
Insights of Fe2O3 and MoO3 Electrodes for Electrocatalytic CO2 Reduction in Aprotic Media

Abstract
:1. Introduction
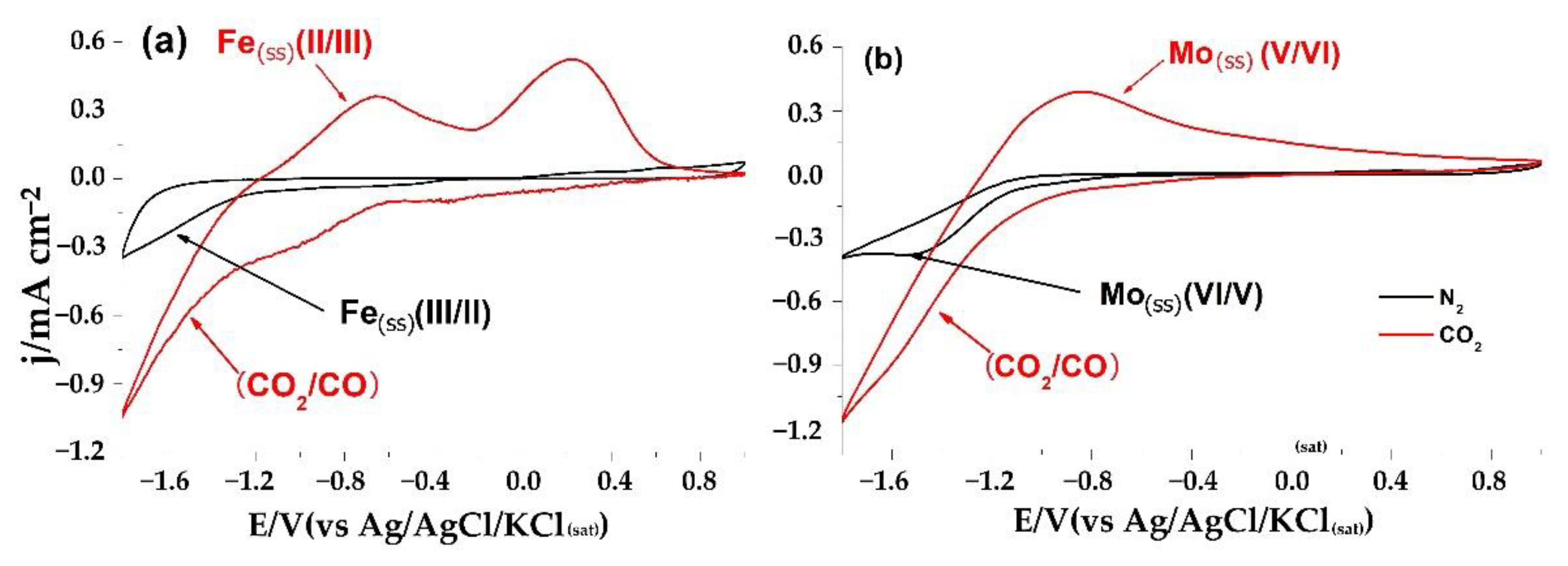
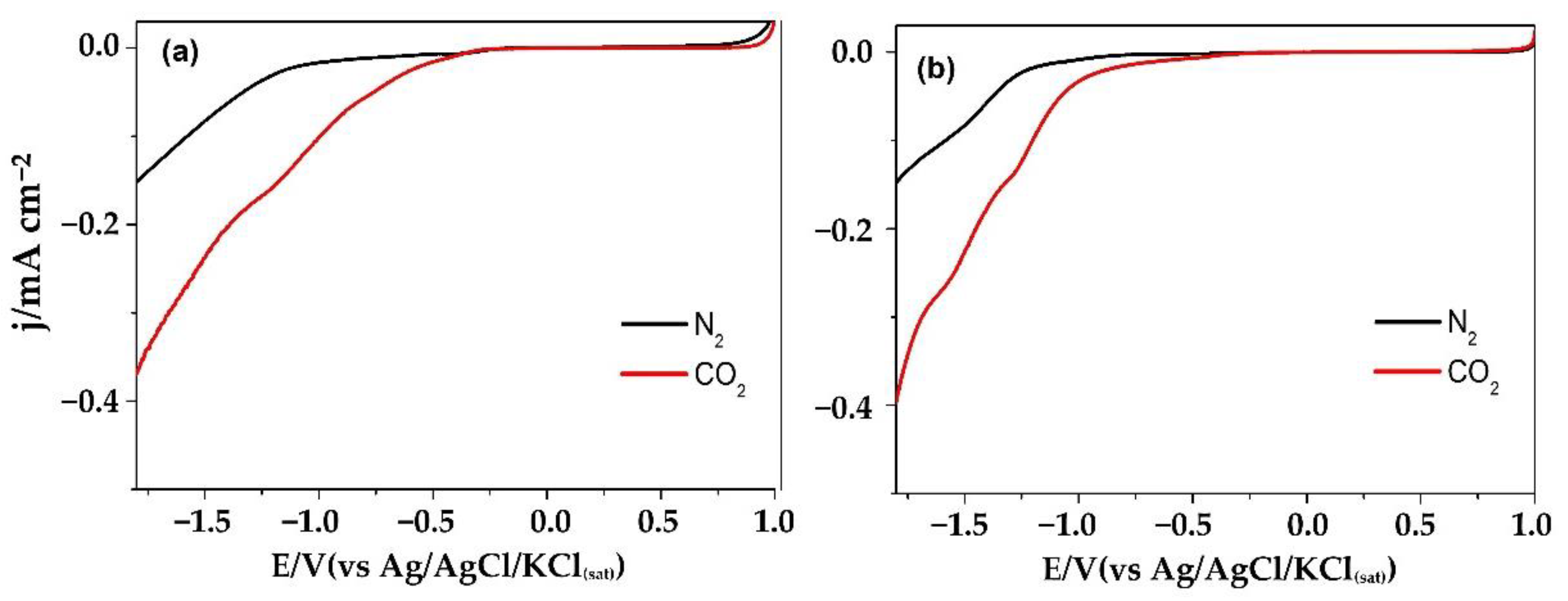
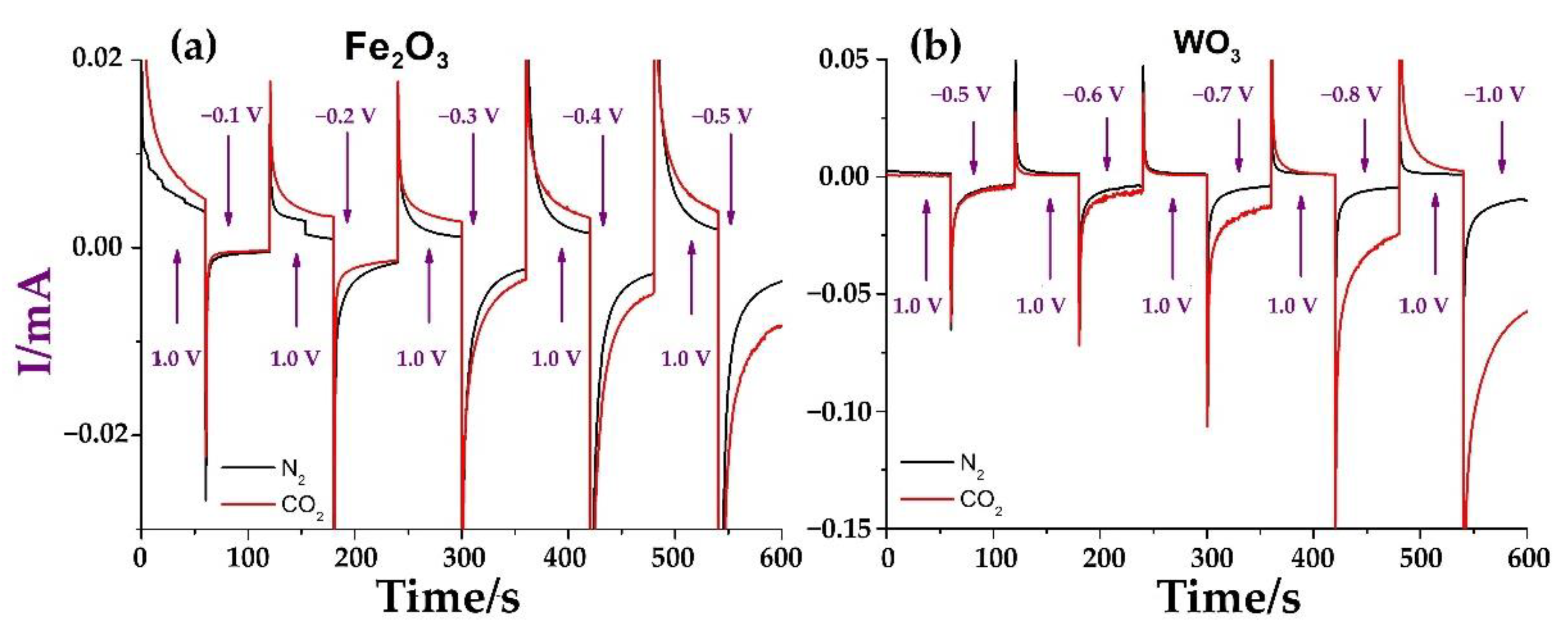
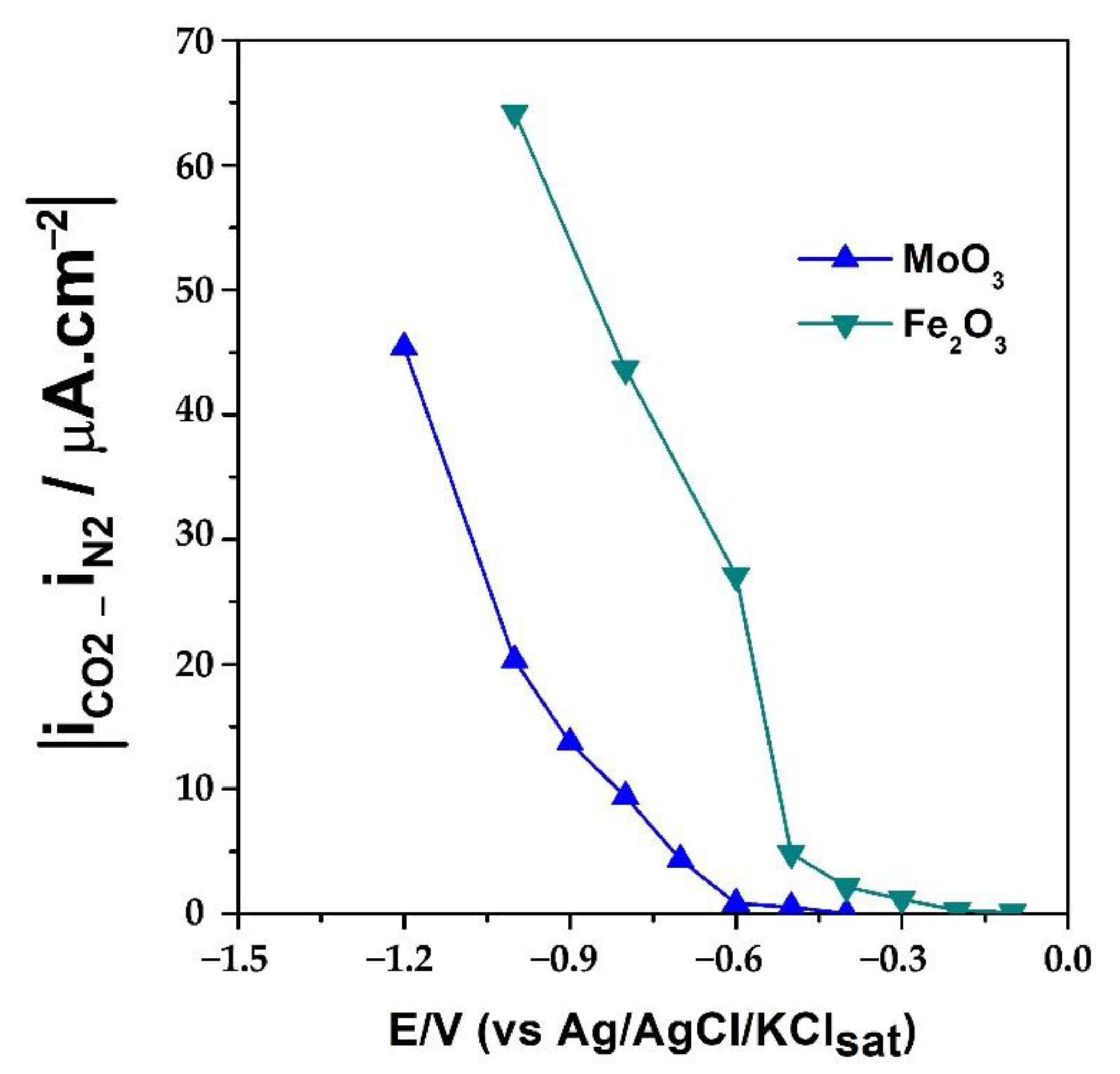
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sinsel, S.R.; Riemke, R.L.; Hoffmann, V.H. Challenges and solution technologies for the integration of variable renewable energy sources—A review. Renew. Energy 2020, 145, 2271–2285. [Google Scholar] [CrossRef]
- Kondratenko, E.V.; Mul, G.; Baltrusaitis, J.; Larrazábal, G.O.; Pérez-Ramírez, J. Status and perspectives of CO2 conversion into fuels and chemicals by catalytic, photocatalytic and electrocatalytic processes. Energy Environ. Sci. 2013, 6, 3112–3135. [Google Scholar] [CrossRef] [Green Version]
- Lv, J.; Yin, R.; Zhou, L.; Li, J.; Kikas, R.; Xu, T.; Wang, Z.; Jin, H.; Wang, X.; Wang, S. Microenvironment Engineering for the Electrocatalytic CO2 Reduction Reaction. Angew. Chem. Int. Ed. 2022, 61, e202207252. [Google Scholar] [CrossRef] [PubMed]
- Moura de Salles Pupo, M.; Kortlever, R. Electrolyte Effects on the Electrochemical Reduction of CO2. ChemPhysChem 2019, 20, 2926–2935. [Google Scholar] [CrossRef] [Green Version]
