
Vulnerable Atherosclerotic Plaque: Is There a Molecular Signature?
 ,
,  , , and
, , and
Abstract
:1. Introduction
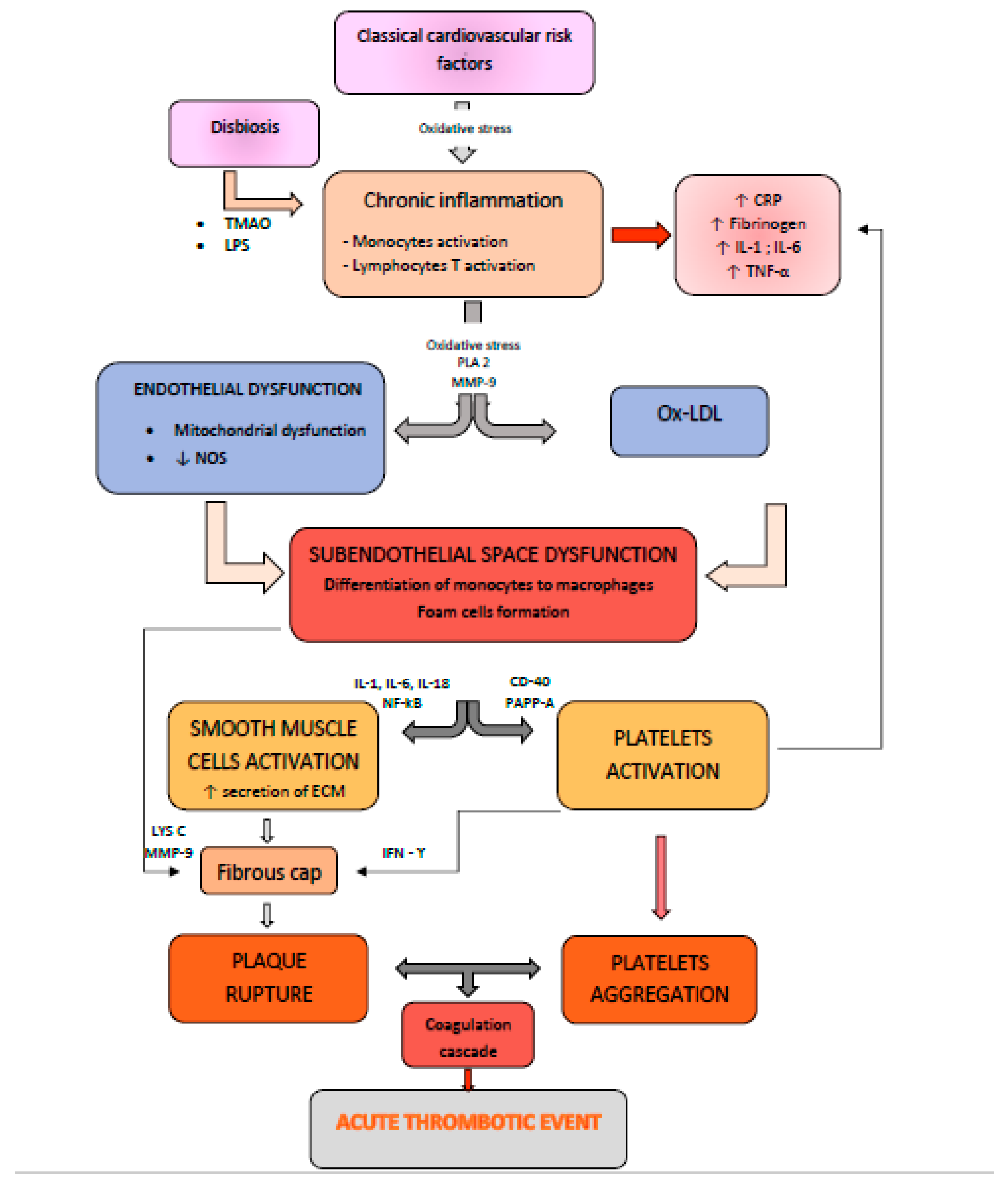
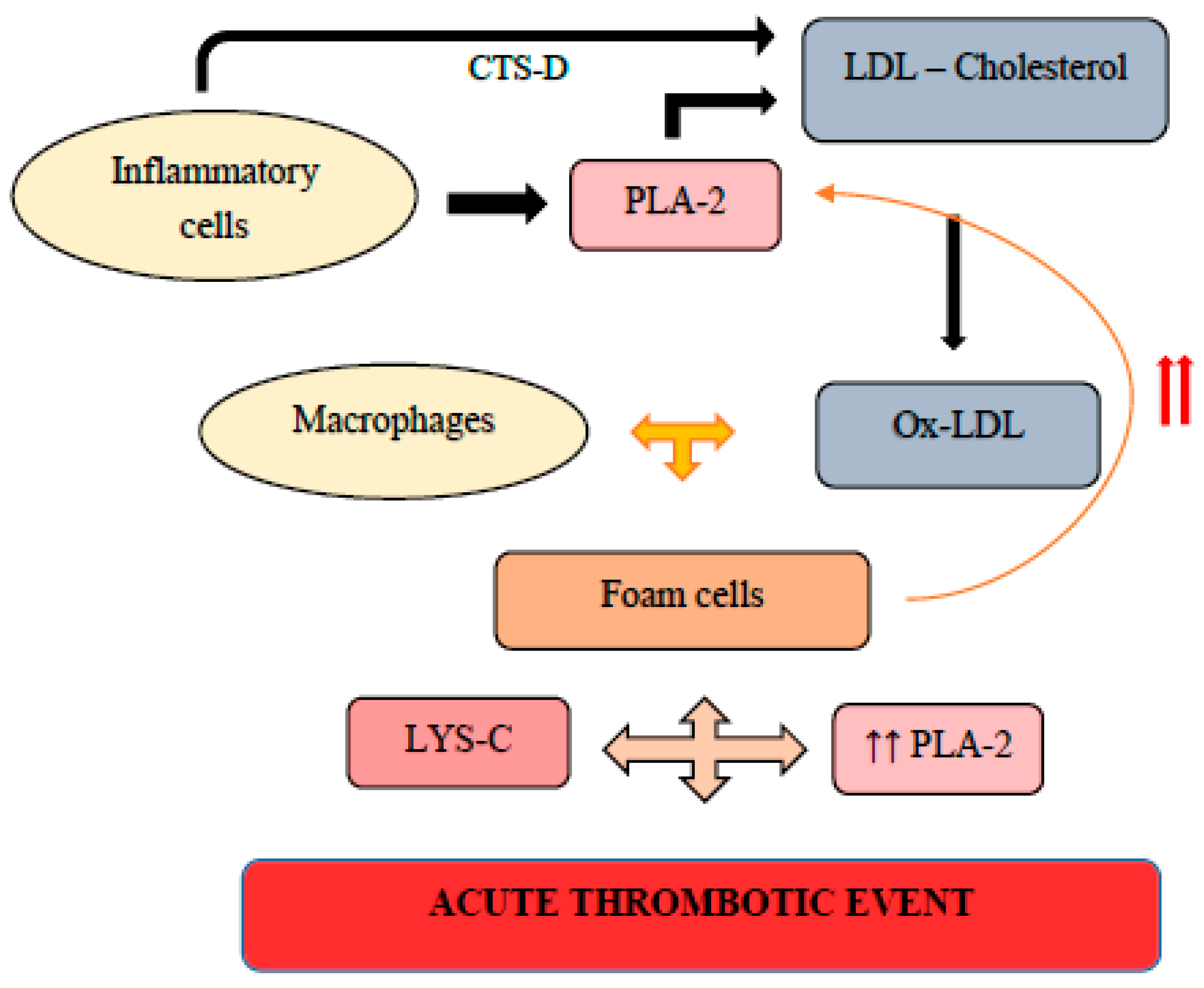
2. Pathophysiological Mechanisms and Molecular Events during the Development of Vulnerable Atherosclerotic Plaques: Plaque Erosion versus Plaque Fissure
3. Imaging Biomarkers of Vulnerable Plaques
4. Circulating Biomarkers of Vulnerable Plaques
4.1. Systemic Inflammatory Biomarkers
4.1.1. C-reactive Protein
4.1.2. Fibrinogen
4.1.3. Cytokines
TNF-α
IFN-γ
IL-1 Family
IL-6 Family
NF-KB
CD40-CD40L
PAPP-A
MMP
MPO
MCP-1/CCL-2
C5b-9
RGC-32
SIRT 1

{kind=link}
{kind=link}
{kind=link}
| Biomarker | Steps in Atherosclerosis in Which the Biomarker Is Involved | Prognosis | Therapy |
|---|---|---|---|
| Hs-CRP [45,117] |
| Associated with AMI [51] | LDL-lowering therapy, including statins ACEI/ARB BB [118,119] Antidiabetic drugs, including Metformin [120,121] |
| Fibrinogen [52] |
| Prediction of all-cause and cardiac mortality [55] | |
| TNF-α [58,59] |
| Associated with the risk of CAD [60] | |
| IFN-γ [61] |
| Associated with lesions prone to rupture [62] | |
| NLRP3/IL-1/IL-18 [64,65,69,73] |
| IL-18- prediction of MACE at 60-day follow-up [70] | |
| IL-6 [76] |
| Progression of atherosclerotic lesions [78] | |
| NF-κB [81] |
| Plaque destabilization [81] | |
| CD40/CD4-L [82,83,84,85,86,87] |
| Prediction of cardiovascular events and plaque progression [87] | |
| PAPP-A [89] |
| Prediction of MACE [93] | |
| MMPs [94] |
| Prediction of major cardiovascular and cerebrovascular events [95,97] | |
| MPO [102] |
| Prediction of MACE [104] | |
| MCP-1 [108] |
| Correlated with symptomatic lesions and a higher risk of MACE [109] |
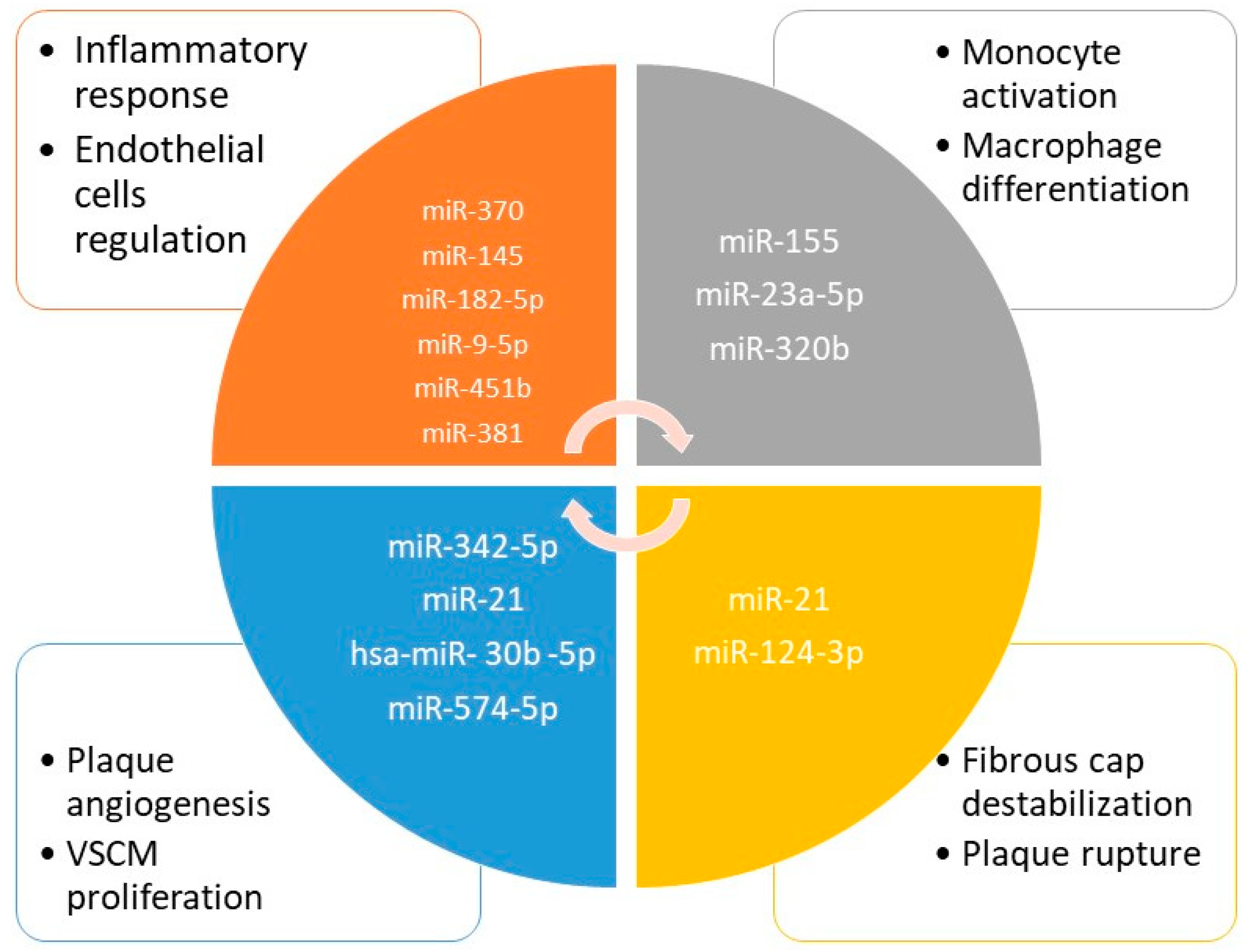
4.2. Epigenetic Biomarkers of Vulnerable Plaques
4.3. Gut Microbiota Biomarkers
4.3.1. TMAO
4.3.2. LPS
4.3.3. SCFA
4.3.4. Pagln
4.4. Oxidative Stress Biomarkers
4.5. Metabolic Biomarkers
5. Proteomics of Vulnerable Plaques
Strengths and Limitations of the Review
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Björkegren, J.L.; Lusis, A.J. Atherosclerosis: Recent developments. Cell 2022, 185, 1630–1645. [Google Scholar] [CrossRef] [PubMed]
- Vivanco, F.; Martín-Ventura, J.L.; Duran, M.C.; Barderas, M.G.; Blanco-Colio, L.; Dardé, V.M.; Mas, S.; Meilhac, O.; Michel, J.B.; Tuñón, J.; et al. Quest for Novel Cardiovascular Biomarkers by Proteomic Analysis. J. Proteome Res. 2005, 4, 1181–1191. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.; Barallobre-Barreiro, J.; Jahangiri, M.; Mayr, M. Vascular proteomics in metabolic and cardiovascular diseases. J. Intern. Med. 2016, 280, 325–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiorescu, R.M.; Mocan, M.; Barta, A.; Stoia, M.A.; Anton, P.; Mocan-Hognogi, D.L.; Cerasela, M.; Goidescu, A.D.F. Hypoalbuminemia—Prognostic factor for the short-term evolution of patients with unstable angina. Hum. Vet. Med. 2017, 11, 160–165. [Google Scholar]
- Hirase, T.; Node, K. Endothelial dysfunction as a cellular mechanism for vascular failure. Am. J. Physiol. Circ. Physiol. 2012, 302, H499–H505. [Google Scholar] [CrossRef] [Green Version]
- Hansson, G.K.; Libby, P. The immune response in atherosclerosis: A double-edged sword. Nat. Rev. Immunol. 2006, 6, 508–519. [Google Scholar] [CrossRef]
- Gimbrone, M.A.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [Green Version]
- Quillard, T.; Araújo, H.A.; Franck, G.; Shvartz, E.; Sukhova, G.; Libby, P. TLR2 and neutrophils potentiate endothelial stress, apoptosis and detachment: Implications for superficial erosion. Eur. Hear. J. 2015, 36, 1394–1404. [Google Scholar] [CrossRef] [Green Version]
- Vlaicu, S.I.; Tatomir, A.; Rus, V.; Mekala, A.P.; Mircea, P.A.; Niculescu, F.; Rus, H. The role of complement activation in atherogenesis: The first 40 years. Immunol. Res. 2015, 64, 1–13. [Google Scholar] [CrossRef]
- Edsfeldt, A.; Swart, M.; Singh, P.; Dib, L.; Sun, J.; E Cole, J.; Park, I.; Al-Sharify, D.; Persson, A.; Nitulescu, M.; et al. Interferon regulatory factor-5-dependent CD11c+ macrophages contribute to the formation of rupture–prone atherosclerotic plaques. Eur. Hear. J. 2022, 43, 1864–1877. [Google Scholar] [CrossRef]
- Bengtsson, E.; Hultman, K.; Edsfeldt, A.; Persson, A.; Nitulescu, M.; Nilsson, J.; Goncalves, I.; Björkbacka, H. CD163+ macrophages are associated with a vulnerable plaque phenotype in human carotid plaques. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Hermansson, A. The immune system in atherosclerosis. Nat. Immunol. 2011, 12, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, D.M.; Rahman, A.H.; Fernandez, N.F.; Chudnovskiy, A.; Amir, E.D.; Amadori, L.; Khan, N.S.; Wong, C.K.; Shamailova, R.; Hill, C.A.; et al. Single-cell immune landscape of human atherosclerotic plaques. Nat. Med. 2019, 25, 1576–1588. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S. Atherosclerosis. Nat. Rev. Dis. Prim. 2019, 5. Available online: http://www.nature.com/articles/s41572-019-0106-z (accessed on 16 August 2019). [CrossRef]
- Hansson, G.K.; Libby, P.; Tabas, I. Inflammation and plaque vulnerability. J. Intern. Med. 2015, 278, 483–493. [Google Scholar] [CrossRef] [Green Version]
- Partida, R.A.; Libby, P.; Crea, F.; Jang, I.-K. Plaque erosion: A new in vivo diagnosis and a potential major shift in the management of patients with acute coronary syndromes. Eur. Heart J. 2018, 39, 2070–2076. [Google Scholar] [CrossRef]
- Goncalves, I.; Sun, J.; Tengryd, C.; Nitulescu, M.; Persson, A.F.; Nilsson, J.; Edsfeldt, A. Plaque Vulnerability Index Predicts Cardiovascular Events: A Histological Study of an Endarterectomy Cohort. J. Am. Hear. Assoc. 2021, 10. [Google Scholar] [CrossRef]
- Libby, P.; Pasterkamp, G.; Crea, F.; Jang, I.K. Reassessing the Mechanisms of Acute Coronary Syndromes: The “vulnerable Plaque” and Superficial Erosion. Circ. Res. 2019, 124, 150–160. [Google Scholar] [CrossRef]
- Buono, M.F.; Slenders, L.; Wesseling, M.; Hartman, R.J.; Monaco, C.; Ruijter, H.M.D.; Pasterkamp, G.; Mokry, M. The changing landscape of the vulnerable plaque: A call for fine-tuning of preclinical models. Vasc. Pharmacol. 2021, 141, 106924. [Google Scholar] [CrossRef]
- Guo, Y.; Tang, Z.; Yan, B.; Yin, H.; Tai, S.; Peng, J.; Cui, Y.; Gui, Y.; Belke, D.; Zhou, S.; et al. PCSK9 (Proprotein Convertase Subtilisin/Kexin Type 9) Triggers Vascular Smooth Muscle Cell Senescence and Apoptosis: Implication of Its Direct Role in Degenerative Vascular Disease. Arter. Thromb. Vasc. Biol. 2022, 42, 67–86. [Google Scholar] [CrossRef]
- Kolodgie, F.D.; Burke, A.P.; Farb, A.; Weber, D.K.; Kutys, R.; Wight, T.N. Differential accumulation of proteoglycans and hyaluronan in culprit lesions: Insights into plaque erosion. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1642–1648. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, T.; Dweck, M.R.; Narula, N.; Pisapia, D.; Narula, J.; Strauss, H.W. Coronary Artery Calcification From Mechanism to Molecular Imaging. JACC Cardiovasc. Imaging 2017, 10, 582–593. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Gao, J.; Lv, Q.; Cai, H.; Wang, F.; Ye, R. Calcification in Atherosclerotic Plaque Vulnerability: Friend or Foe? Front Physiol. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Buffinton, C.M.; Ebenstein, D. Effect of Calcification Modulus and Geometry on Stress in Models of Calcified Atherosclerotic Plaque. Cardiovasc. Eng. Technol. 2014, 5, 244–260. [Google Scholar] [CrossRef]
