

Non-Muscle MLCK Contributes to Endothelial Cell Hyper-Proliferation through the ERK Pathway as a Mechanism for Vascular Remodeling in Pulmonary Hypertension

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Upregulated Protein Expression of nmMLCK and ERK in BMPR2 Silenced Human Pulmonary Artery Endothelial Cells
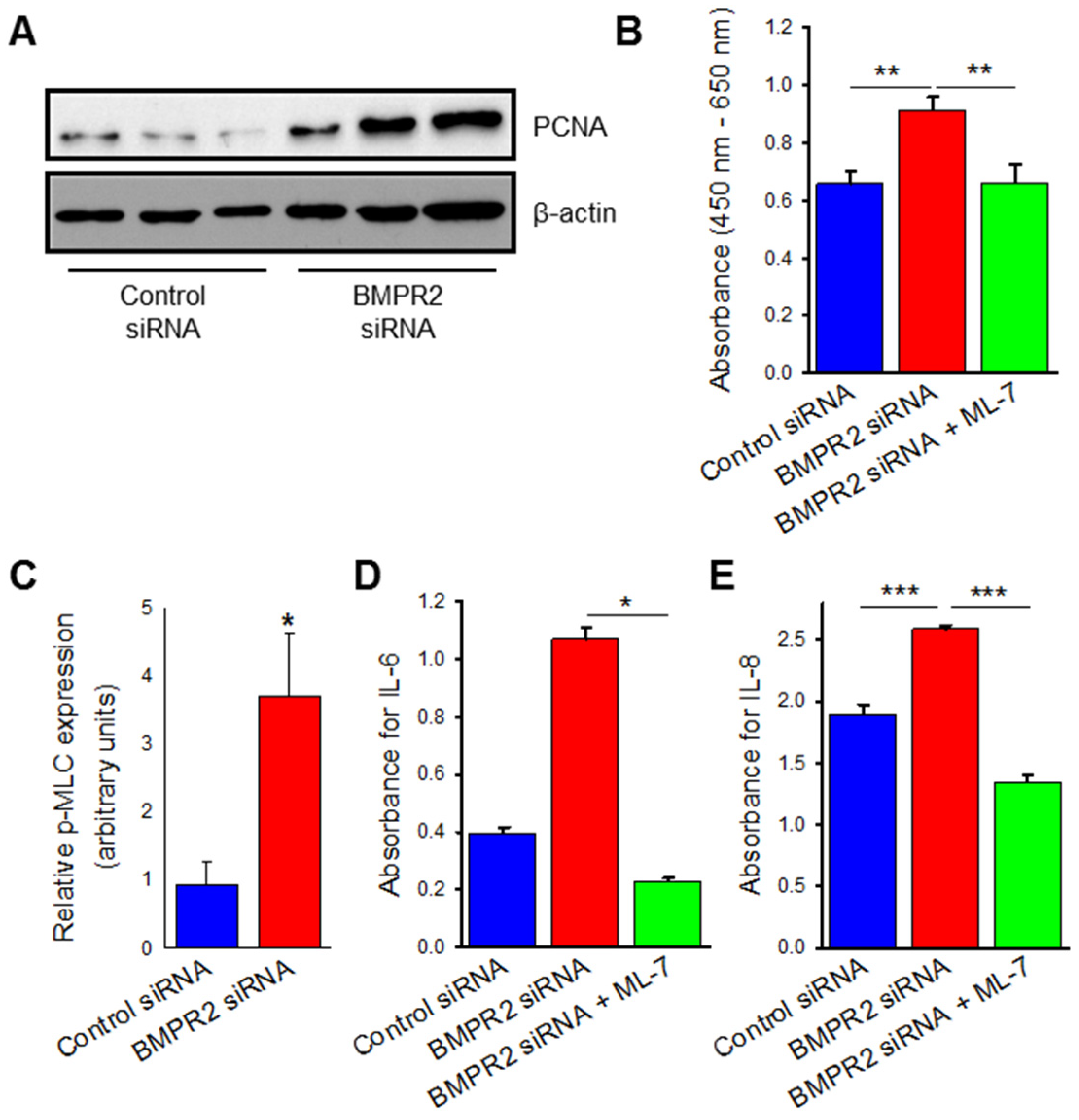
2.2. BMPR2 Silencing Induces Increased Endothelial Cell Viability, Proliferation, and Cytokine Release Which Are Attenuated by MLCK Inhibition
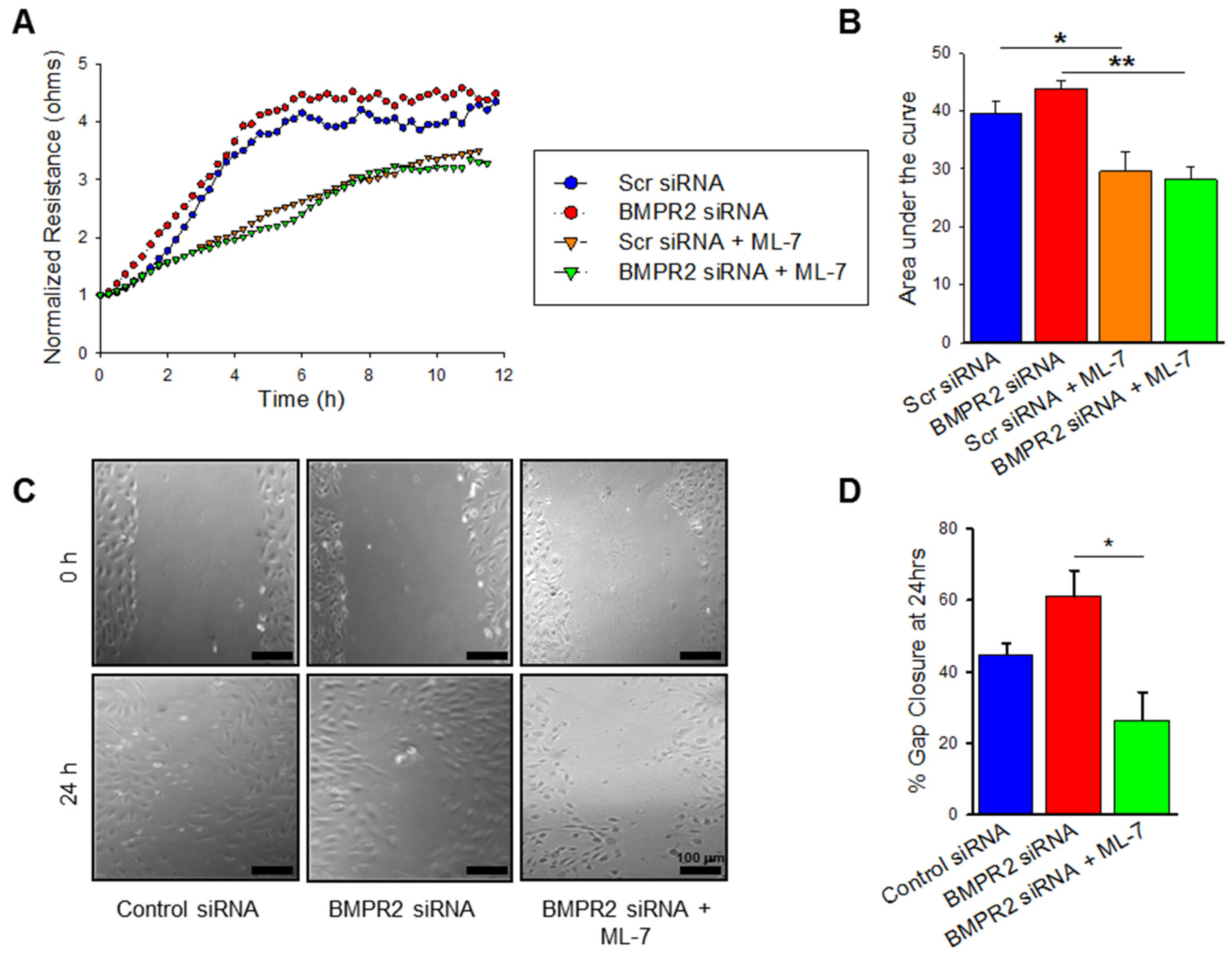
2.3. BMPR2 Silencing Significantly Increases nmMLCK Dependant HPAEC Migration
2.4. VEGF Treatment Leads to Increased nmMLCK Activity
2.5. VEGF Treatment Increases Endothelial Cell Proliferation and Cytokine Release Which Is Negated by MLCK Inhibition
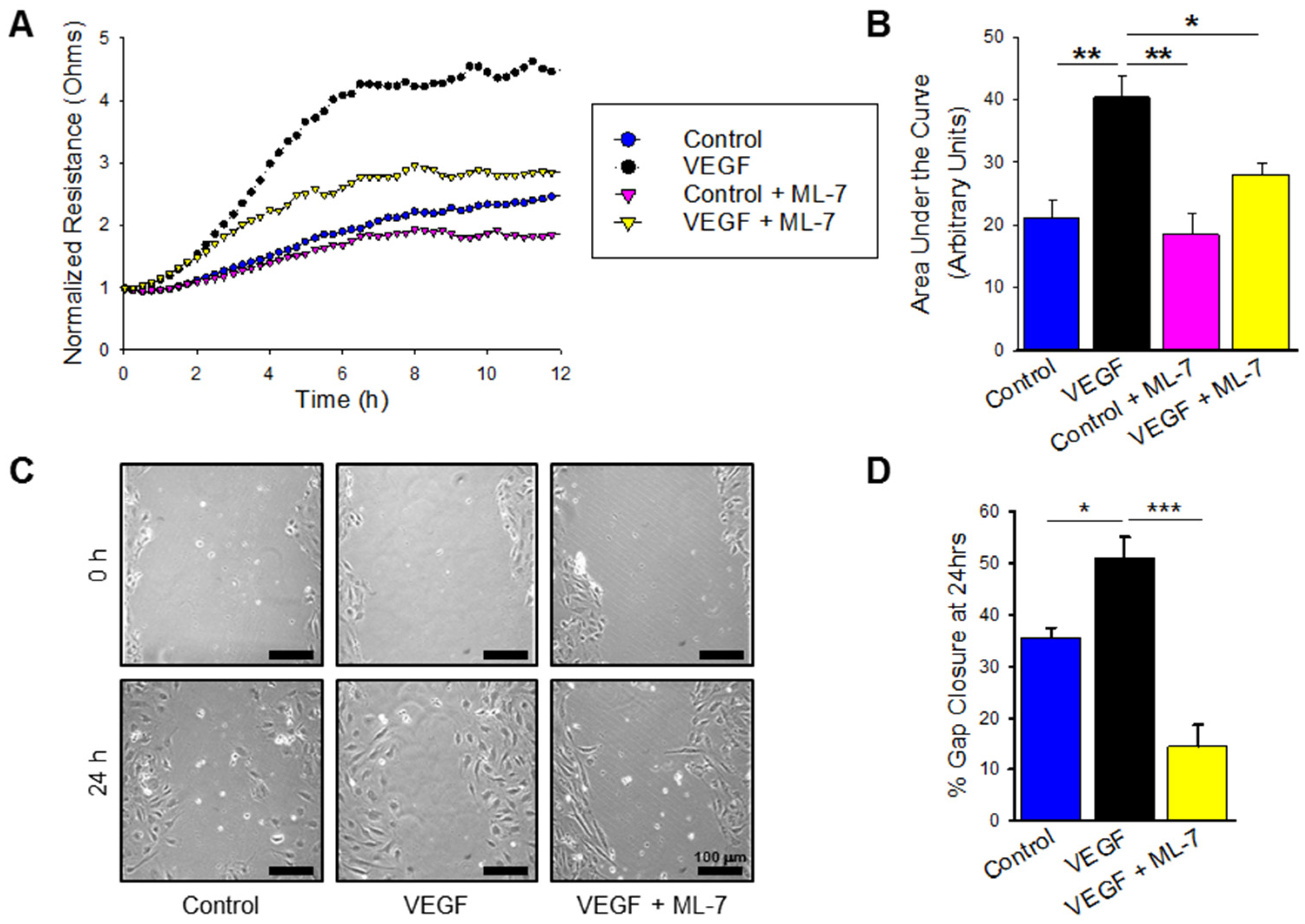
2.6. VEGF Treatment Significantly Increases nmMLCK Dependant HPAEC Migration
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. BMPR2 Transfection
4.3. Western Blotting
4.4. Cell Proliferation and Viability Assay
4.5. Enzyme Linked Immunosorbent Assay (ELISA)
4.6. Electric Cell-Substrate Impedance Sensing (ECIS) Wound Assay
4.7. Scratch Assay
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hoeper, M.M.; Bogaard, H.J.; Condliffe, R.; Frantz, R.; Khanna, D.; Kurzyna, M.; Langleben, D.; Manes, A.; Satoh, T.; Torres, F.; et al. Definitions and diagnosis of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D42–D50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, K.Y.; Duval, S.; Badesch, D.B.; Bull, T.M.; Chakinala, M.M.; de Marco, T.; Frantz, R.P.; Hemnes, A.; Mathai, S.C.; Rosenzweig, E.B.; et al. Mortality in pulmonary arterial hypertension in the modern era: Early insights from the pulmonary hypertension association registry. J. Am. Heart Assoc. 2022, 11, e024969. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.J.; Shenoy, V.; Yamazato, Y.; Sriramula, S.; Francis, J.; Yuan, L.; Castellano, R.K.; Ostrov, D.A.; Oh, S.P.; Katovich, M.J.; et al. Evidence for angiotensin-converting enzyme 2 as a therapeutic target for the prevention of pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2009, 179, 1048–1054. [Google Scholar] [CrossRef] [Green Version]
- International PPH Consortium; Lane, K.B.; Machado, R.D.; Pauciulo, M.W.; Thomson, J.R.; Phillips, J.A., 3rd; Loyd, J.E.; Nichols, W.C.; Trembath, R.C. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat. Genet. 2000, 26, 81–84. [Google Scholar]