- Ait Ahsaine, H.; Zbair, M.; BaQais, A.; Arab, M. CO2 Electroreduction over Metallic Oxide, Carbon-Based, and Molecular Catalysts: A Mini-Review of the Current Advances. Catalysts 2022, 12, 450. [Google Scholar] [CrossRef]
- Yang, J.H.; Park, S.J.; Rhee, C.K.; Sohn, Y. Photocatalytic CO2 Reduction and Electrocatalytic H2 Evolution over Pt(0,II,IV)-Loaded Oxidized Ti Sheets. Nanomaterials 2020, 10, 1909. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, Y.; Zhu, H.; Suo, B. High-Performance of Electrocatalytic CO2 Reduction on Defective Graphene-Supported Cu4S2 Cluster. Catalysts 2022, 12, 454. [Google Scholar] [CrossRef]
- Tackett, B.M.; Gomez, E.; Chen, J.G. Net reduction of CO2 via its thermocatalytic and electrocatalytic transformation reactions in standard and hybrid processes. Nat. Catal. 2019, 2, 381–386. [Google Scholar] [CrossRef]
- Xu, S.; Carter, E.A. Theoretical Insights into Heterogeneous (Photo)electrochemical CO2 Reduction. Chem. Rev. 2018, 119, 6631–6669. [Google Scholar] [CrossRef]
- Freund, H.J.; Roberts, M. Surface chemistry of carbon dioxide. Surf. Sci. Rep. 1996, 25, 225–273. [Google Scholar] [CrossRef]
- Indrakanti, V.P.; Kubicki, J.D.; Schobert, H.H. Photoinduced activation of CO2 on Ti-based heterogeneous catalysts: Current state, chemical physics-based insights and outlook. Energy Environ. Sci. 2009, 2, 745–758. [Google Scholar] [CrossRef]