- Lee, K.Y.; Chang, K. Understanding Vulnerable Plaques: Current Status and Future Directions. Korean Circ. J. 2019, 49, 1115–1122. [Google Scholar] [CrossRef]
- Libby, P. The changing landscape of atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef]
- Syed, M.B.; Fletcher, A.; O Forsythe, R.; Kaczynski, J.; E Newby, D.; Dweck, M.R.; Van Beek, E.J. Emerging techniques in atherosclerosis imaging. Br. J. Radiol. 2019, 92, 20180309. [Google Scholar] [CrossRef]
- Milzi, A.; Lemma, E.D.; Dettori, R.; Burgmaier, K.; Marx, N.; Reith, S.; Burgmaier, M. Coronary plaque composition influences biomechanical stress and predicts plaque rupture in a morpho-mechanic oct analysis. Elife 2021, 10, 1–16. [Google Scholar] [CrossRef]
- Prati, F.; Romagnoli, E.; Gatto, L.; Manna ALa Burzotta, F.; Ozaki, Y. Relationship between coronary plaque morphology of the left anterior descending artery and 12 months clinical outcome: The CLIMA study. Eur. Heart J. 2020, 41, 383–391. [Google Scholar] [CrossRef] [Green Version]
- Muller, J.; Madder, R. OCT-NIRS Imaging for Detection of Coronary Plaque Structure and Vulnerability. Front. Cardiovasc. Med. 2020, 7. [Google Scholar] [CrossRef]
- Tomaniak, M.; Katagiri, Y.; Modolo, R.; de Silva, R.; Khamis, R.Y.; Bourantas, C.V.; Torii, R.; Wentzel, J.J.; Gijsen, F.J.H.; van Soest, G.; et al. Vulnerable plaques and patients: State-of-the-art. Eur. Hear. J. 2020, 41, 2997–3004. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Cai, J.; He, Y.; Sun, C.; Lian, X. Usability of Ultrasonic MicroPure Imaging for Evaluating the Vulnerability of Carotid Atherosclerotic Plaques. J. Ultrasound Med. 2021, 40, 2727–2734. [Google Scholar] [CrossRef] [PubMed]
- Puchner, S.B.; Liu, T.; Mayrhofer, T.; Truong, Q.A.; Lee, H.; Fleg, J.L. High-risk plaque detected on coronary CT angiography predicts acute coronary syndromes independent of significant stenosis in acute chest pain: Results from the ROMICAT-II trial. J. Am. Coll. Cardiol. 2014, 64, 684–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Maurovich-Horvat, P.; Mayrhofer, T.; Puchner, S.B.; Lu, M.T.; Ghemigian, K.; Kitslaar, P.H.; Broersen, A.; Pursnani, A.; Hoffmann, U.; et al. Quantitative coronary plaque analysis predicts high-risk plaque morphology on coronary computed tomography angiography: Results from the ROMICAT II trial. Int. J. Cardiovasc. Imaging 2017, 34, 311–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canu, M.; Broisat, A.; Riou, L.; Vanzetto, G.; Fagret, D.; Ghezzi, C.; Djaileb, L.; Barone-Rochette, G. Non-invasive Multimodality Imaging of Coronary Vulnerable Patient. Front. Cardiovasc. Med. 2022, 9. [Google Scholar] [CrossRef] [PubMed]
- Oikonomou, E.K.; Williams, M.C.; Kotanidis, C.P.; Desai, M.Y.; Marwan, M.; Antonopoulos, A.S. A novel machine learning-derived radiotranscriptomic signature of perivascular fat improves cardiac risk prediction using coronary CTangiography. Eur. Heart J. 2019, 40, 3529–3543. [Google Scholar] [CrossRef] [Green Version]
- Hyafil, F.; Vigne, J. Imaging inflammation in atherosclerotic plaques: Just make it easy! J. Nucl. Cardiol. 2018, 26, 1705–1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varasteh, Z.; De Rose, F.; Mohanta, S.; Li, Y.; Zhang, X.; Miritsch, B.; Scafetta, G.; Yin, C.; Sager, H.B.; Glasl, S.; et al. Imaging atherosclerotic plaques by targeting Galectin-3 and activated macrophages using (89Zr)-DFO- Galectin3-F(ab’)2 mAb. Theranostics 2021, 11, 1864–1876. [Google Scholar] [CrossRef]
- Meester, E.J.; Krenning, B.J.; de Blois, E.; de Jong, M.; van der Steen, A.F.W.; Bernsen, M.R. Imaging inflammation in atherosclerotic plaques, targeting SST2 with [111In]In-DOTA-JR. J. Nucl. Cardiol. 2021, 28, 2506–2513. [Google Scholar] [CrossRef] [Green Version]
- Karakatsanis, N.A.; Abgral, R.; Trivieri, M.G.; Dweck, M.R.; Robson, P.M.; Calcagno, C. Hybrid PET- and MR-driven attenuation correction for enhanced 18F-NaF and 18F-FDG quantification in cardiovascular PET/MR imaging. J Nucl. Cardiol. 2020, 27, 1126–1141. [Google Scholar] [CrossRef]
- Htun, N.M.; Chen, Y.C.; Lim, B.; Schiller, T.; Maghzal, G.J.; Huang, A.L.; Elgass, K.D.; Rivera, J.; Schneider, H.G.; Wood, B.R.; et al. Near-infrared autofluorescence induced by intraplaque hemorrhage and heme degradation as marker for high-risk atherosclerotic plaques. Nat. Commun. 2017, 8, 75. [Google Scholar] [CrossRef] [PubMed]
- Giannakou, S.; Angelidis, G.; Tsougos, I.; Valotassiou, V.; Kappas, K.; Georgoulias, P. Pet tracers for vulnerable plaque imaging. Ann. Nucl. Med. 2020, 34, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Oikonomou, E.K.; Siddique, M.; Antoniades, C. Artificial intelligence in medical imaging: A radiomic guide to precision phenotyping of cardiovascular disease. Cardiovasc. Res. 2020, 116, 2040–2054. [Google Scholar] [CrossRef] [Green Version]
- Fedewa, R.; Puri, R.; Fleischman, E.; Lee, J.; Prabhu, D.; Wilson, D.L.; Vince, D.G.; Fleischman, A. Artificial Intelligence in Intracoronary Imaging. Curr. Cardiol. Rep. 2020, 22, 1–15. [Google Scholar] [CrossRef]
- Badimon, L.; Peña, E.; Arderiu, G.; Padró, T.; Slevin, M.; Vilahur, G.; Chiva-Blanch, G. C-Reactive Protein in Atherothrombosis and Angiogenesis. Front. Immunol. 2018, 9, 430. [Google Scholar] [CrossRef] [Green Version]
- Grad, E.; Danenberg, H.D. C-reactive protein and atherothrombosis: Cause or effect? Blood Rev. 2013, 27, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Wu, Y.; Liu, E. C-reactive protein and cardiovascular disease: From animal studies to the clinic (Review). Exp. Ther. Med. 2020, 20, 1211–1219. [Google Scholar] [CrossRef]
- Stancel, N.; Chen, C.-C.; Ke, L.-Y.; Chu, C.-S.; Lu, J.; Sawamura, T.; Chen, C.-H. Interplay between CRP, Atherogenic LDL, and LOX-1 and Its Potential Role in the Pathogenesis of Atherosclerosis. Clin. Chem. 2016, 62, 320–327. [Google Scholar] [CrossRef] [Green Version]
- Mocan, M.; Vesa, S.; Suciu, S.; Blaga, S.N. Systemic markers of oxidative stress in relation to metabolic syndrome components. Clujul Med 2013, 86, 227–234. [Google Scholar]
- Mocan, M.; Anton, F.; Suciu, Š.; Rǎhian, R.; Blaga, S.N.; Fǎrcaş, A.D. Multimarker Assessment of Diastolic Dysfunction in Metabolic Syndrome Patients. Metab. Syndr. Relat. Disord. 2017, 15, 507–514. [Google Scholar] [CrossRef]
- Wang, J.; Tang, B.; Liu, X.; Wu, X.; Wang, H.; Xu, D.; Guo, Y. Increased monomeric CRP levels in acute myocardial infarction: A possible new and specific biomarker for diagnosis and severity assessment of disease. Atherosclerosis 2015, 239, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Surma, S.; Banach, M. Fibrinogen and Atherosclerotic Cardiovascular Diseases—Review of the Literature and Clinical Studies. Int. J. Mol. Sci. 2021, 23, 193. [Google Scholar] [CrossRef] [PubMed]