- Lane, K.B.; Blackwell, T.R.; Runo, J.; Wheeler, L.; Phillips, J.A., 3rd; Loyd, J.E. Aberrant signal transduction in pulmonary hypertension. Chest 2005, 128, 564S–565S. [Google Scholar] [CrossRef] [PubMed]
- Machado, R.D.; Aldred, M.A.; James, V.; Harrison, R.E.; Patel, B.; Schwalbe, E.C.; Gruenig, E.; Janssen, B.; Koehler, R.; Seeger, W.; et al. Mutations of the TGF-beta type II receptor BMPR2 in pulmonary arterial hypertension. Hum. Mutat. 2006, 27, 121–132. [Google Scholar] [CrossRef]
- Rajkumar, R.; Konishi, K.; Richards, T.J.; Ishizawar, D.C.; Wiechert, A.C.; Kaminski, N.; Ahmad, F. Genomewide RNA expression profiling in lung identifies distinct signatures in idiopathic pulmonary arterial hypertension and secondary pulmonary hypertension. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1235–H1248. [Google Scholar] [CrossRef] [Green Version]
- West, J.; Harral, J.; Lane, K.; Deng, Y.; Ickes, B.; Crona, D.; Albu, S.; Stewart, D.; Fagan, K. Mice expressing BMPR2R899X transgene in smooth muscle develop pulmonary vascular lesions. Am. J. Physiol.-Lung Cell Mol. Physiol. 2008, 295, L744–L755. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, C.; Stewart, S.; Upton, P.D.; Machado, R.; Thomson, J.R.; Trembath, R.C.; Morrell, N.W. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation 2002, 105, 1672–1678. [Google Scholar] [CrossRef] [Green Version]
- Thomson, J.R.; Machado, R.D.; Pauciulo, M.W.; Morgan, N.V.; Humbert, M.; Elliott, G.C.; Ward, K.; Yacoub, M.; Mikhail, G.; Rogers, P.; et al. Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF-beta family. J. Med. Genet. 2000, 37, 741–745. [Google Scholar] [CrossRef]
- Humbert, M.; Morrell, N.W.; Archer, S.L.; Stenmark, K.R.; MacLean, M.R.; Lang, I.M.; Christman, B.W.; Weir, E.K.; Eickelberg, O.; Voelkel, N.F.; et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2004, 43, 13S–24S. [Google Scholar] [CrossRef] [PubMed]
- Foletta, V.C.; Lim, M.A.; Soosairajah, J.; Kelly, A.P.; Stanley, E.G.; Shannon, M.; He, W.; Das, S.; Massague, J.; Bernard, O. Direct signaling by the BMP type II receptor via the cytoskeletal regulator LIMK1. J. Cell Biol. 2003, 162, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Machado, R.D.; Rudarakanchana, N.; Atkinson, C.; Flanagan, J.A.; Harrison, R.; Morrell, N.W.; Trembath, R.C. Functional interaction between BMPR-II and Tctex-1, a light chain of Dynein, is isoform-specific and disrupted by mutations underlying primary pulmonary hypertension. Hum. Mol. Genet. 