- Lamy, E.; Nadjo, L.; Savéant, J.M. Standard potential and kinetic parameters of the electrochemical reduction of carbon dioxide in dimethylformamide. J. Electroanal. Chem. Interfacial Electrochem. 1977, 78, 403–407. [Google Scholar] [CrossRef]
- Koppenol, W. Oxyradical reactions: From bond-dissociation energies to reduction potentials. FEBS Lett. 1990, 264, 165–167. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.S.A.; Sufyan Javed, M.; Najam, T.; Molochas, C.; Khan, N.A.; Nazir, M.A.; Xu, M.; Tsiakaras, P.; Bao, S.J. Metal oxides for the electrocatalytic reduction of carbon dioxide: Mechanism of active sites, composites, interface and defect engineering strategies. Coord. Chem. Rev. 2022, 471, 214716. [Google Scholar] [CrossRef]
- Rasko, J.; Solymosi, F. Infrared Spectroscopic Study of the Photoinduced Activation of CO2 on TiO2 and Rh/TiO2 Catalysts. J. Phys. Chem. 1994, 98, 7147–7152. [Google Scholar] [CrossRef]
- Perissinotti, L.L.; Brusa, M.A.; Grela, M.A. Yield of Carboxyl Anion Radicals in the Photocatalytic Degradation of Formate over TiO2 Particles. Langmuir 2001, 17, 8422–8427. [Google Scholar] [CrossRef]
- Habisreutinger, S.N.; Schmidt-Mende, L.; Stolarczyk, J.K. Photocatalytic Reduction of CO2 on TiO2 and Other Semiconductors. Angew. Chem. Int. Ed. 2013, 52, 7372–7408. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, T.; Singh, A.R.; Nørskov, J.K. Acetonitrile Transition Metal Interfaces from First Prin-ciples. J. Phys. Chem. Lett. 2020, 11, 9802–9811. [Google Scholar] [CrossRef]