- Tabakcı, M.M.; Gerin, F.; Sunbul, M.; Toprak, C.; Durmuş, H.I.; Demir, S.; Arslantaş, U.; Cerşit, S.; Batgerel, U.; Kargın, R. Relation of Plasma Fibrinogen Level With the Presence, Severity, and Complexity of Coronary Artery Disease. Clin. Appl. Thromb. 2016, 23, 638–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Xia, T.-L.; Li, Y.-M.; Huang, F.-Y.; Chai, H.; Wang, P.-J.; Liu, W.; Zhang, C.; Pu, X.-B.; Chen, S.-J.; et al. Fibrinogen is related to long-term mortality in Chinese patients with acute coronary syndrome but failed to enhance the prognostic value of the GRACE score. Oncotarget 2017, 8, 20622–20629. [Google Scholar] [CrossRef] [Green Version]
- Yuan, D.; Jiang, P.; Zhu, P.; Jia, S.; Zhang, C.; Liu, Y. Prognostic value of fibrinogen in patients with coronary artery disease and prediabetes or diabetes following percutaneous coronary intervention: 5-year findings from a large cohort study. Cardiovasc. Diabetol. 2021, 20, 1–13. [Google Scholar] [CrossRef]
- Moss, J.W.; Ramji, D.P. Cytokines: Roles in atherosclerosis disease progression and potential therapeutic targets. Futur. Med. Chem. 2016, 8, 1317–1330. [Google Scholar] [CrossRef] [Green Version]
- Imanishi, T.; Akasaka, T. Biomarkers associated with vulnerable atheromatous plaque. Curr. Med. Chem. 2012, 19, 2588–2596. [Google Scholar] [CrossRef]
- Ohta, H.; Wada, H.; Niwa, T.; Kirii, H.; Iwamoto, N.; Fujii, H.; Saito, K.; Sekikawa, K.; Seishima, M. Disruption of tumor necrosis factor-α gene diminishes the development of atherosclerosis in ApoE-deficient mice. Atherosclerosis 2005, 180, 11–17. [Google Scholar] [CrossRef]
- Zhang, L.; Peppel, K.; Sivashanmugam Perumal Orman, E.S.; Brian, L.; Exum, S.T.; Freedman, N.J. Expression of Tumor Necrosis Factor Receptor-1 in Arterial Wall Cells Promotes Atherosclerosis. Arter. Thromb. Vasc. Biol. 2007, 27, 1087–1094. [Google Scholar] [CrossRef] [Green Version]
- Kaptoge, S.; Seshasai, S.R.K.; Jørgensen, T.; Danesh, J.; Gao, P.; Freitag, D.F.; Butterworth, A.S.; Borglykke, A.; Di Angelantonio, E.; Gudnason, V.; et al. Inflammatory cytokines and risk of coronary heart disease: New prospective study and updated meta-analysis. Eur. Heart J. 2014, 35, 578–589. [Google Scholar] [CrossRef] [Green Version]
- Elyasi, A.; Voloshyna, I.; Ahmed, S.; Kasselman, L.J.; Behbodikhah, J.; De Leon, J.; Reiss, A.B. The role of interferon-γ in cardiovascular disease: An update. Agents Actions 2020, 69, 975–988. [Google Scholar] [CrossRef] [PubMed]
- Voloshyna, I.; Littlefield, M.J.; Reiss, A.B. Atherosclerosis and interferon-γ: New insights and therapeutic targets. Trends Cardiovasc. Med. 2013, 24, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The Interleukin-1 Family: Back to the Future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef] [Green Version]
- Grebe, A.; Hoss, F.; Latz, E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ. Res. 2018, 122, 1722–1740. [Google Scholar] [CrossRef]
- Khan, R.; Rheaume, E.; Tardif, J.-C. Examining the Role of and Treatment Directed at IL-1β in Atherosclerosis. Curr. Atheroscler. Rep. 2018, 20, 53. [Google Scholar] [CrossRef]
- Vromman, A.; Ruvkun, V.; Shvartz, E.; Wojtkiewicz, G.; Masson, G.S.; Tesmenitsky, Y.; Folco, E.; Gram, H.; Nahrendorf, M.; Swirski, F.K.; et al. Stage-dependent differential effects of interleukin-1 isoforms on experimental atherosclerosis. Eur. Hear. J. 2019, 40, 2482–2491. [Google Scholar] [CrossRef] [PubMed]
- Ørn, S.; Ueland, T.; Manhenke, C.; Sandanger Godang, K.; Yndestad, A. Increased interleukin-1β levels are associated with left ventricular hypertrophy and remodelling following acute ST segment elevation myocardial infarction treated by primary percutaneous coronary intervention. J. Int. Med. 2012, 272, 267–276. [Google Scholar] [CrossRef]
- Orrem, H.L.; Shetelig, C.; Ueland, T.; Limalanathan, S.; Nilsson, P.; Husebye, T.; Aukrust, P.; Seljeflot, I.; Hoffmann, P.; Eritsland, J.; et al. Soluble IL-1 receptor 2 is associated with left ventricular remodelling in patients with ST-elevation myocardial infarction. Int. J. Cardiol. 2018, 268, 187–192. [Google Scholar] [CrossRef] [Green Version]
- Bhat, O.M.; Kumar, P.U.; Giridharan, N.V.; Kaul, D.; Kumar, M.J.M.; Dhawan, V. Interleukin-18-induced atherosclerosis involves CD36 and NF-κB crosstalk in Apo E-/- mice. J. Cardiol. 2015, 66, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Tong, G.-X.; Zhang, X.-W.; Leng, J.-H.; Jin, J.-F.; Wang, N.-F.; Yang, J.-M. Interleukin-18 Levels on Admission Are Associated With Mid-Term Adverse Clinical Events in Patients With ST-Segment Elevation Acute Myocardial Infarction Undergoing Percutaneous Coronary Intervention. Int. Hear. J. 2010, 51, 75–81. [Google Scholar] [CrossRef] [Green Version]
- Toldo, S.; Abbate, A. The NLRP3 inflammasome in acute myocardial infarction. Nat. Rev. Cardiol. 2017, 15, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Xie, S.; Yang, G.; Wang, N. Spotlight on NLRP3 Inflammasome: Role in Pathogenesis and Therapies of Atherosclerosis. J. Inflamm. Res. 2021, ume 14, 7143–7172. [Google Scholar] [CrossRef]
- Baldrighi, M.; Mallat, Z.; Li, X. NLRP3 inflammasome pathways in atherosclerosis. 2018. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, S.; Yutani, C.; Takahashi, S.; Takewa, M.; Ohara, T.; Hirayama, A.; Kodama, K. Debris collected in-situ from spontaneously ruptured atherosclerotic plaque invariably contains large cholesterol crystals and evidence of activation of innate inflammation: Insights from non-obstructive general angioscopy. Atherosclerosis 2022, 352, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Mauro, A.G.; Bonaventura, A.; Mezzaroma, E.; Quader, M.; Toldo, S. NLRP3 Inflammasome in Acute Myocardial Infarction. J. Cardiovasc. Pharmacol. 2019, 74, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Ye, D.; Wang, Z.; Pan, H.; Lu, X.; Wang, M.; Xu, Y.; Yu, J.; Zhang, J.; Zhao, M.; et al. The Role of Interleukin-6 Family Members in Cardiovascular Diseases. Front. Cardiovasc. Med. 2022, 9. [Google Scholar] [CrossRef]
- Wu, M.-Y.; Li, C.-J.; Hou, M.-F.; Chu, P.-Y. New Insights into the Role of Inflammation in the Pathogenesis of Atherosclerosis. Int. J. Mol. Sci. 2017, 18, 2034. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, S.; Sakaguchi, M.; Miwa, K.; Furukado, S.; Yamagami, H.; Yagita, Y. Association of interleukin-6 with the progression of carotid atherosclerosis: A 9-year follow-up study. Stroke 2014, 45, 2924–2929. [Google Scholar] [CrossRef] [Green Version]
- Groot, H.E.; Al Ali, L.; van der Horst, I.C.C.; Schurer, R.A.J.; van der Werf, H.W.; Lipsic, E.; van Veldhuisen, D.J.; Karper, J.C.; van der Harst, P. Plasma interleukin 6 levels are associated with cardiac function after ST-elevation myocardial infarction. Clin. Res. Cardiol. 2018, 108, 612–621. [Google Scholar] [CrossRef] [Green Version]