2003, 12, 3277–3286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, W.K.; Knowles, J.A.; Morse, J.H. Bone morphogenetic protein receptor type II C-terminus interacts with c-Src: Implication for a role in pulmonary arterial hypertension. Am. J. Respir. Cell Mol. Biol. 2005, 33, 438–446. [Google Scholar] [CrossRef] [Green Version]
- Burton, V.J.; Ciuclan, L.I.; Holmes, A.M.; Rodman, D.M.; Walker, C.; Budd, D.C. Bone morphogenetic protein receptor II regulates pulmonary artery endothelial cell barrier function. Blood 2011, 117, 333–341. [Google Scholar] [CrossRef] [Green Version]
- Gamell, C.; Osses, N.; Bartrons, R.; Ruckle, T.; Camps, M.; Rosa, J.L.; Ventura, F. BMP2 induction of actin cytoskeleton reorganization and cell migration requires PI3-kinase and Cdc42 activity. J. Cell Sci. 2008, 121 Pt 23, 3960–3970. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Erzurum, S.C. Endothelial cell energy metabolism, proliferation, and apoptosis in pulmonary hypertension. Compr. Physiol. 2011, 1, 357–372. [Google Scholar]
- Guignabert, C.; Tu, L.; Girerd, B.; Ricard, N.; Huertas, A.; Montani, D.; Humbert, M. New molecular targets of pulmonary vascular remodeling in pulmonary arterial hypertension: Importance of endothelial communication. Chest 2015, 147, 529–537. [Google Scholar] [CrossRef]
- Voelkel, N.F.; Vandivier, R.W.; Tuder, R.M. Vascular endothelial growth factor in the lung. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2006, 290, L209–L221. [Google Scholar] [CrossRef]
- Voelkel, N.F.; Gomez-Arroyo, J. The role of vascular endothelial growth factor in pulmonary arterial hypertension. The angiogenesis paradox. Am. J. Respir. Cell Mol. Biol. 2014, 51, 474–484. [Google Scholar] [CrossRef]
- Claesson-Welsh, L.; Welsh, M. VEGFA and tumour angiogenesis. J. Intern. Med. 2013, 273, 114–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuder, R.M.; Zhen, L.; Cho, C.Y.; Taraseviciene-Stewart, L.; Kasahara, Y.; Salvemini, D.; Voelkel, N.F.; Flores, S.C. Oxidative stress and apoptosis interact and cause emphysema due to vascular endothelial growth factor receptor blockade. Am. J. Respir. Cell Mol. Biol. 2003, 29, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Asai, K.; Kanazawa, H.; Kamoi, H.; Shiraishi, S.; Hirata, K.; Yoshikawa, J. Increased levels of vascular endothelial growth factor in