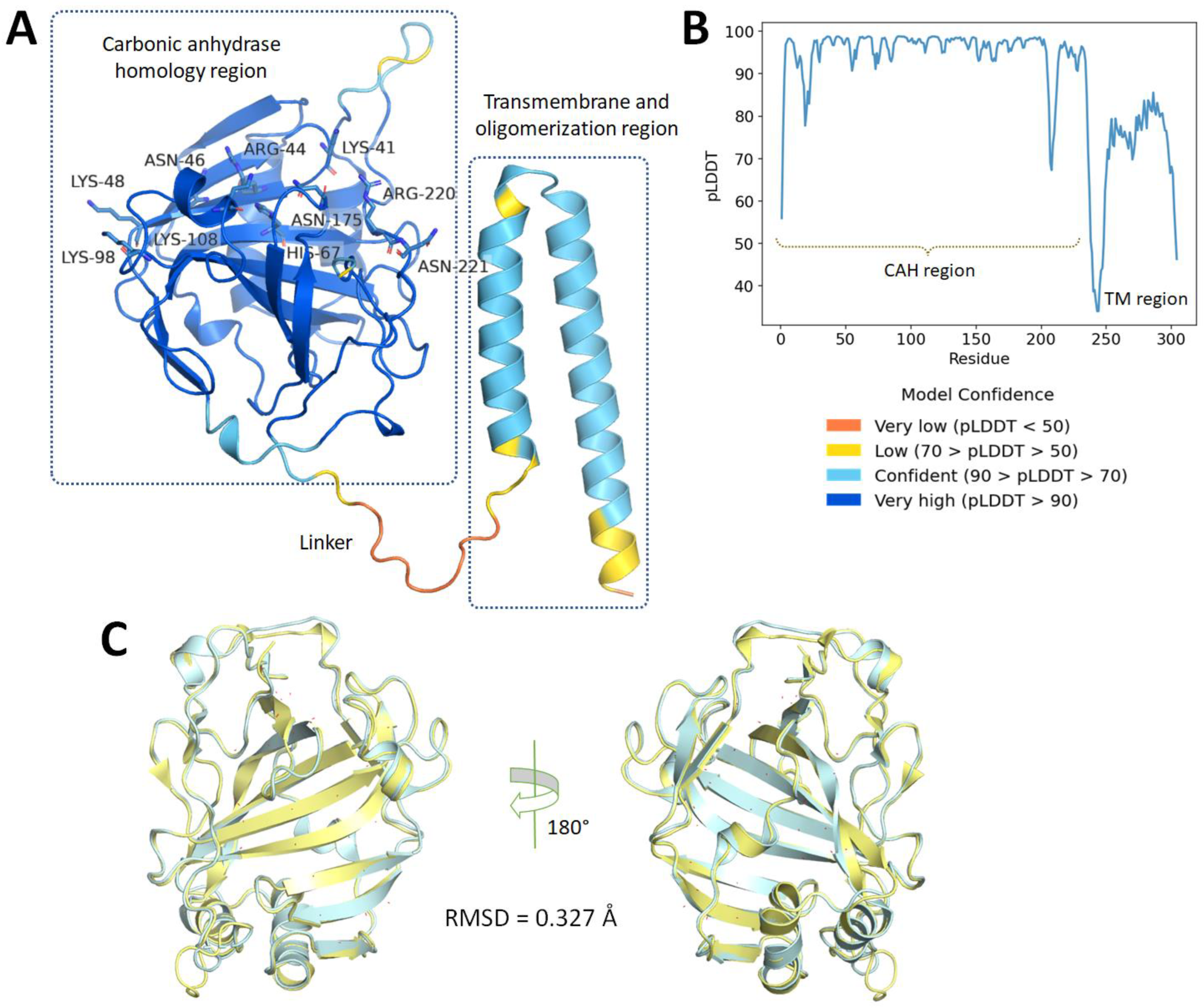
2.1. Protein Homology Modeling Using Deep Learning Approach
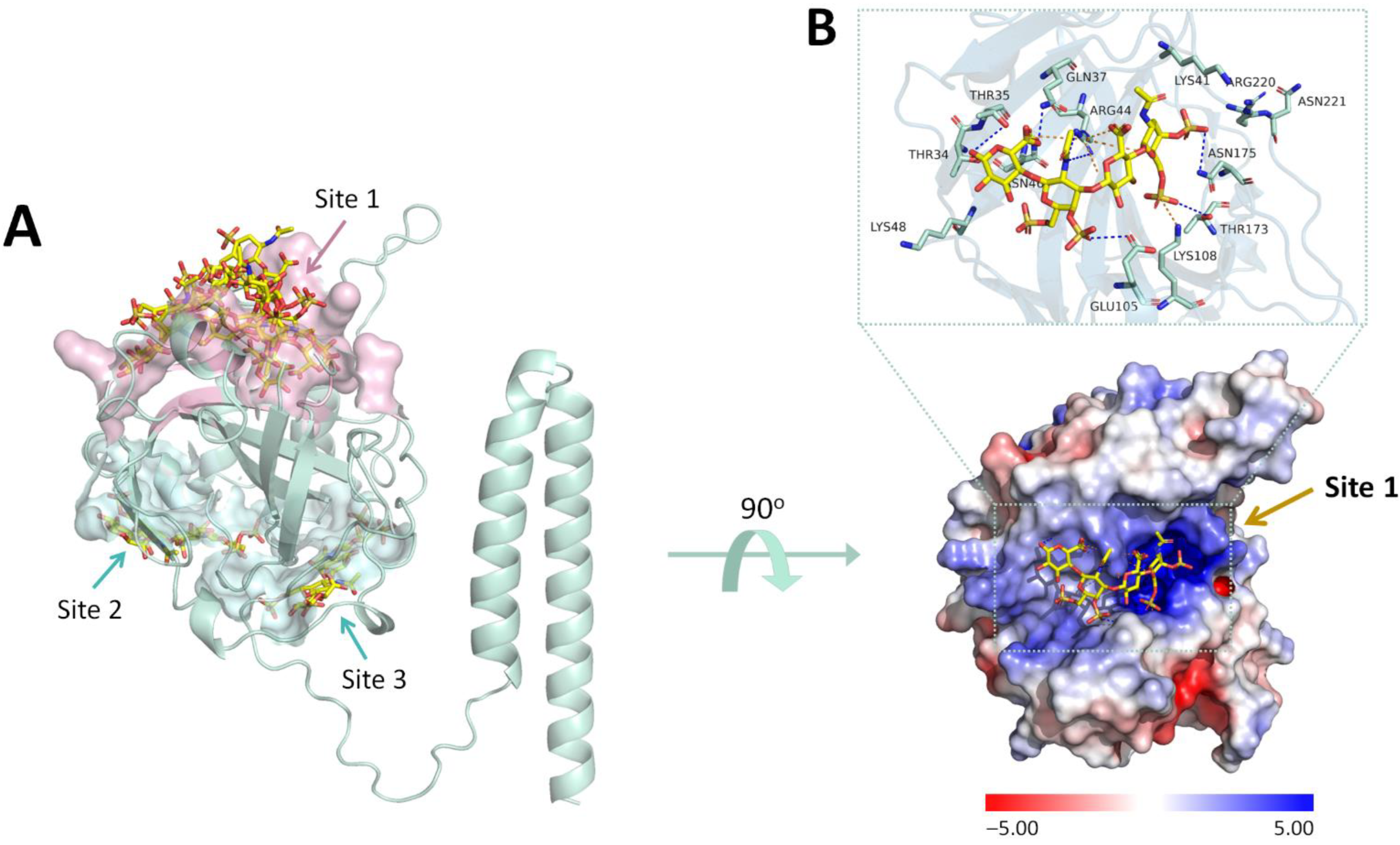
2.2. Identification of Chondroitin Sulfate-E Binding Pocket in the MPXV E8 Protein Using Blind Docking Approach
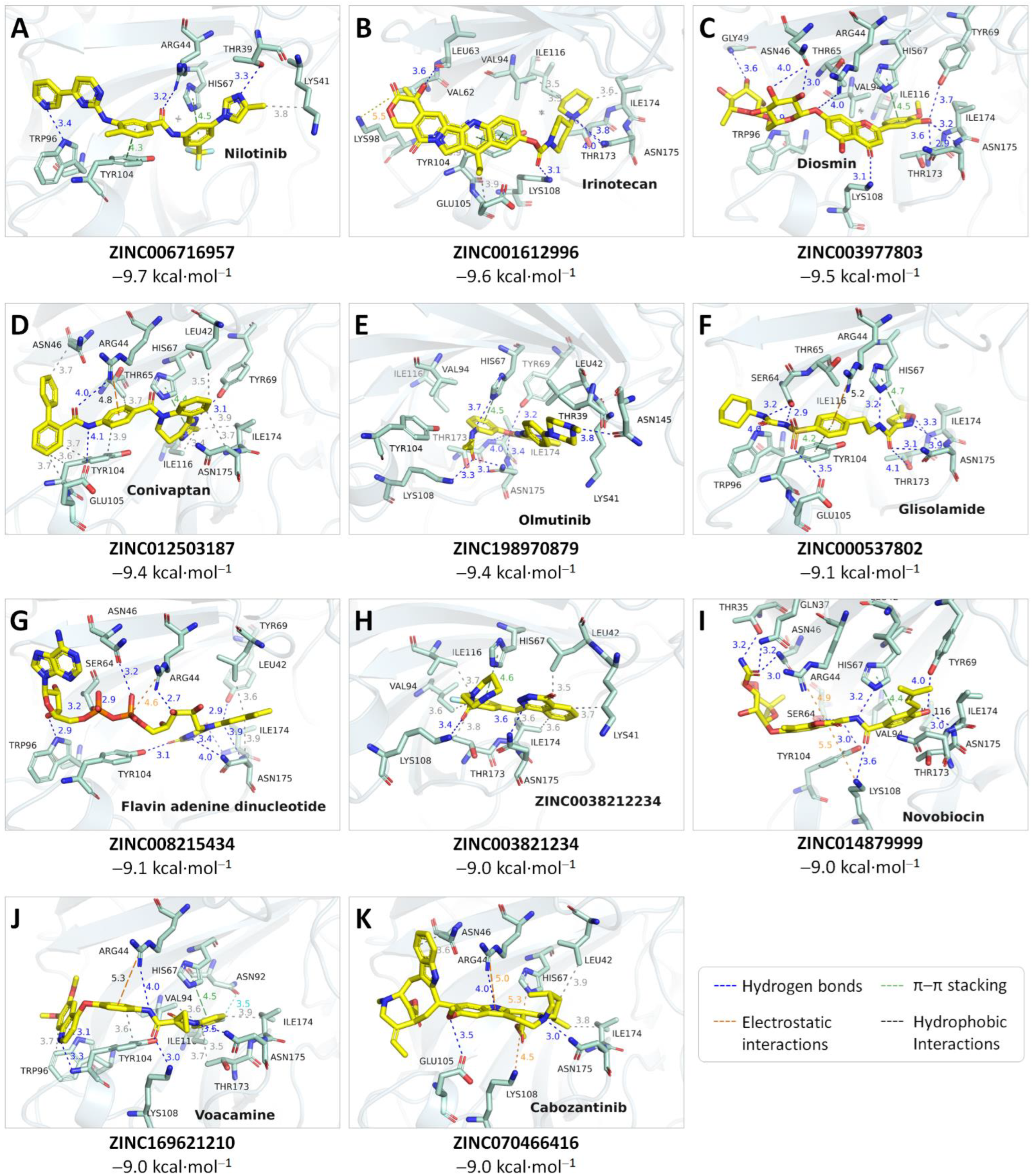
2.3. Virtual Screening Using Focused Docking Approach
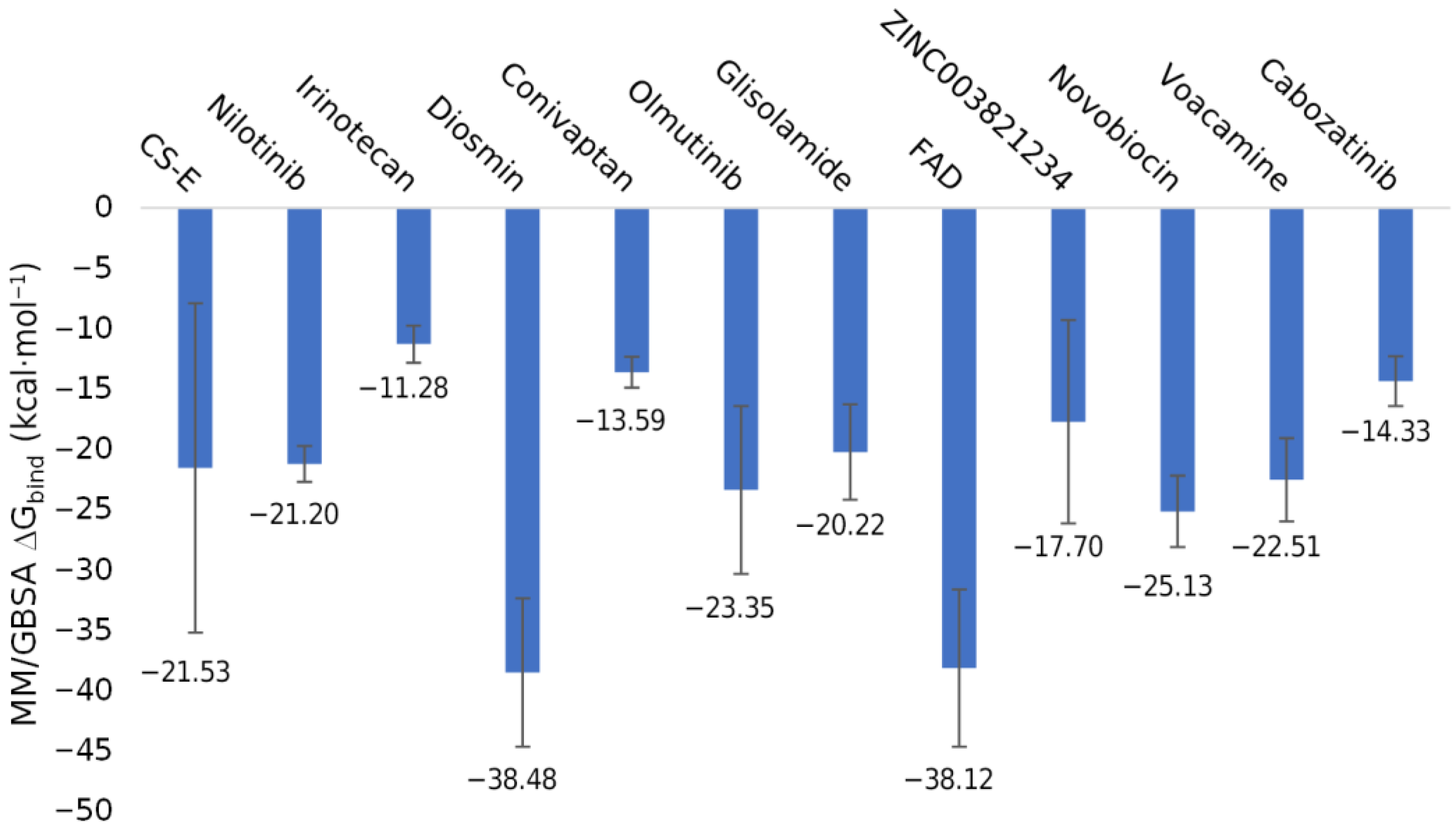
2.4. Molecular Dynamics Simulations and Binding Free Energy Calculation
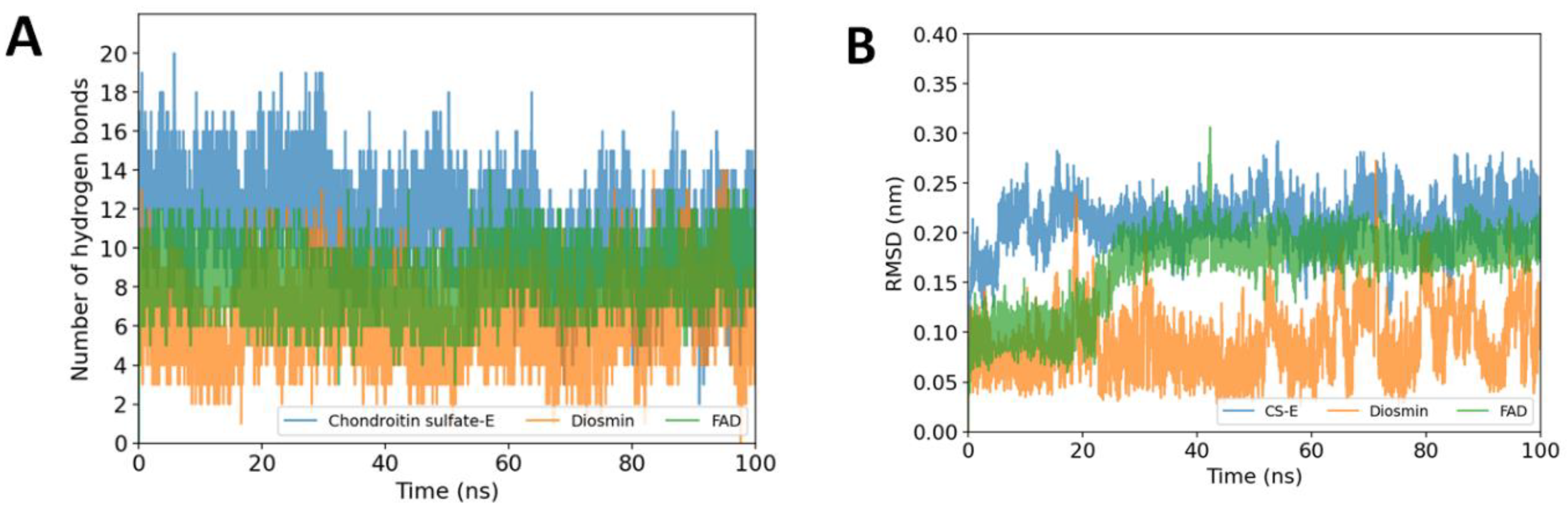
2.4.1. Stability of the Protein-Ligand Complexes
2.4.2. Ligand Stability and Their Binding Free Energy with the MPXV E8 Protein
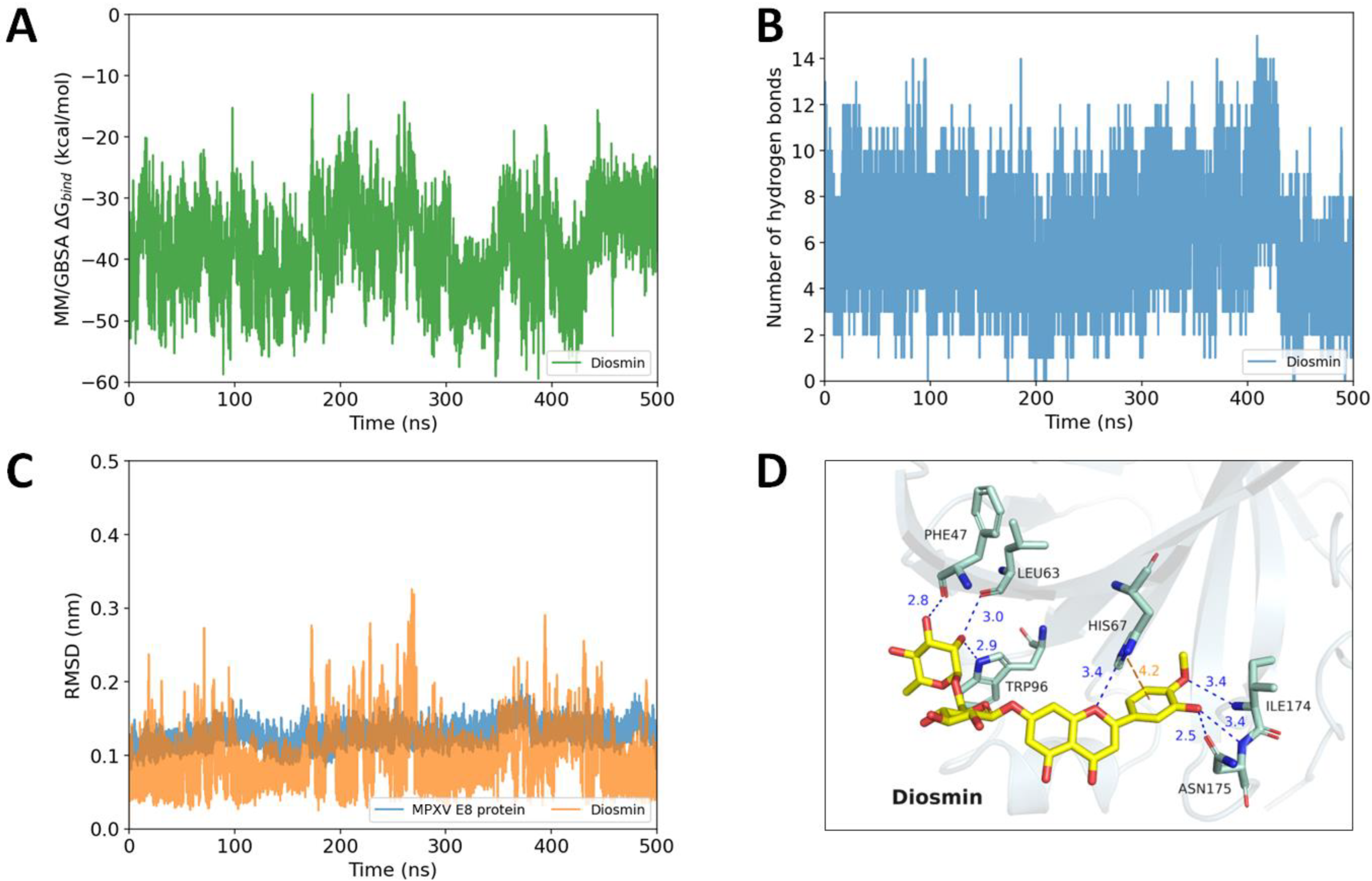
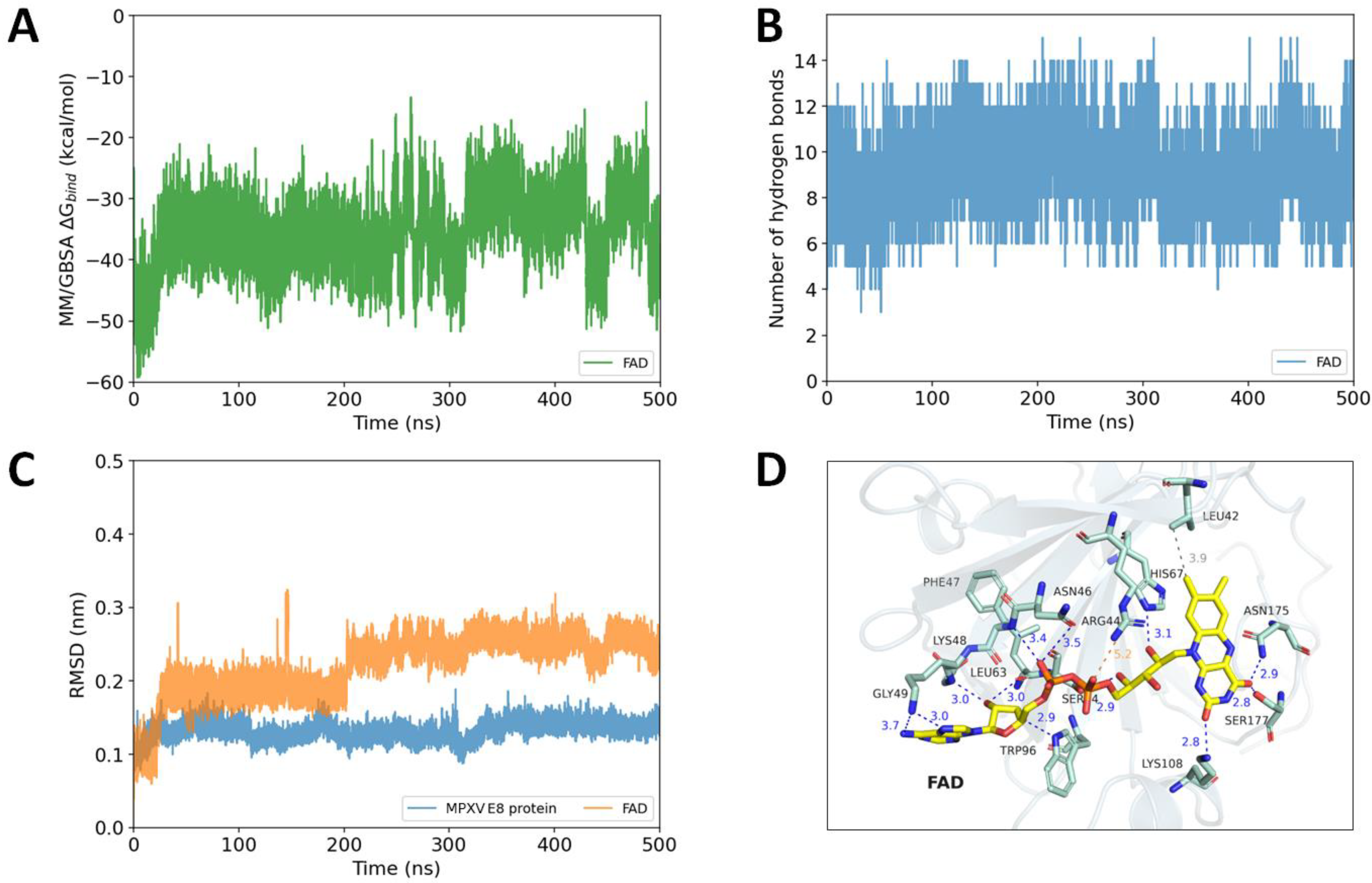
2.5. Diosmin and FAD as Potential MPXV E8 Protein Inhibitors Proposed by Long Molecular Dynamics Simulations
3. Discussion