- Gomes, R.J.; Birch, C.; Cencer, M.M.; Li, C.; Son, S.B.; Bloom, I.D.; Assary, R.S.; Amanchukwu, C.V. Probing Electrolyte Influence on CO2 Reduction in Aprotic Solvents. J. Phys. Chem. C 2022, 126, 13595–13606. [Google Scholar] [CrossRef]
- Kuntyi, R.; Zozulya, G.; Shepida, M. CO2 Electroreduction in Organic Aprotic Solvents: A Mini Review. J. Chem. 2022, 2022, 1306688. [Google Scholar] [CrossRef]
- Bandi, A. Electrochemical Reduction of Carbon Dioxide on Conductive Metallic Oxides. J. Electrochem. Soc. 1990, 137, 2157–2160. [Google Scholar] [CrossRef]
- Chu, S.; Yan, X.; Choi, C.; Hong, S.; Robertson, A.W.; Masa, J.; Han, B.; Jung, Y.; Sun, Z. Stabilization of Cu+ by tuning a CuO–CeO2 interface for selective electrochemical CO2 reduction to ethylene. Green Chem. 2020, 22, 6540–6546. [Google Scholar] [CrossRef]
- Lum, Y.; Ager, J.W. Stability of Residual Oxides in Oxide-Derived Copper Catalysts for Electrochemical CO2 Reduction Investigated with 18O Labeling. Angew. Chem. Int. Ed. 2017, 57, 551–554. [Google Scholar] [CrossRef]
- Zou, Y.; Wang, S. An Investigation of Active Sites for electrochemical CO2 Reduction Reactions: From In Situ Characterization to Rational Design. Adv. Sci. 2021, 8, 2003579. [Google Scholar] [CrossRef]