induced sputum in asthmatic patients. Clin. Exp. Allergy 2003, 33, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Thickett, D.R.; Armstrong, L.; Christie, S.J.; Millar, A.B. Vascular endothelial growth factor may contribute to increased vascular permeability in acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2001, 164, 1601–1605. [Google Scholar] [CrossRef]
- Kumpers, P.; Nickel, N.; Lukasz, A.; Golpon, H.; Westerkamp, V.; Olsson, K.M.; Jonigk, D.; Maegel, L.; Bockmeyer, C.L.; David, S.; et al. Circulating angiopoietins in idiopathic pulmonary arterial hypertension. Eur. Heart J. 2010, 31, 2291–2300. [Google Scholar] [CrossRef]
- Papaioannou, A.I.; Zakynthinos, E.; Kostikas, K.; Kiropoulos, T.; Koutsokera, A.; Ziogas, A.; Koutroumpas, A.; Sakkas, L.; Gourgoulianis, K.I.; Daniil, Z.D. Serum VEGF levels are related to the presence of pulmonary arterial hypertension in systemic sclerosis. BMC Pulm. Med. 2009, 9, 18. [Google Scholar] [CrossRef] [Green Version]
- Selimovic, N.; Bergh, C.H.; Andersson, B.; Sakiniene, E.; Carlsten, H.; Rundqvist, B. Growth factors and interleukin-6 across the lung circulation in pulmonary hypertension. Eur. Respir. J. 2009, 34, 662–668. [Google Scholar] [CrossRef] [Green Version]
- Tuder, R.M.; Chacon, M.; Alger, L.; Wang, J.; Taraseviciene-Stewart, L.; Kasahara, Y.; Cool, C.D.; Bishop, A.E.; Geraci, M.; Semenza, G.L.; et al. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: Evidence for a process of disordered angiogenesis. J. Pathol. 2001, 195, 367–374. [Google Scholar] [CrossRef]
- Hirose, S.; Hosoda, Y.; Furuya, S.; Otsuki, T.; Ikeda, E. Expression of vascular endothelial growth factor and its receptors correlates closely with formation of the plexiform lesion in human pulmonary hypertension. Pathol. Int. 2000, 50, 472–479. [Google Scholar] [CrossRef]
- Tuder, R.M.; Flook, B.E.; Voelkel, N.F. Increased gene-expression for Vegf and the Vegf Receptors Kdr/Flk and Flt in lungs exposed to acute or to chronic hypoxia. Modulation of gene-expression by nitric-oxide. J. Clin. Investig. 1995, 95, 1798–1807. [Google Scholar] [CrossRef] [Green Version]
- Partovian, C.; Adnot, S.; Eddahibi, S.; Teiger, E.; Levame, M.; Dreyfus, P.; Raffestin, B.; Frelin, C. Heart and lung VEGF mRNA expression in rats with monocrotaline- or hypoxia-induced pulmonary hypertension. Am. J. Physiol.-Heart Circ. Physiol. 1998, 275, H1948–H1956. [Google Scholar] [CrossRef] [PubMed]