- Fiordelisi, A.; Iaccarino, G.; Morisco, C.; Coscioni, E.; Sorriento, D. NFkappaB is a Key Player in the Crosstalk between Inflammation and Cardiovascular Diseases. Int. J. Mol. Sci. 2019, 20, 1599. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.H.; Zheng, X.L.; Tang, C.K. Nuclear Factor-κB Activation as a Pathological Mechanism of Lipid Metabolism and Atherosclerosis, 1st ed.; Advances in Clinical Chemistry; Elsevier Inc.: Amsterdam, The Netherlands, 2015; Volume 70, pp. 1–30. [Google Scholar]
- Daub, S.; Lutgens, E.; Münzel, T.; Daiber, A. CD40/CD40L and Related Signaling Pathways in Cardiovascular Health and Disease — The Pros and Cons for Cardioprotection. Int. J. Mol. Sci. 2020, 21, 8533. [Google Scholar] [CrossRef] [PubMed]
- Gissler, M.C.; Scherrer, P.; Anto-Michel, N.; Pennig, J.; Hoppe, N.; Füner, L.; Härdtner, C.; Stachon, P.; Li, X.; Mitre, L.S.; et al. Deficiency of Endothelial CD40 Induces a Stable Plaque Phenotype and Limits Inflammatory Cell Recruitment to Atherosclerotic Lesions in Mice. Thromb. Haemost. 2021, 121, 1530–1540. [Google Scholar] [CrossRef] [PubMed]
- Bosmans, L.A.; van Tiel, C.M.; Aarts, S.A.B.M.; Willemsen, L.; Baardman, J.; van Os, B.W.; Toom, M.D.; Beckers, L.; Ahern, D.J.; Levels, J.H.; et al. Myeloid CD40 deficiency reduces atherosclerosis by impairing macrophages’ transition into a pro-inflammatory state. Cardiovasc. Res. 2022. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, N.; Seijkens, T.; Lievens, D.; Kuijpers, M.J.; Winkels, H.; Projahn, D.; Hartwig, H.; Beckers, L.; Megens, R.T.; Boon, L.; et al. Platelet CD40 Exacerbates Atherosclerosis by Transcellular Activation of Endothelial Cells and Leukocytes. Arter. Thromb. Vasc. Biol. 2016, 36, 482–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacy, M.; Bürger, C.; Shami, A.; Ahmadsei, M.; Winkels, H.; Nitz, K.; van Tiel, C.M.; Seijkens, T.T.P.; Kusters, P.J.H.; Karshovka, E.; et al. Cell-specific and divergent roles of the CD40L-CD40 axis in atherosclerotic vascular disease. Nat. Commun. 2021, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Shami, A.; Edsfeldt, A.; Bengtsson, E.; Nilsson, J.; Shore, A.C.; Natali, A.; Khan, F.; Lutgens, E.; Gonçalves, I. Soluble CD40 Levels in Plasma Are Associated with Cardiovascular Disease and in Carotid Plaques with a Vulnerable Phenotype. J. Stroke 2021, 23, 367–376. [Google Scholar] [CrossRef]
- Li, W.; Li, H.; Zhou, L.; Wang, Z.; Hua, B. Pregnancy-Associated Plasma Protein A Induces Inflammatory Cytokine Expression by Activating IGF-I/PI3K/Akt Pathways. Mediat. Inflamm. 2019, 2019, 8436985. [Google Scholar] [CrossRef]
- Yu, X.-H.; He, L.-H.; Gao, J.-H.; Zhang, D.-W.; Zheng, X.-L.; Tang, C.-K. Pregnancy-associated plasma protein-A in atherosclerosis: Molecular marker, mechanistic insight, and therapeutic target. Atherosclerosis 2018, 278, 250–258. [Google Scholar] [CrossRef]
- Wang, G.; Liu, X.; Li, X.; Zhao, Y. Suppression of PAPP-A mitigates atherosclerosis by mediating macrophage polarization via STAT3 signaling. Biochem. Biophys. Res. Commun. 2021, 543, 29–37. [Google Scholar] [CrossRef]
- Tang, S.-L.; Zhao, Z.-W.; Liu, S.-M.; Wang, G.; Yu, X.-H.; Zou, J.; Wang, S.-Q.; Dai, X.-Y.; Fu, M.-G.; Zheng, X.-L.; et al. Pregnancy-Associated Plasma Protein-A Accelerates Atherosclerosis by Regulating Reverse Cholesterol Transport and Inflammation. Circ. J. 2019, 83, 515–523. [Google Scholar] [CrossRef] [Green Version]
- Cirillo, P.; Conte, S.; Pellegrino, G.; Ziviello, F.; Barra, G.; De Palma, R. Pregnancy-associated plasma protein-A promotes TF procoagulant activity in human endothelial cells by Akt–NF-κB axis. J. Thromb. Thrombolysis. 2016, 42, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-P.; Neradilek, M.B.; Gu, F.-S.; Isquith, D.A.; Sun, Z.-J.; Wu, X.; Li, H.-W.; Zhao, X.-Q. Pregnancy-associated plasma protein-A is a stronger predictor for adverse cardiovascular outcomes after acute coronary syndrome in type-2 diabetes mellitus. Cardiovasc. Diabetol. 2017, 16, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Myasoedova, V.A.; Chistiakov, D.A.; Grechko, A.; Orekhov, A.N. Matrix metalloproteinases in pro-atherosclerotic arterial remodeling. J. Mol. Cell. Cardiol. 2018, 123, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Wei, R.; Wang, L.; Lu, J.; Liu, H.; Zhang, W. Correlations of MMP-1, MMP-3, and MMP-12 with the degree of atherosclerosis, plaque stability and cardiovascular and cerebrovascular events. Exp. Ther. Med. 2017, 15, 1994–1998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Zhang, B.; Yan, Y.; Gao, S.; Liu, J.; Xu, L.; Hui, P. Specific matrix metalloproteinases and calcification factors are associated with the vulnerability of human carotid plaque. Exp. Ther. Med. 2018, 16, 2071–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.M.; Akkerhuis, K.M.; Meilhac, O.; Oemrawsingh, R.M.; Garcia-Garcia, H.M.; van Geuns, R.-J.; Piquer, D.; Merle, D.; du Paty, E.; Galéa, P.; et al. Circulating Osteoglycin and NGAL/MMP9 Complex Concentrations Predict 1-Year Major Adverse Cardiovascular Events After Coronary Angiography. Arter. Thromb. Vasc. Biol. 2014, 34, 1078–1084. [Google Scholar] [CrossRef] [Green Version]
- Eilenberg, W.; Stojkovic, S.; Kaider, A.; Kozakowski, N.; Domenig, C.M.; Burghuber, C. NGAL and MMP-9/NGAL as biomarkers of plaque vulnerability and targets of statins in patients with carotid atherosclerosis. Clin. Chem. Lab Med. 2017, 56, 147–156. [Google Scholar] [CrossRef]
- Opincariu, D.; Rodean, I.; Rat, N.; Hodas, R.; Benedek, I.; Benedek, T. Systemic Vulnerability, as Expressed by I-CAM and MMP-9 at Presentation, Predicts One Year Outcomes in Patients with Acute Myocardial Infarction—Insights from the VIP Clinical Study. J. Clin. Med. 2021, 10, 3435. [Google Scholar] [CrossRef]
- Macarie, R.D.; Vadana, M.; Ciortan, L.; Tucureanu, M.M.; Ciobanu, A.; Vinereanu, D. The expression of MMP-1 and MMP-9 is up-regulated by smooth muscle cells after their cross-talk with macrophages in high glucose conditions. J. Cell Mol. Med. 2018, 22, 4366–4376. [Google Scholar]
- Kremastiotis, G.; Handa, I.; Jackson, C.; George, S.; Johnson, J. Disparate effects of MMP and TIMP modulation on coronary atherosclerosis and associated myocardial fibrosis. Sci. Rep. 2021, 11, 1–16. [Google Scholar] [CrossRef]
- Maiocchi, S.L.; Ku, J.; Thai, T.; Chan, E.; Rees, M.D.; Thomas, S.R. Myeloperoxidase: A versatile mediator of endothelial dysfunction and therapeutic target during cardiovascular disease. Pharmacol. Ther. 2021, 221, 107711. [Google Scholar] [CrossRef] [PubMed]
- Döring, Y.; Libby, P.; Soehnlein, O. Neutrophil Extracellular Traps Participate in Cardiovascular Diseases: Recent Experimental and Clinical Insights. Circ. Res. 2020, 126, 1228–1241. [Google Scholar] [CrossRef] [PubMed]
- Kacprzak, M.; Zielinska, M. Prognostic value of myeloperoxidase concentration in patients with ST-segment elevation myocardial infarction treated with primary percutaneous coronary intervention. Int. J. Cardiol. 2016, 223, 452–457. [Google Scholar] [CrossRef]