- Figueiredo, M.C.; Ledezma-Yanez, I.; Koper, M.T.M. In Situ Spectroscopic Study of CO2 Electroreduction at Copper Electrodes in Acetonitrile. ACS Catal. 2016, 6, 2382–2392. [Google Scholar] [CrossRef]
- Rudnev, A.V.; Zhumaev, U.E.; Kuzume, A.; Vesztergom, S.; Furrer, J.; Broekmann, P.; Wandlowski, T. (The promoting effect of water on the electroreduction of CO2 in acetonitrile. Electrochim. Acta 2016, 189, 38–44. [Google Scholar] [CrossRef]
- de Castro, I.A.; Datta, R.S.; Ou, J.Z.; Castellanos-Gomez, A.; Sriram, S.; Daeneke, T.; Kalantar-Zadeh, K. Molybdenum Oxides—From Fundamentals to Functionality. Adv. Mater. 2017, 29, 1701619. [Google Scholar] [CrossRef]
- Mendieta-Reyes, N.E.; Díaz-García, A.K.; Gómez, R. Simultaneous Electrocatalytic CO2 Reduction and Enhanced Electrochromic Effect at WO3 Nanostructured Electrodes in Acetonitrile. ACS Catal. 2018, 8, 1903–1912. [Google Scholar] [CrossRef] [Green Version]
- Masetti, E.; Dini, D.; Decker, F. The electrochromic response of tungsten bronzes MxWO3 with different ions and insertion rates. Sol. Energy Mater. Sol. Cells 1995, 39, 301–307. [Google Scholar] [CrossRef]
- Foley, J.K.; Korzeniewski, C.; Pons, S. Anodic and cathodic reactions in acetonitrile/tetra-n-butylammonium tetrafluoroborate: An electrochemical and infrared spectroelectrochemical study. Can. J. Chem. 1988, 66, 201–206. [Google Scholar] [CrossRef]