- Christou, H.; Yoshida, A.; Arthur, V.; Morita, T.; Kourembanas, S. Increased vascular endothelial growth factor production in the lungs of rats with hypoxia-induced pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 1998, 18, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.H.; Popel, A.S.; Gabhann, F.M. Computational model of VEGFR2 pathway to ERK activation and modulation through receptor trafficking. Cell. Signal. 2013, 25, 2496–2510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.; Finley, S.D. Mechanistic insight into activation of MAPK signaling by pro-angiogenic factors. BMC Syst. Biol. 2018, 12, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Awad, K.S.; Elinoff, J.M.; Wang, S.; Gairhe, S.; Ferreyra, G.A.; Cai, R.; Sun, J.; Solomon, M.A.; Danner, R.L. Raf/ERK drives the proliferative and invasive phenotype of BMPR2-silenced pulmonary artery endothelial cells. Am. J. Physiol.-Lung Cell Mol. Physiol. 2016, 310, L187–L201. [Google Scholar] [CrossRef] [Green Version]
- Barnes, E.A.; Chen, C.H.; Sedan, O.; Cornfield, D.N. Loss of smooth muscle cell hypoxia inducible factor-1alpha underlies increased vascular contractility in pulmonary hypertension. FASEB J. 2017, 31, 650–662. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Zhou, T.; Saadat, L.; Garcia, J.G. A MYLK variant regulates asthmatic inflammation via alterations in mRNA secondary structure. Eur. J. Hum. Genet. 2015, 23, 874–876. [Google Scholar] [CrossRef] [Green Version]
- Garcia, J.G.; Davis, H.W.; Patterson, C.E. Regulation of endothelial cell gap formation and barrier dysfunction: Role of myosin light chain phosphorylation. J. Cell Physiol. 1995, 163, 510–522. [Google Scholar] [CrossRef]
- Lazar, V.; Garcia, J.G. A single human myosin light chain kinase gene (MLCK; MYLK) transcribes multiple nonmuscle isoforms. Genomics 1999, 57, 256–267. [Google Scholar] [CrossRef]
- Sawada, H.; Saito, T.; Nickel, N.P.; Alastalo, T.P.; Glotzbach, J.P.; Chan, R.; Haghighat, L.; Fuchs, G.; Januszyk, M.; Cao, A.; et al. Reduced BMPR2 expression induces GM-CSF translation and macrophage recruitment in humans and mice to exacerbate pulmonary hypertension. J. Exp. Med. 2014, 211, 263–280. [Google Scholar] [CrossRef] [Green Version]