- Rashid, I.; Maghzal, G.J.; Chen, Y.-C.; Cheng, D.; Talib, J.; Newington, D.; Ren, M.; Vajandar, S.; Searle, A.; Maluenda, A.; et al. Myeloperoxidase is a potential molecular imaging and therapeutic target for the identification and stabilization of high-risk atherosclerotic plaque. Eur. Hear. J. 2018, 39, 3301–3310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewing, B.; Parathath, S.; Barrett, T.; Chung, W.K.K.; Astudillo, Y.M.; Hamada, T.; Ramkhelawon, B.; Tallant, T.C.; Yusufishaq, M.S.S.; DiDonato, J.A.; et al. Effects of Native and Myeloperoxidase-Modified Apolipoprotein A-I on Reverse Cholesterol Transport and Atherosclerosis in Mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; DiDonato, J.A.; Levison, B.S.; Schmitt, D.; Li, L.; Wu, Y.; Buffa, J.; Kim, T.; Gerstenecker, G.S.; Gu, X.; et al. An abundant dysfunctional apolipoprotein A1 in human atheroma. Nat. Med. 2014, 20, 193–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Thakur, M.; van der Vorst, E.P.C.; Weber, C.; Döring, Y. Targeting the chemokine network in atherosclerosis. Atherosclerosis 2021, 330, 95–106. [Google Scholar] [CrossRef]
- Georgakis, M.K.; van der Laan, S.W.; Asare, Y.; Mekke, J.M.; Haitjema, S.; Schoneveld, A.H.; de Jager, S.C.; Nurmohamed, N.S.; Kroon, J.; Stroes, E.S.; et al. Monocyte-Chemoattractant Protein-1 Levels in Human Atherosclerotic Lesions Associate With Plaque Vulnerability. Arter. Thromb. Vasc. Biol. 2021, 41, 2038–2048. [Google Scholar] [CrossRef]
- Li, M.; Chen, Y.; Zhang, Y.; Li, D.; Liu, J. Correlation between monocyte chemoattractant protein-1/chemokine (C-C motif) ligand 2 and coronary plaque characteristics. Exp. Biol. Med. 2020, 245, 1335–1343. [Google Scholar] [CrossRef]
- Bot, I.; Zacarías, N.V.O.; De Witte, W.E.A.; De Vries, H.; Van Santbrink, P.J.; Van Der Velden, D.; Kröner, M.J.; Van Der Berg, D.-J.; Stamos, D.; De Lange, E.C.M.; et al. A novel CCR2 antagonist inhibits atherogenesis in apoE deficient mice by achieving high receptor occupancy. Sci. Rep. 2017, 7, 52. [Google Scholar] [CrossRef]
- Kiss, M.G.; Binder, C.J. The multifaceted impact of complement on atherosclerosis. Atherosclerosis 2022, 351, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Si, W.; He, P.; Wang, Y.; Fu, Y.; Li, X.; Lin, X.; Chen, F.; Cao, G.; Zhang, H. Complement Complex C5b-9 Levels Are Associated with the Clinical Outcomes of Acute Ischemic Stroke and Carotid Plaque Stability. Transl. Stroke Res. 2018, 10, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.-B.; Chen, S.-Y. Response Gene to Complement 32 in Vascular Diseases. Front. Cardiovasc. Med. 2018, 5, 128. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.-B.; Luan, J.-N.; Dong, K.; Chen, S.; Wang, Y.; Watford, W.T. RGC-32 deficiency protects endothelial cell from inflammation and attenuates atherosclerosis. Arter. Thromb. Vasc. Biol. 2018, 38, 36–47. [Google Scholar]
- Vlaicu, S.I.; Tatomir, A.; Fosbrink, M.; Nguyen, V.; Boodhoo, D.; Cudrici, C.; Badea, T.C.; Rus, V.; Rus, H. RGC-32′ dual role in smooth muscle cells and atherogenesis. Clin. Immunol. 2022, 238. [Google Scholar] [CrossRef]
- Scimeca, M.; Montanaro, M.; Cardellini, M.; Bonfiglio, R.; Anemona, L.; Urbano, N.; Bonanno, E.; Menghini, R.; Casagrande, V.; Martelli, E.; et al. High Sensitivity C-Reactive Protein Increases the Risk of Carotid Plaque Instability in Male Dyslipidemic Patients. Diagnostics 2021, 11, 2117. [Google Scholar] [CrossRef]
- Hafiane, A. Vulnerable plaque, characteristics, detection, and potential therapies. J. Cardiovasc. Dev. Dis. 2019, 6, 26. [Google Scholar] [CrossRef] [Green Version]
- Kowara, M.; Cudnoch-jedrzejewska, A. Different approaches in therapy aiming to stabilize an unstable atherosclerotic plaque. Int. J. Mol. Sci. 2021, 22, 9. [Google Scholar]
- Pereira, C.A.; Carneiro, F.S.; Matsumoto, T.; Tostes, R.C. Bonus Effects of Antidiabetic Drugs: Possible Beneficial Effects on Endothelial Dysfunction, Vascular Inflammation and Atherosclerosis. Basic Clin. Pharmacol. Toxicol. 2018, 123, 523–538. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Chen, W.; Ni, X.; Little, P.J.; Xu, S.; Tang, L.; Weng, J. Metformin, Macrophage Dysfunction and Atherosclerosis. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef]
- Sumi, M.P.; Mahajan, B.; Sattar, R.S.A.; Nimisha; Apurva; Kumar, A.; Sharma, A.K.; Ahmad, E.; Ali, A.; Saluja, S.S. Elucidation of Epigenetic Landscape in Coronary Artery Disease: A Review on Basic Concept to Personalized Medicine. Epigenetics Insights 2021, 14. [Google Scholar] [CrossRef] [PubMed]
- Carballo-Perich, L.; Puigoriol-Illamola, D.; Bashir, S.; Terceño, M.; Silva, Y.; Gubern-Mérida, C.; Serena, J. Clinical Parameters and Epigenetic Biomarkers of Plaque Vulnerability in Patients with Carotid Stenosis. Int. J. Mol. Sci. 2022, 23, 5149. [Google Scholar] [CrossRef] [PubMed]
- Aavik, E.; Babu, M.; Ylä-Herttuala, S. DNA methylation processes in atherosclerotic plaque. Atherosclerosis 2019, 281, 168–179. [Google Scholar] [CrossRef]
- Wang, X.; Liu, A.-H.; Jia, Z.-W.; Pu, K.; Chen, K.-Y.; Guo, H. Genome-wide DNA methylation patterns in coronary heart disease. Herz 2017, 43, 656–662. [Google Scholar] [CrossRef]
- Li, J.; Zhang, X.; Yang, M.; Yang, H.; Xu, N.; Fan, X. DNA methylome profiling reveals epigenetic regulation of lipoprotein-associated phospholipase A2 in human vulnerable atherosclerotic plaque. Clin. Epigenet. 2021, 13, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Choi, B.-G.; Jelinek, J.; Kim, D.H.; Lee, S.H.; Cho, K.; Rha, S.H.; Lee, Y.H.; Jin, H.S.; Choi, D.-K.; et al. Promoter methylation changes in ALOX12 and AIRE1: Novel epigenetic markers for atherosclerosis. Clin. Epigenet. 2020, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Carbone, F.; Montecucco, F.; Xu, S.; Banach, M.; Jamialahmadi, T.; Sahebkar, A. Epigenetics in atherosclerosis: Key features and therapeutic implications. Expert Opin. Ther. Targets 2020, 24, 719–721. [Google Scholar] [CrossRef]
- Wei, X.; Zhang, Y.; Xie, L.; Wang, K.; Wang, X. Pharmacological inhibition of EZH2 by GSK126 decreases atherosclerosis by modulating foam cell formation and monocyte adhesion in apolipoprotein E-deficient mice. Exp. Ther. Med. 2021, 22, 1–9. [Google Scholar] [CrossRef]
- Gorabi, A.M.; Penson, P.; Banach, M.; Motallebnezhad, M.; Jamialahmadi, T.; Sahebkar, A. Epigenetic control of atherosclerosis via DNA methylation: A new therapeutic target? Life Sci. 2020, 253, 117682. [Google Scholar] [CrossRef]
- Soler-Botija, C.; Gálvez-Montón, C.; Bayes-Genis, A. Epigenetic Biomarkers in Cardiovascular Diseases. Front. Genet. 2019, 10, 950. [Google Scholar] [CrossRef]
- Zhang, H.; Lang, Z.; Zhu, J.-K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 2018, 19, 489–506. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Kamato, D.; Little, P.J.; Nakagawa, S.; Pelisek, J.; Jin, Z.G. Targeting epigenetics and non-coding RNAs in atherosclerosis: From mechanisms to therapeutics. Pharmacol. Ther. 2018, 196, 15–43. [Google Scholar] [CrossRef]