- Hepel, M.; Wickham, D. Large Cation Model of Dissociative Reduction of WO3−x Lattice Studied by EQCN and AFM. ECS Trans. 2009, 19, 11–23. [Google Scholar] [CrossRef]
- Chava, R.K.; Son, N.; Kang, M. Band structure alignment transitioning strategy for the fabrication of efficient photocatalysts for solar fuel generation and environmental remediation applications. J. Colloid Interface Sci. 2022, 627, 247–260. [Google Scholar] [CrossRef]
- Jian, J.; Sun, J. A Review of Recent Progress on Silicon Carbide for Photoelectrochemical Water Splitting. Sol. RRL 2020, 4, 2000111. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Liu, Z.P.; Yang, W.M. Proton-Promoted Electron Transfer in Photocatalysis: Key Step for Photocatalytic Hydrogen Evolution on Metal/Titania Composites. ACS Catal. 2017, 7, 2744–2752. [Google Scholar] [CrossRef]
- Ismael, M. Highly effective ruthenium-doped TiO2 nanoparticles photocatalyst for visible-light-driven photocatalytic hydrogen production. New J. Chem. 2019, 43, 9596–9605. [Google Scholar] [CrossRef]
- Breyer, C.; Reichert, D.; Seidel, J.; Hüttl, R.; Mertens, F.; Kureti, S. Kinetic modeling of the adsorption and desorption of CO2 on α-Fe2O3. Phys. Chem. Chem. Phys. 2015, 17, 27011–27018. [Google Scholar] [CrossRef]
- Liu, L.; Lin, M.; Liu, Z.; Sun, H.; Zhao, X. Density functional theory study of CO2 and H2O adsorption on a monoclinic WO3(001) surface. Chem. Res. Chin. Univ. 2017, 33, 255–260. [Google Scholar] [CrossRef]