- Diebold, I.; Hennigs, J.K.; Miyagawa, K.; Li, C.G.; Nickel, N.P.; Kaschwich, M.; Cao, A.; Wang, L.; Reddy, S.; Chen, P.I.; et al. BMPR2 preserves mitochondrial function and DNA during reoxygenation to promote endothelial cell survival and reverse pulmonary hypertension. Cell Metab. 2015, 21, 596–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Jesus Perez, V.A.; Alastalo, T.P.; Wu, J.C.; Axelrod, J.D.; Cooke, J.P.; Amieva, M.; Rabinovitch, M. Bone morphogenetic protein 2 induces pulmonary angiogenesis via Wnt-beta-catenin and Wnt-RhoA-Rac1 pathways. J. Cell Biol. 2009, 184, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Alastalo, T.P.; Li, M.; Vde, J.P.; Pham, D.; Sawada, H.; Wang, J.K.; Koskenvuo, M.; Wang, L.; Freeman, B.A.; Chang, H.Y.; et al. Disruption of PPARgamma/beta-catenin-mediated regulation of apelin impairs BMP-induced mouse and human pulmonary arterial EC survival. J. Clin. Investig. 2011, 121, 3735–3746. [Google Scholar] [CrossRef] [Green Version]
- Massague, J. Integration of Smad and MAPK pathways: A link and a linker revisited. Genes Dev. 2003, 17, 2993–2997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansmann, G.; Perez, V.A.d.; Alastalo, T.P.; Alvira, C.M.; Guignabert, C.; Bekker, J.M.; Schellong, S.; Urashima, T.; Wang, L.; Morrell, N.W.; et al. An antiproliferative BMP-2/PPARgamma/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J. Clin. Investig. 2008, 118, 1846–1857. [Google Scholar] [CrossRef] [Green Version]
- West, J.D.; Austin, E.D.; Gaskill, C.; Marriott, S.; Baskir, R.; Bilousova, G.; Jean, J.C.; Hemnes, A.R.; Menon, S.; Bloodworth, N.C.; et al. Identification of a common Wnt-associated genetic signature across multiple cell types in pulmonary arterial hypertension. Am. J. Physiol. Cell Physiol. 2014, 307, C415–C430. [Google Scholar] [CrossRef] [Green Version]
- Cui, W.J.; Liu, Y.; Zhou, X.L.; Wang, F.Z.; Zhang, X.D.; Ye, L.H. Myosin light chain kinase is responsible for high proliferative ability of breast cancer cells via anti-apoptosis involving p38 pathway. Acta Pharmacol. Sin. 2010, 31, 725–732. [Google Scholar] [CrossRef] [Green Version]
- Hopper, R.K.; Feinstein, J.A.; Manning, M.A.; Benitz, W.; Hudgins, L. Neonatal pulmonary arterial hypertension and Noonan syndrome: Two fatal cases with a specific RAF1 mutation. Am. J. Med. Genet. A 2015, 167A, 882–885. [Google Scholar] [CrossRef]
- Chamorro-Jorganes, A.; Lee, M.Y.; Araldi, E.; Landskroner-Eiger, S.; Fernandez-Fuertes, M.; Sahraei, M.; del Rey, M.Q.; van Solingen, C.; Yu, J.; Fernandez-Hernando, C.; et al. VEGF-Induced Expression of miR-17-92 Cluster in Endothelial Cells Is Mediated by ERK/ELK1 Activation and Regulates Angiogenesis. Circ. Res. 2016, 118, 38–47. [Google Scholar] [CrossRef]