- de Gonzalo-Calvo, D.; Iglesias-Gutiérrez, E.; Llorente-Cortés, V. Epigenetic Biomarkers and Cardiovascular Disease: Circulating MicroRNAs. Rev. Esp. Cardiol. 2017, 70, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Kaudewitz, D.; Zampetaki, A.; Mayr, M. MicroRNA Biomarkers for Coronary Artery Disease? Curr. Atheroscler. Rep. 2015, 17, 12. [Google Scholar] [CrossRef] [Green Version]
- Cipollone, F.; Felicioni, L.; Sarzani, R.; Ucchino, S.; Spigonardo, F.; Mandolini, C.; Malatesta, S.; Bucci, M.; Mammarella, C.; Santovito, D.; et al. A Unique MicroRNA Signature Associated With Plaque Instability in Humans. Stroke 2011, 42, 2556–2563. [Google Scholar] [CrossRef] [PubMed]
- Kumric, M.; Borovac, J.A.; Martinovic, D.; Kurir, T.T.; Bozic, J. Circulating biomarkers reflecting destabilization mechanisms of coronary artery plaques: Are we looking for the impossible? Biomolecules 2021, 11, 6. [Google Scholar] [CrossRef]
- Guo, J.; Ning, Y.; Su, Z.; Guo, L.; Gu, Y. Identification of hub genes and regulatory networks in histologically unstable carotid atherosclerotic plaque by bioinformatics analysis. BMC Med Genom. 2022, 15, 1–11. [Google Scholar] [CrossRef]
- Theofilis, P.; Sagris, M.; Antonopoulos, A.S.; Oikonomou, E.; Tsioufis, K.; Tousoulis, D. Non-Invasive Modalities in the Assessment of Vulnerable Coronary Atherosclerotic Plaques. Tomography 2022, 8. [Google Scholar] [CrossRef]
- Kong, A.S.-Y.; Lai, K.-S.; Lim, S.-H.E.; Sivalingam, S.; Loh, J.-Y.; Maran, S. miRNA in Ischemic Heart Disease and Its Potential as Biomarkers: A Comprehensive Review. Int. J. Mol. Sci. 2022, 23, 9001. [Google Scholar] [CrossRef]
- Taraldsen, M.D.; Wiseth, R.; Videm, V.; Bye, A.; Madssen, E. Associations between circulating microRNAs and coronary plaque characteristics: Potential impact from physical exercise. Physiol. Genom. 2022, 54, 129–140. [Google Scholar] [CrossRef]
- He, W.; Zhu, L.; Huang, Y.; Zhang, Y.; Shen, W.; Fang, L.; Li, J.; Wang, Z.; Xie, Q. The relationship of MicroRNA-21 and plaque stability in acute coronary syndrome. Medicine 2019, 98, e18049. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; de Ronde, M.W.J.; Kok, M.G.M.; Beijk, M.A.; De Winter, R.J.; van der Wal, A.C.; Sondermeijer, B.M.; Meijers, J.C.M.; Creemers, E.; Pinto-Sietsma, S.-J. MiR-223-3p and miR-122-5p as circulating biomarkers for plaque instability. Open Hear. 2020, 7, e001223. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Ma, T.; Zhang, Y.; Zhang, F.; Cui, B. Overexpressed miR-335-5p reduces atherosclerotic vulnerable plaque formation in acute coronary syndrome. J. Clin. Lab. Anal. 2020, 35, e23608. [Google Scholar] [CrossRef]
- Yu, D.; Wang, T.; Huang, J.; Fang, X.; Fan, H.; Yi, G.; Liu, Q.; Zhang, Y.; Zeng, X.; Liu, Q. MicroRNA-9 overexpression suppresses vulnerable atherosclerotic plaque and enhances vascular remodeling through negative regulation of the p38MAPK pathway via OLR1 in acute coronary syndrome. J. Cell. Biochem. 2019, 121, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Mokry, M.; Boltjes, A.; Cui, K.; Slenders, L.; Mekke, J.M.; Depuydt, M.A.C. Transcriptomic-based clustering of advanced atherosclerotic plaques identifies subgroups of plaques with differential underlying biology that associate with clinical presentation. medRxiv 2021, 2021, 21266855. [Google Scholar]
- Eberhardt, N.; Giannarelli, C. How Single-Cell Technologies Have Provided New Insights Into Atherosclerosis. Arter. Thromb. Vasc. Biol. 2022, 42, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.A.; Fernandez, D.M.; Giannarelli, C. Single cell analyses to understand the immune continuum in atherosclerosis. Atherosclerosis 2021, 330, 85–94. [Google Scholar] [CrossRef]
- Kim, K.; Shim, D.; Lee, J.S.; Zaitsev, K.; Williams, J.; Kim, K.-W.; Jang, M.-Y.; Jang, H.S.; Yun, T.J.; Lee, S.H.; et al. Transcriptome Analysis Reveals Nonfoamy Rather Than Foamy Plaque Macrophages Are Proinflammatory in Atherosclerotic Murine Models. Circ. Res. 2018, 123, 1127–1142. [Google Scholar] [CrossRef]
- Hajkarim, M.C.; Won, K.J. Single Cell RNA-Sequencing for the Study of Atherosclerosis. J. Lipid Atheroscler. 2019, 8, 152–161. [Google Scholar] [CrossRef]
- Slenders, L.; Tessels, D.E.; van der Laan, S.W.; Pasterkamp, G.; Mokry, M. The Applications of Single-Cell RNA Sequencing in Atherosclerotic Disease. Front. Cardiovasc. Med. 2022, 9. [Google Scholar] [CrossRef]
- Shen, X.; Li, L.; Sun, Z.; Zang, G.; Zhang, L.; Shao, C.; Wang, Z. Gut Microbiota and Atherosclerosis—Focusing on the Plaque Stability. Front. Cardiovasc. Med. 2021, 8, 668532. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Chang, M.; Guo, Y.; Zhang, L.; Xue, C.; Yanagita, T.; Zhang, T.; Wang, Y. Trimethylamine-N-oxide (TMAO)-induced atherosclerosis is associated with bile acid metabolism. Lipids Health Dis. 2018, 17, 21–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seldin, M.M.; Meng, Y.; Qi, H.; Zhu, W.F.; Wang, Z.; Hazen, S.L. Trimethylamine N-oxide promotes vascular inflammation through signaling of mitogen-activated protein kinase and nuclear factor-κb. J. Am. Heart Assoc. 2016, 5, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.L.; Zhu, X.H.; Ran, L.; Lang, H.D.; Yi, L.; Mi, M.T. Trimethylamine-N-oxide induces vascular inflammation by activating the NLRP3 inflammasome through the SIRT3-SOD2-mtROS signaling pathway. J. Am. Heart Assoc. 2017, 6, 9. [Google Scholar] [CrossRef]
- Yang, S.; Li, X.; Yang, F.; Zhao, R.; Pan, X.; Liang, J.; Tian, L.; Li, X.; Liu, L.; Xing, Y.; et al. Gut Microbiota-Dependent Marker TMAO in Promoting Cardiovascular Disease: Inflammation Mechanism, Clinical Prognostic, and Potential as a Therapeutic Target. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef]
- Eyileten, C.; Jarosz-Popek, J.; Jakubik, D.; Gasecka, A.; Wolska, M.; Ufnal, M.; Postula, M.; Toma, A.; Lang, I.M.; Siller-Matula, J.M. Plasma Trimethylamine-N-Oxide Is an Independent Predictor of Long-Term Cardiovascular Mortality in Patients Undergoing Percutaneous Coronary Intervention for Acute Coronary Syndrome. Front. Cardiovasc. Med. 2021, 8. [Google Scholar] [CrossRef]
- Li, X.S.; Obeid, S.; Klingenberg, R.; Gencer, B.; Mach, F.; Räber, L. Gutmicrobiota-dependent trimethylamine N-oxide in acute coronary syndromes: A prognostic marker for incident cardiovascular events beyond traditional risk factors. Eur. Heart J. 2017, 38, 814–824. [Google Scholar]
- Li, X.S.; Obeid, S.; Wang, Z.; Hazen, B.J.; Li, L.; Wu, Y. Trimethyllysine, a trimethylamine N-oxide precursor, provides near-and long-term prognostic value in patients presenting with acute coronary syndromes. Eur. Heart J. 2019, 40, 2700–2709. [Google Scholar] [CrossRef]
- Koay, Y.C.; Chen, Y.C.; Wali, J.A.; Luk, A.W.S.; Li, M.; Doma, H. Plasma levels of trimethylamine-N-oxide can be increased with “healthy” and “unhealthy” diets and do not correlate with the extent of atherosclerosis but with plaque instability. Cardiovasc. Res. 2021, 117, 435–449. [Google Scholar] [CrossRef]