- Wicke, E.J.R., Anderson, M., Boudart. (Eds.): Catalysis, Science and Technology. Vol. 4, Springer-Verlag, Berlin, Heidelberg, New York 1983. 289 Seiten, Preis: DM 132,-. Ber. Bunsenges. Phys. Chem. 1984, 88, 586. [CrossRef]
- Rao, C.N.R. Transition Metal Oxides. Annu. Rev. Phys. Chem. 1989, 40, 291–326. [Google Scholar] [CrossRef]
- Di Valentin, C.; Pacchioni, G.; Selloni, A. Reduced and n-Type Doped TiO2: Nature of Ti3+ Species. J. Phys. Chem. C 2009, 113, 20543–20552. [Google Scholar] [CrossRef]
- Cheng, H.; Selloni, A. Energetics and diffusion of intrinsic surface and subsurface defects on anatase TiO2 (101). J. Chem. Phys. 2009, 131, 054703. [Google Scholar] [CrossRef] [PubMed]
- Le, H.M.; Vu, N.H.; Phan, B.T. Migrations of oxygen vacancy in tungsten oxide (WO3): A density functional theory study. Comput. Mater. Sci. 2014, 90, 171–176. [Google Scholar] [CrossRef]
- Yin, W.J.; Wen, B.; Zhou, C.; Selloni, A.; Liu, L.M. Excess electrons in reduced rutile and anatase TiO2. Surf. Sci. Rep. 2018, 73, 58–82. [Google Scholar] [CrossRef] [Green Version]
- Hasani-Sadrabadi, M.M.; Dashtimoghadam, E.; Nasseri, R.; Karkhaneh, A.; Majedi, F.S.; Mokarram, N.; Renaud, P.; Jacob, K.I. Cellulose nanowhiskers to regulate the microstructure of perfluorosulfonate ionomers for high-performance fuel cells. J. Mater. Chem. A 2014, 2, 11334. [Google Scholar] [CrossRef]