- Shen, Q.; Rigor, R.R.; Pivetti, C.D.; Wu, M.H.; Yuan, S.Y. Myosin light chain kinase in microvascular endothelial barrier function. Cardiovasc. Res. 2010, 87, 272–280. [Google Scholar] [CrossRef] [Green Version]
- Masri, F.A.; Xu, W.; Comhair, S.A.; Asosingh, K.; Koo, M.; Vasanji, A.; Drazba, J.; Anand-Apte, B.; Erzurum, S.C. Hyperproliferative apoptosis-resistant endothelial cells in idiopathic pulmonary arterial hypertension. Am. J. Physiol.-Lung Cell Mol. Physiol. 2007, 293, L548–L554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toshner, M.; Voswinckel, R.; Southwood, M.; Al-Lamki, R.; Howard, L.S.; Marchesan, D.; Yang, J.; Suntharalingam, J.; Soon, E.; Exley, A.; et al. Evidence of dysfunction of endothelial progenitors in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2009, 180, 780–787. [Google Scholar] [CrossRef] [PubMed]
- Teichert-Kuliszewska, K.; Kutryk, M.J.; Kuliszewski, M.A.; Karoubi, G.; Courtman, D.W.; Zucco, L.; Granton, J.; Stewart, D.J. Bone morphogenetic protein receptor-2 signaling promotes pulmonary arterial endothelial cell survival: Implications for loss-of-function mutations in the pathogenesis of pulmonary hypertension. Circ. Res. 2006, 98, 209–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.S.; Munn, L.L.; Hillsley, M.V.; Dull, R.O.; Yuan, J.; Lakshminarayanan, S.; Gardner, T.W.; Jain, R.K.; Tarbell, J.M. Effect of vascular endothelial growth factor on cultured endothelial cell monolayer transport properties. Microvasc. Res. 2000, 59, 265–277. [Google Scholar] [CrossRef]
- Ferrara, N. Vascular endothelial growth factor: Basic science and clinical progress. Endocr. Rev. 2004, 25, 581–611. [Google Scholar] [CrossRef]
- Geiger, R.; Berger, R.M.; Hess, J.; Bogers, A.J.; Sharma, H.S.; Mooi, W.J. Enhanced expression of vascular endothelial growth factor in pulmonary plexogenic arteriopathy due to congenital heart disease. J. Pathol. 2000, 191, 202–207. [Google Scholar] [CrossRef]
- Saitoh, M.; Ishikawa, T.; Matsushima, S.; Naka, M.; Hidaka, H. Selective inhibition of catalytic activity of smooth muscle myosin light chain kinase. J. Biol. Chem. 1987, 262, 7796–7801. [Google Scholar] [CrossRef]
- Ma, T.Y.; Nguyen, D.; Bui, V.; Nguyen, H.; Hoa, N. Ethanol modulation of intestinal epithelial tight junction barrier. Am. J. Physiol. 1999, 276, G965–G974. [Google Scholar] [CrossRef]