4. Materials and Methods
4.1. MPXV E8 Protein Homology Modeling
4.2. Preparation of the Target Protein
4.3. Preparation of Small-Molecule Ligands
4.4. Virtual Screening Based on Blind Docking and Focused Docking
4.5. Molecular Dynamics Simulations
4.6. End-State Binding Free Energy Calculation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CAH | Carbonic anhydrase homology |
| CS-E | Chondroitin sulfate-E |
| FAD | Flavin adenine dinucleotide |
| GAG | Glycosaminoglycan |
| MPXV | Monkeypox virus |
| MSA | Multiple sequence alignment |
| Rg | Radius of gyration |
| RMSD | Root mean square deviation |
| SASA | Solvent accessible surface area |
| MDs | Molecular dynamics simulation |
| VACV | Vaccinia virus |
| WHO | World Health Organization |
| MM/GBSA | Molecular mechanics/Generalized Born Surface Area |
| FDA | Food and Drug Administration |
References
- Nuzzo, J.B.; Borio, L.L.; Gostin, L.O. The WHO Declaration of Monkeypox as a Global Public Health Emergency. JAMA 2022, 328, 615–617. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Multi-Country Outbreak of Monkeypox, External Situation Report #5–7 September 2022. Available online: https://www.who.int/publications/m/item/multi-country-outbreak-of-monkeypox-external-situation-report-5–7-September-2022 (accessed on 8 September 2022).
- World Health Organization. Monkeypox. Available online: https://www.who.int/news-room/fact-sheets/detail/monkeypox (accessed on 8 September 2022).
- Titanji, B.K.; Tegomoh, B.; Nematollahi, S.; Konomos, M.; Kulkarni, P.A. Monkeypox: A Contemporary Review for Healthcare Professionals. Open Forum Infect. Dis. 2022, 9, ofac310. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.L.; Vanderplasschen, A.; Law, M. The formation and function of extracellular enveloped vaccinia virus. J. Gen. Virol. 2002, 83, 2915–2931. [Google Scholar] [CrossRef] [PubMed]
- Gong, Q.; Wang, C.; Chuai, X.; Chiu, S. Monkeypox virus: A re-emergent threat to humans. Virol. Sin. 2022, 37, 477–482. [Google Scholar] [CrossRef]
- Senkevich, T.G.; Ojeda, S.; Townsley, A.; Nelson, G.E.; Moss, B. Poxvirus multiprotein entry–fusion complex. Proc. Natl. Acad. Sci. USA 2005, 102, 18572–18577. [Google Scholar] [CrossRef]
- Carter, G.C.; Law, M.; Hollinshead, M.; Smith, G.L. Entry of the vaccinia virus intracellular mature virion and its interactions with glycosaminoglycans. J. Gen. Virol. 2005, 86, 1279–1290. [Google Scholar] [CrossRef]
- Lin, C.L.; Chung, C.S.; Heine, H.G.; Chang, W. Vaccinia virus envelope H3L protein binds to cell surface heparan sulfate and is important for intracellular mature virion morphogenesis and virus infection in vitro and in vivo. J. Virol. 2000, 74, 3353–3365. [Google Scholar] [CrossRef]
- Chung, C.S.; Hsiao, J.C.; Chang, Y.S.; Chang, W. A27L protein mediates vaccinia virus interaction with cell surface heparan sulfate. J. Virol. 1998, 72, 1577–1585. [Google Scholar] [CrossRef]
- Hsiao, J.C.; Chung, C.S.; Chang, W. Vaccinia virus envelope D8L protein binds to cell surface chondroitin sulfate and mediates the adsorption of intracellular mature virions to cells. J. Virol. 1999, 73, 8750–8761. [Google Scholar] [CrossRef]
- Bartlett, A.H.; Park, P.W. Proteoglycans in host-pathogen interactions: Molecular mechanisms and therapeutic implications. Expert Rev. Mol. Med. 2010, 12, e5. [Google Scholar] [CrossRef] [Green Version]
- Matho, M.H.; Maybeno, M.; Benhnia, M.R.; Becker, D.; Meng, X.; Xiang, Y.; Crotty, S.; Peters, B.; Zajonc, D.M. Structural and biochemical characterization of the vaccinia virus envelope protein D8 and its recognition by the antibody LA5. J. Virol. 2012, 86, 8050–8058. [Google Scholar] [CrossRef] [PubMed]
- Matho, M.H.; de Val, N.; Miller, G.M.; Brown, J.; Schlossman, A.; Meng, X.; Crotty, S.; Peters, B.; Xiang, Y.; Hsieh-Wilson, L.C.; et al. Murine Anti-vaccinia Virus D8 Antibodies Target Different Epitopes and Differ in Their Ability to Block D8 Binding to CS-E. PLoS Pathog. 2014, 10, e1004495. [Google Scholar] [CrossRef] [PubMed]
- FDA. Smallpox Preparedness and Response Updates from FDA. Available online: https://www.fda.gov/emergency-preparedness-and-response/mcm-issues/smallpox-preparedness-and-response-updates-fda (accessed on 29 July 2022).