- Liu, X.; Xie, Z.; Sun, M.; Wang, X.; Li, J.; Cui, J.; Zhang, F.; Yin, L.; Huang, D.; Hou, J.; et al. Plasma trimethylamine N-oxide is associated with vulnerable plaque characteristics in CAD patients as assessed by optical coherence tomography. Int. J. Cardiol. 2018, 265, 18–23. [Google Scholar] [CrossRef]
- Anto, L.; Blesso, C.N. Interplay between diet, the gut microbiome, and atherosclerosis: Role of dysbiosis and microbial metabolites on inflammation and disordered lipid metabolism. J. Nutr. Biochem. 2022, 105, 108991. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, R.; Nocella, C.; Petrozza, V.; Cammisotto, V.; Pacini, L.; Sorrentino, V.; Martinelli, O.; Irace, L.; Sciarretta, S.; Frati, G.; et al. Localization of lipopolysaccharide from Escherichia Coli into human atherosclerotic plaque. Sci. Rep. 2018, 8, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, N.; Emoto, T.; Yamashita, T.; Watanabe, H.; Hayashi, T.; Tabata, T.; Hoshi, N.; Hatano, N.; Ozawa, G.; Sasaki, N.; et al. Bacteroides vulgatus and Bacteroides dorei Reduce Gut Microbial Lipopolysaccharide Production and Inhibit Atherosclerosis. Circulation 2018, 138, 2486–2498. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, E.; Leonel, A.; Teixeira, L.; Silva, A.; Silva, J.; Pelaez, J.; Capettini, L.; Lemos, V.; Santos, R.; Alvarez-Leite, J. Butyrate impairs atherogenesis by reducing plaque inflammation and vulnerability and decreasing NFκB activation. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, E.C.; dos Santos, L.C.; Leonel, A.J.; de Oliveira, J.S.; Santos, E.A.; Navia-Pelaez, J.M.; da Silva, J.F.; Mendes, B.P.; Capettini, L.S.; Teixeira, L.G.; et al. Oral butyrate reduces oxidative stress in atherosclerotic lesion sites by a mechanism involving NADPH oxidase down-regulation in endothelial cells. J. Nutr. Biochem. 2016, 34, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Nemet, I.; Saha, P.P.; Gupta, N.; Zhu, W.; Romano, K.A.; Skye, S.M.; Cajka, T.; Mohan, M.L.; Li, L.; Wu, Y.; et al. A Cardiovascular Disease-Linked Gut Microbial Metabolite Acts via Adrenergic Receptors. Cell 2020, 180, 862–877.e22. [Google Scholar] [CrossRef]
- Salnikova, D.; Orekhova, V.; Grechko, A.; Starodubova, A.; Bezsonov, E.; Popkova, T.; Orekhov, A. Mitochondrial Dysfunction in Vascular Wall Cells and Its Role in Atherosclerosis. Int. J. Mol. Sci. 2021, 22, 8990. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Orekhov, A.N.; Poznyak, A.; Sobenin, I.A.; Nikifirov, N.N.; Ivanova, E.A. Mitochondrion as a Selective Target for the Treatment of Atherosclerosis: Role of Mitochondrial DNA Mutations and Defective Mitophagy in the Pathogenesis of Atherosclerosis and Chronic Inflammation. Curr. Neuropharmacol. 2020, 18, 1064–1075. [Google Scholar] [CrossRef]
- Marchio, P.; Guerra-Ojeda, S.; Vila, J.M.; Aldasoro, M.; Victor, V.M.; Mauricio, M.D. Targeting Early Atherosclerosis: A Focus on Oxidative Stress and Inflammation. Oxid. Med. Cell Longev. 2019, 2019, 8563845. [Google Scholar] [CrossRef] [Green Version]
- Mollazadeh, H.; Tavana, E.; Fanni, G.; Bo, S.; Banach, M.; Pirro, M.; von Haehling, S.; Jamialahmadi, T.; Sahebkar, A. Effects of statins on mitochondrial pathways. J. Cachex- Sarcopenia Muscle 2021, 12, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Bharath, L.P.; Nikolajczyk, B.S. The intersection of metformin and inflammation. Am. J. Physiol. -Cell Physiol. 2021, 320, C873–C879. [Google Scholar] [CrossRef] [PubMed]
- Ben-Meir, A.; Burstein, E.; Borrego-Alvarez, A.; Chong, J.; Wong, E.; Yavorska, T.; Naranian, T.; Chi, M.; Wang, Y.; Bentov, Y.; et al. Coenzyme Q10 restores oocyte mitochondrial function and fertility during reproductive aging. Aging Cell 2015, 14, 887–895. [Google Scholar] [CrossRef] [Green Version]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siasos, G.; Tousoulis, D.; Antoniades, C.; Stefanadi, E.; Stefanadis, C. l-Arginine, the substrate for NO synthesis: An alternative treatment for premature atherosclerosis? Int. J. Cardiol. 2007, 116, 300–308. [Google Scholar] [CrossRef]
- Fernando, V.; Zheng, X.; Walia, Y.; Sharma, V.; Letson, J.; Furuta, S. S-Nitrosylation: An Emerging Paradigm of Redox Signaling. Antioxidants 2019, 8, 404. [Google Scholar] [CrossRef] [Green Version]
- Bibli, S.I.; Hu, J.; Leisegang, M.S.; Wittig, J.; Zukunft, S.; Kapasakalidi, A. Shear stress regulates cystathionine γ lyase expression to preserve endothelial redox balance and reduce membrane lipid peroxidation: Regulation of CSE by KLF2 and miR-27b. Redox. Biol. 2020, 28, 101379. [Google Scholar] [CrossRef]
- Ciccone, V.; Genah, S.; Morbidelli, L. Endothelium as a Source and Target of H2S to Improve Its Trophism and Function. Antioxidants 2021, 10, 486. [Google Scholar] [CrossRef]
- Bibli, S.-I.; Fleming, I. Oxidative Post-Translational Modifications: A Focus on Cysteine S-Sulfhydration and the Regulation of Endothelial Fitness. Antioxidants Redox Signal. 2021, 35, 1494–1514. [Google Scholar] [CrossRef]
- Navas-Carrillo, D.; Marín, F.; Valdés, M.; Orenes-Piñero, E. Deciphering acute coronary syndrome biomarkers: High-resolution proteomics in platelets, thrombi and microparticles. Crit. Rev. Clin. Lab. Sci. 2016, 54, 49–58. [Google Scholar] [CrossRef]
- Ward, L.J.; Olausson, P.; Li, W.; Yuan, X.-M. Proteomics and multivariate modelling reveal sex-specific alterations in distinct regions of human carotid atheroma. Biol. Sex Differ. 2018, 9, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholls, S.J.; Kastelein, J.J.P.; Schwartz, G.G.; Bash, D.; Rosenson, R.S.; Cavender, M.A. Varespladib and cardiovascular events in patients with an acute coronary syndrome: The VISTA-16 randomized clinical trial. JAMA -J. Am. Med. Assoc. 2014, 311, 252–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langley, S.R.; Willeit, K.; Didangelos, A.; Matic, L.P.; Skroblin, P.; Barallobre-Barreiro, J. Extracellular matrix proteomics identifies molecular signature of symptomatic carotid plaques. J. Clin. Investig. 2017, 127, 1546–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.F.; Herrington, D.M. The function of cathepsins B, D, and X in atherosclerosis. Am. J. Cardiovasc. Dis. 2016, 6, 163–170. [Google Scholar] [PubMed]
- Mohammadpour, A.H.; Salehinejad, Z.; Elyasi, S.; Mouhebati, M.; Mirhafez, S.R.; Samadi, S.; Ghayour-Mobarhan, M.; Ferns, G.; Sahebkar, A. Evaluation of serum cathepsin D concentrations in coronary artery disease. Indian Hear. J. 2018, 70, 471–475. [Google Scholar] [CrossRef]
- Gonçalves, I.; Hultman, K.; Dunér, P.; Edsfeldt, A.; Hedblad, B.; Fredrikson, G.N.; Björkbacka, H.; Nilsson, J.; Bengtsson, E. High levels of cathepsin D and cystatin B are associated with increased risk of coronary events. Open Hear. 2016, 3, e000353. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Chen, B.; Zhang, X.; Tan, L.; Wang, D.W. Increased Cathepsin D Correlates with Clinical Parameters in Newly Diagnosed Type 2 Diabetes. Dis. Markers 2017, 2017, 1–6. [Google Scholar] [CrossRef]
- Li, T.; Li, X.; Feng, Y.; Dong, G.; Wang, Y.; Yang, J. The Role of Matrix Metalloproteinase-9 in Atherosclerotic Plaque Instability. Mediat. Inflamm. 2020, 2020, 1–13. [Google Scholar] [CrossRef]