- Usatyuk, P.V.; Singleton, P.A.; Pendyala, S.; Kalari, S.K.; He, D.; Gorshkova, I.A.; Camp, S.M.; Moitra, J.; Dudek, S.M.; Garcia, J.G.; et al. Novel role for non-muscle myosin light chain kinase (MLCK) in hyperoxia-induced recruitment of cytoskeletal proteins, NADPH oxidase activation, and reactive oxygen species generation in lung endothelium. J. Biol. Chem. 2012, 287, 9360–9375. [Google Scholar] [CrossRef] [Green Version]
- Gonzales, J.; Holbert, K.; Czysz, K.; George, J.; Fernandes, C.; Fraidenburg, D.R. Hemin-induced endothelial dysfunction and endothelial to mesenchymal transition in the pathogenesis of pulmonary hypertension due to chronic hemolysis. Int. J. Mol. Sci. 2022, 23, 4763. [Google Scholar] [CrossRef]
- Trepat, X.; Chen, Z.; Jacobson, K. Cell migration. Compr. Physiol. 2012, 2, 2369–2392. [Google Scholar] [PubMed] [Green Version]
- Guo, S.; Lok, J.; Liu, Y.; Hayakawa, K.; Leung, W.; Xing, C.; Ji, X.; Lo, E.H. Assays to examine endothelial cell migration, tube formation, and gene expression profiles. Methods Mol. Biol. 2014, 1135, 393–402. [Google Scholar] [PubMed]
- Liang, C.C.; Park, A.Y.; Guan, J.L. In Vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in Vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anis, M.; Gonzales, J.; Halstrom, R.; Baig, N.; Humpal, C.; Demeritte, R.; Epshtein, Y.; Jacobson, J.R.; Fraidenburg, D.R. Non-Muscle MLCK Contributes to Endothelial Cell Hyper-Proliferation through the ERK Pathway as a Mechanism for Vascular Remodeling in Pulmonary Hypertension. Int. J. Mol. Sci. 2022, 23, 13641. https://doi.org/10.3390/ijms232113641
Anis M, Gonzales J, Halstrom R, Baig N, Humpal C, Demeritte R, Epshtein Y, Jacobson JR, Fraidenburg DR. Non-Muscle MLCK Contributes to Endothelial Cell Hyper-Proliferation through the ERK Pathway as a Mechanism for Vascular Remodeling in Pulmonary Hypertension. International Journal of Molecular Sciences. 2022; 23(21):13641. https://doi.org/10.3390/ijms232113641
Chicago/Turabian StyleAnis, Mariam, Janae Gonzales, Rachel Halstrom, Noman Baig, Cat Humpal, Regaina Demeritte, Yulia Epshtein, Jeffrey R. Jacobson, and Dustin R. Fraidenburg. 2022. "Non-Muscle MLCK Contributes to Endothelial Cell Hyper-Proliferation through the ERK Pathway as a Mechanism for Vascular Remodeling in Pulmonary Hypertension" International Journal of Molecular Sciences 23, no. 21: 13641. https://doi.org/10.3390/ijms232113641