- Chang, C.C.; Hsu, H.J.; Wu, T.Y.; Liou, J.W. Computer-aided discovery, design, and investigation of COVID-19 therapeutics. Tzu Chi Med. J. 2022, 34, 276–286. [Google Scholar] [CrossRef]
- Kamal, L.; Ramadan, A.; Farraj, S.; Bahig, L.; Ezzat, S. The pill of recovery; Molnupiravir for treatment of COVID-19 patients; a systematic review. Saudi Pharm. J. 2022, 30, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.A.; Hassan, M.I.; Islam, A.; Ahmad, F. A review of methods available to estimate solvent-accessible surface areas of soluble proteins in the folded and unfolded states. Curr. Protein Pept. Sci. 2014, 15, 456–476. [Google Scholar] [CrossRef] [PubMed]
- Sneha, P.; Doss, C.G. Molecular Dynamics: New Frontier in Personalized Medicine. Adv. Protein Chem. Struct. Biol. 2016, 102, 181–224. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Cazaubon, M.; Benigni, J.P.; Steinbruch, M.; Jabbour, V.; Gouhier-Kodas, C. Is There a Difference in the Clinical Efficacy of Diosmin and Micronized Purified Flavonoid Fraction for the Treatment of Chronic Venous Disorders? Review of Available Evidence. Vasc. Health Risk Manag. 2021, 17, 591–600. [Google Scholar] [CrossRef]
- Huwait, E.; Mobashir, M. Potential and Therapeutic Roles of Diosmin in Human Diseases. Biomedicines 2022, 10, 1076. [Google Scholar] [CrossRef]
- Alzaabi, M.M.; Hamdy, R.; Ashmawy, N.S.; Hamoda, A.M.; Alkhayat, F.; Khademi, N.N.; Al Joud, S.M.A.; El-Keblawy, A.A.; Soliman, S.S.M. Flavonoids are promising safe therapy against COVID-19. Phytochem. Rev. 2022, 21, 291–312. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Sharma, P.P.; Upadhyay, C.; Kempaiah, P.; Rathi, B.; Poonam. Multi-targeting approach for nsp3, nsp9, nsp12 and nsp15 proteins of SARS-CoV-2 by Diosmin as illustrated by molecular docking and molecular dynamics simulation methodologies. Methods 2021, 195, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Yokochi, S.; Ishiwata, Y.; Saito, H.; Ebinuma, H.; Tsuchiya, M.; Ishii, H. Stimulation of antiviral activities of interferon by a liver extract preparation. Arzneimittelforschung 1997, 47, 968–974. [Google Scholar] [PubMed]
- Wei, T.-Z.; Wang, H.; Wu, X.-Q.; Lu, Y.; Guan, S.-H.; Dong, F.-Q.; Dong, C.-L.; Zhu, G.-L.; Bao, Y.-Z.; Zhang, J.; et al. In Silico Screening of Potential Spike Glycoprotein Inhibitors of SARS-CoV-2 with Drug Repurposing Strategy. Chin. J. Integr. Med. 2020, 26, 663–669. [Google Scholar] [CrossRef]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef]
- Anwaar, M.U.; Adnan, F.; Abro, A.; Khan, R.A.; Rehman, A.U.; Osama, M.; Rainville, C.; Kumar, S.; Sterner, D.E.; Javed, S.; et al. Combined deep learning and molecular docking simulations approach identifies potentially effective FDA approved drugs for repurposing against SARS-CoV-2. Comput. Biol. Med. 2022, 141, 105049. [Google Scholar] [CrossRef]
- Huang, F.; Zhang, C.; Liu, Q.; Zhao, Y.; Zhang, Y.; Qin, Y.; Li, X.; Li, C.; Zhou, C.; Jin, N.; et al. Identification of amitriptyline HCl, flavin adenine dinucleotide, azacitidine and calcitriol as repurposing drugs for influenza A H5N1 virus-induced lung injury. PLoS Pathog. 2020, 16, e1008341. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Irwin, J.J.; Tang, K.G.; Young, J.; Dandarchuluun, C.; Wong, B.R.; Khurelbaatar, M.; Moroz, Y.S.; Mayfield, J.; Sayle, R.A. ZINC20—A free ultralarge-scale chemical database for ligand discovery. J. Chem. Inf. Model. 2020, 60, 6065–6073. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Kienetz, M.; Cherney, M.M.; James, M.N.G.; Brömme, D. The Crystal and Molecular Structures of a Cathepsin K:Chondroitin Sulfate Complex. J. Mol. Biol. 2008, 383, 78–91. [Google Scholar] [CrossRef] [PubMed]
- PyMOL, Version 2.5.2; Schrödinger LLC: New York, NY, USA, 2020.