- Pavlishchuk, V.V.; Addison, A.W. Conversion constants for redox potentials measured versus different reference electrodes in acetonitrile solutions at 25 °C. Inorg. Chim. Acta 2000, 298, 97–102. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
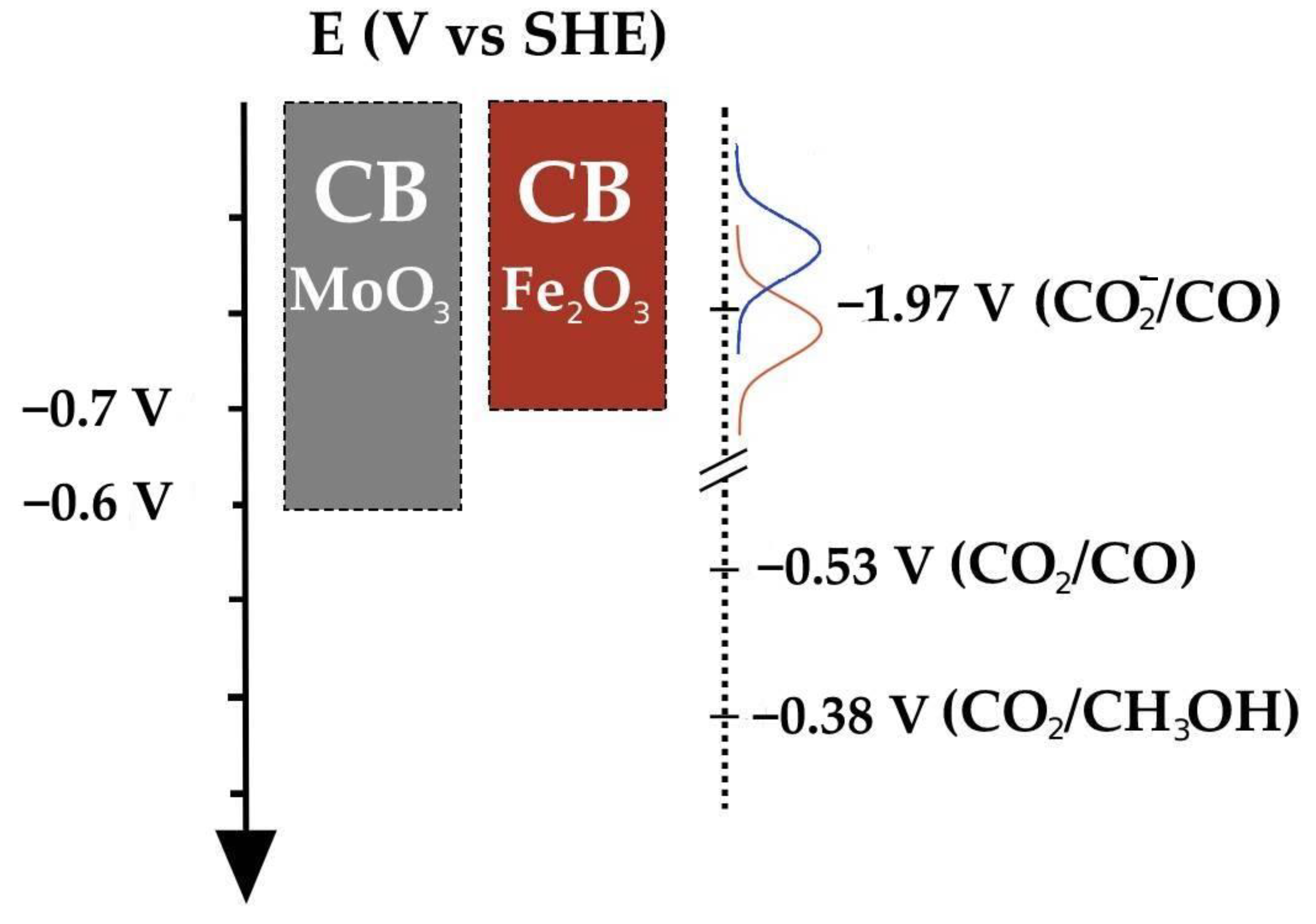{kind=link}
| Electrode | E/V (Efb) | E/V (Efi) |
|---|---|---|
| Fe2O3 | −0.70 | −0.4 |
| WO3 | −0.50 | −0.6 |
| MoO3 | −0.60 | −0.6 |
| TiO2 | −1.55 | −1.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendieta-Reyes, N.E.; Lozano-Pérez, A.S.; Guerrero-Fajardo, C.A. Insights of Fe2O3 and MoO3 Electrodes for Electrocatalytic CO2 Reduction in Aprotic Media. Int. J. Mol. Sci. 2022, 23, 13367. https://doi.org/10.3390/ijms232113367
Mendieta-Reyes NE, Lozano-Pérez AS, Guerrero-Fajardo CA. Insights of Fe2O3 and MoO3 Electrodes for Electrocatalytic CO2 Reduction in Aprotic Media. International Journal of Molecular Sciences. 2022; 23(21):13367. https://doi.org/10.3390/ijms232113367
Chicago/Turabian StyleMendieta-Reyes, Néstor E., Alejandra S. Lozano-Pérez, and Carlos A. Guerrero-Fajardo. 2022. "Insights of Fe2O3 and MoO3 Electrodes for Electrocatalytic CO2 Reduction in Aprotic Media" International Journal of Molecular Sciences 23, no. 21: 13367. https://doi.org/10.3390/ijms232113367