- Konstantino, Y.; Nguyen, T.T.; Wolk, R.; Aiello, R.J.; Terra, S.G.; Fryburg, D.A. Potential implications of matrix metalloproteinase-9 in assessment and treatment of coronary artery disease. Biomarkers 2009, 14, 118–129. [Google Scholar] [CrossRef]
- Gao, Z.; Liu, Z.; Wang, R.; Zheng, Y.; Li, H.; Yang, L. Galectin-3 Is a Potential Mediator for Atherosclerosis. J. Immunol. Res. 2020, 2020, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Gullestad, L.; Ueland, T.; Kjekshus, J.; Nymo, S.H.; Hulthe, J.; Muntendam, P.; Adourian, A.; Böhm, M.; van Veldhuisen, D.J.; Komajda, M.; et al. Galectin-3 predicts response to statin therapy in the Controlled Rosuvastatin Multinational Trial in Heart Failure (CORONA). Eur. Hear. J. 2012, 33, 2290–2296. [Google Scholar] [CrossRef] [Green Version]
- Chiorescu, R.M.; Lazar, R.-D.; Buksa, S.-B.; Mocan, M.; Blendea, D. Biomarkers of Volume Overload and Edema in Heart Failure With Reduced Ejection Fraction. Front. Cardiovasc. Med. 2022, 9. [Google Scholar] [CrossRef]
- Lax, A.; Sanchez-Mas, J.; Asensio-Lopez, M.C.; Fernandez-Del Palacio, M.J.; Caballero, L.; Garrido, I.P. Mineralocorticoid receptor antagonists modulate galectin-3 and interleukin-33/ST2 signaling in left ventricular systolic dysfunction after acute myocardial infarction. JACC Hear Fail. 2015, 3, 50–58. [Google Scholar] [CrossRef]
- Watson, A.M.; Li, J.; Samijono, D.; Bierhaus, A.; Thomas, M.C.; Jandeleit-Dahm, K.A.; Cooper, M.E. Quinapril treatment abolishes diabetes-associated atherosclerosis in RAGE/apolipoprotein E double knockout mice. Atherosclerosis 2014, 235, 444–448. [Google Scholar] [CrossRef]
- Qian, X.; Li, M.; Wagner, M.B.; Chen, G.; Song, X. Doxazosin Stimulates Galectin-3 Expression and Collagen Synthesis in HL-1 Cardiomyocytes Independent of Protein Kinase C Pathway. Front. Pharmacol. 2016, 7, 495. [Google Scholar] [CrossRef] [Green Version]
- Aukrust, P.; Halvorsen, B.; Ueland, T.; E Michelsen, A.; Skjelland, M.; Gullestad, L.; Yndestad, A.; Otterdal, K. Activated platelets and atherosclerosis. Expert Rev. Cardiovasc. Ther. 2010, 8, 1297–1307. [Google Scholar] [CrossRef]



| Mechanism of Plaque Vulnerability | CTCA | PET Techniques |
|---|---|---|
| Inflammatory cell infiltration (macrophage activation) | Fat attenuation index (FAI) CTCA Radiomics [36] Radiotranscriptomics | 18F-fluorodeoxyglucose (18F-FDG) |
| 68Ga-somatostatin receptor subtype 2 (68Ga-DOTATATE) [37] | ||
| 11C-translocator protein (11C-PK11195) | ||
| 18F-fluorocholine (18F–FCH) | ||
| FRP CTCA | 89Zr-DFO-Gal3-F(ab’)2 mAb [38] | |
| In-DOTA-JR11PET/CT [39] | ||
| Neo-angiogenesis | 18F-glycoprotein IIb/IIIa platelet receptor (18F-GP1) | |
| Hypoxia | 18F-fluoromisonidazole (18F-FMISO) | |
| 18F-HX4 | ||
| Apoptosis | ‘Napkin-ring sign’ on CCTA | 18F-annexin V |
| Calcification and microcalcification | Spotty calcification on CCTA | 18F-NaF and 18F-FDG hybrid PET and MR [40] |
| Hemodynamics and shear stress | CFD CCTA | 18F-NaF PET [40] |
| Intraplaque hemorrhage | FLECT NIR-AF [41] | 18F-NaF PET [40] |
| CCTA, coronary computed tomography angiography; CFD, computational flow dynamics; FLECT, fluorescence emission computed tomography; NIR-AF, Near infrared auto-fluorescence; PET, positron emission tomography; 11c, carbon-11; 68Ga, gallium-68; 18F, fluorine-18; 18F-NaF, 18F-sodium fluoride; 124i, iodine-124. | ||
| PLATELET ACTIVATION | Upregulated with high affinity to collagenous structures in the core region of the atherosclerotic plaque |
| Glycoprotein VI receptor | |
| αIIbβ3 receptor | |
| SIGNAL PATHWAYS | Over-expressed—inducing cell growth, survival, adhesion, invasion, and migration |
| Integrin-linked kinase | |
| Sarcoma protooncogene tyrosine-protein kinase | |
| CYTOSKELETON-ASSOCIATED PROTEINS | Downregulated in human atherosclerotic plaques, which indicates the tendency to limit the platelet activity |
| Talin | |
| Vinculin | |
| A and β tubulins | |
| Vimentin | |
| F- actin capping | |
| ENERGY METABOLISM-RELATED PROTEINS | |
| Glyceraldehyde-3-phosphate dehydrogenase | |
| Pyruvate kinase | |
| Lactat dehydrogenase | |
| ANTIOXIDANT PROTEINS | Lower expression that facilitates platelet activation and allows advanced oxidation protein products to maintain chronic inflammation |
| Glutathione-S-transferase | |
| Manganese superoxidase dismutase |
| Biomarker. | Study Conclusion | References |
|---|---|---|
| hs-CRP | Hs-CRP levels correlated with carotid plaque instability in dyslipidemic male patients. | Scimeca M. 2021 [117] |
| sCD40/CD40-L | SCD40 levels were correlated with the severity of carotid atherosclerosis and future cardiovascular events; intra-plaque levels of sCD40 were associated with vulnerable plaque properties. | Shami A. 2021 [87] |
| MMPs | MMP-1, MMP-3, and MMP-12 levels were positively associated with vulnerable plaque phenotype and the occurrence of major acute vascular events in patients with carotid atherosclerotic disease. Circulating MMP-9/NGAL complex levels and NGAL levels were associated with vulnerable atherosclerotic plaques in patients with carotid artery stenosis. | Hu w. 2017 [95] Eilenberg W. 2017 [98] |
| MPO | MPO activity was associated with features of vulnerable plaques in Apo E−/− mice. MPO was suggested to be a pharmacological target against atherosclerosis and a marker for non-invasive imaging of high-risk unstable plaques. | Rashid I. 2018 [105] |
| MCP-1 | MCP-1 plaque levels were associated with histopathological, molecular, and clinical characteristics of vulnerable plaques in carotid endarterectomy samples. | Georgakis MK. 2021 [109] |
| C5b-9 | C5b-9 correlated with the stability, severity, and outcome of carotid lesions in patients with acute ischemic stroke. | Si W. 2019. [113] |
| TMAO | In a tandem stenosis mouse model, TMAO levels were correlated with several features of plaque instability regarding inflammation, platelets activations, and intraplaque hemorrhage. | Koay YC. 2021 [160] |
| Cathepsin D | Cathepsin D serum concentrations were associated with the presence of coronary artery disease. Among other makers, cathepsin D was correlated with the high-risk profile of atherosclerotic plaque from endarterectomy samples. | Mohammadpour AH. 2018. [186] Langley SR. 2018 [184] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiorescu, R.M.; Mocan, M.; Inceu, A.I.; Buda, A.P.; Blendea, D.; Vlaicu, S.I. Vulnerable Atherosclerotic Plaque: Is There a Molecular Signature? Int. J. Mol. Sci. 2022, 23, 13638. https://doi.org/10.3390/ijms232113638
Chiorescu RM, Mocan M, Inceu AI, Buda AP, Blendea D, Vlaicu SI. Vulnerable Atherosclerotic Plaque: Is There a Molecular Signature? International Journal of Molecular Sciences. 2022; 23(21):13638. https://doi.org/10.3390/ijms232113638
Chicago/Turabian StyleChiorescu, Roxana Mihaela, Mihaela Mocan, Andreea Ioana Inceu, Andreea Paula Buda, Dan Blendea, and Sonia Irina Vlaicu. 2022. "Vulnerable Atherosclerotic Plaque: Is There a Molecular Signature?" International Journal of Molecular Sciences 23, no. 21: 13638. https://doi.org/10.3390/ijms232113638