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the scope of the protein–ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 2021, 49, W530–W534. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef]
- Lemkul, J.A. From Proteins to Perturbed Hamiltonians: A Suite of Tutorials for the GROMACS-2018 Molecular Simulation Package [Article v1.0]. LiveCoMS 2018, 1, 5068. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. Model. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Tran, Q.-H.; Nguyen, Q.-T.; Vo, N.-Q.-H.; Mai, T.T.; Tran, T.-T.-N.; Tran, T.-D.; Le, M.-T.; Trinh, D.-T.T.; Thai, K.-M. Structure-based 3D-Pharmacophore modeling to discover novel interleukin 6 inhibitors: An in silico screening, molecular dynamics simulations and binding free energy calculations. PLoS ONE 2022, 17, e0266632. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A new tool to perform end-state free energy calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef] [PubMed]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins Struct. Funct. Genet. 2004, 55, 383–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ZINC ID | INN | MPXV E8 Protein Backbone | Ligand Heavy Atoms | ||
|---|---|---|---|---|---|
| RMSD (nm) | SASA (nm2) | Rg (nm) | RMSD (nm) | ||
| Chondroitin sulfate-E | 0.132 ± 0.015 | 124.882 ± 1.071 | 1.737 ± 0.006 | 0.211 ± 0.025 | |
| ZINC006716957 | Nilotinib | 0.117 ± 0.011 | 124.921 ± 0.991 | 1.743 ± 0.006 | 0.190 ± 0.034 |
| ZINC001612996 | Irinotecan | 0.149 ± 0.036 | 125.139 ± 1.295 | 1.743 ± 0.012 | 0.095 ± 0.029 |
| ZINC003977803 | Diosmin | 0.119 ± 0.012 | 124.627 ± 1.043 | 1.731 ± 0.005 | 0.090 ± 0.032 |
| ZINC012503187 | Conivaptan | 0.126 ± 0.014 | 125.343 ± 0.967 | 1.742 ± 0.007 | 0.194 ± 0.045 |
| ZINC198970879 | Olmutinib | 0.125 ± 0.015 | 125.203 ± 1.221 | 1.739 ± 0.007 | 0.290 ± 0.086 |
| ZINC000537802 | Glisolamide | 0.140 ± 0.017 | 124.362 ± 0.997 | 1.737 ± 0.006 | 0.193 ± 0.034 |
| ZINC008215434 | FAD | 0.131 ± 0.014 | 124.941 ± 1.020 | 1.743 ± 0.007 | 0.166 ± 0.039 |
| ZINC003821234 | Unnamed | 0.146 ± 0.028 | 124.595 ± 1.124 | 1.742 ± 0.006 | 0.196 ± 0.056 |
| ZINC014879999 | Novobiocin | 0.105 ± 0.011 | 124.598 ± 0.981 | 1.741 ± 0.006 | 0.212 ± 0.081 |
| ZINC070466416 | Cabozantinib | 0.126 ± 0.016 | 125.367 ± 0.992 | 1.743 ± 0.007 | 0.203 ± 0.045 |
| ZINC169621210 | Voacamine | 0.167 ± 0.039 | 124.785 ± 1.036 | 1.740 ± 0.006 | 0.076 ± 0.020 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lam, T.-P.; Tran, V.-H.; Mai, T.T.; Lai, N.V.-T.; Dang, B.-T.N.; Le, M.-T.; Tran, T.-D.; Trinh, D.-T.T.; Thai, K.-M. Identification of Diosmin and Flavin Adenine Dinucleotide as Repurposing Treatments for Monkeypox Virus: A Computational Study. Int. J. Mol. Sci. 2022, 23, 11570. https://doi.org/10.3390/ijms231911570
Lam T-P, Tran V-H, Mai TT, Lai NV-T, Dang B-TN, Le M-T, Tran T-D, Trinh D-TT, Thai K-M. Identification of Diosmin and Flavin Adenine Dinucleotide as Repurposing Treatments for Monkeypox Virus: A Computational Study. International Journal of Molecular Sciences. 2022; 23(19):11570. https://doi.org/10.3390/ijms231911570
Chicago/Turabian StyleLam, Thua-Phong, Viet-Hung Tran, Tan Thanh Mai, Nghia Vo-Trong Lai, Bao-Tran Ngoc Dang, Minh-Tri Le, Thanh-Dao Tran, Dieu-Thuong Thi Trinh, and Khac-Minh Thai. 2022. "Identification of Diosmin and Flavin Adenine Dinucleotide as Repurposing Treatments for Monkeypox Virus: A Computational Study" International Journal of Molecular Sciences 23, no. 19: 11570. https://doi.org/10.3390/ijms231911570