
The Role of Tβ4-POP-Ac-SDKP Axis in Organ Fibrosis

{kind=link}
{kind=link}
Abstract
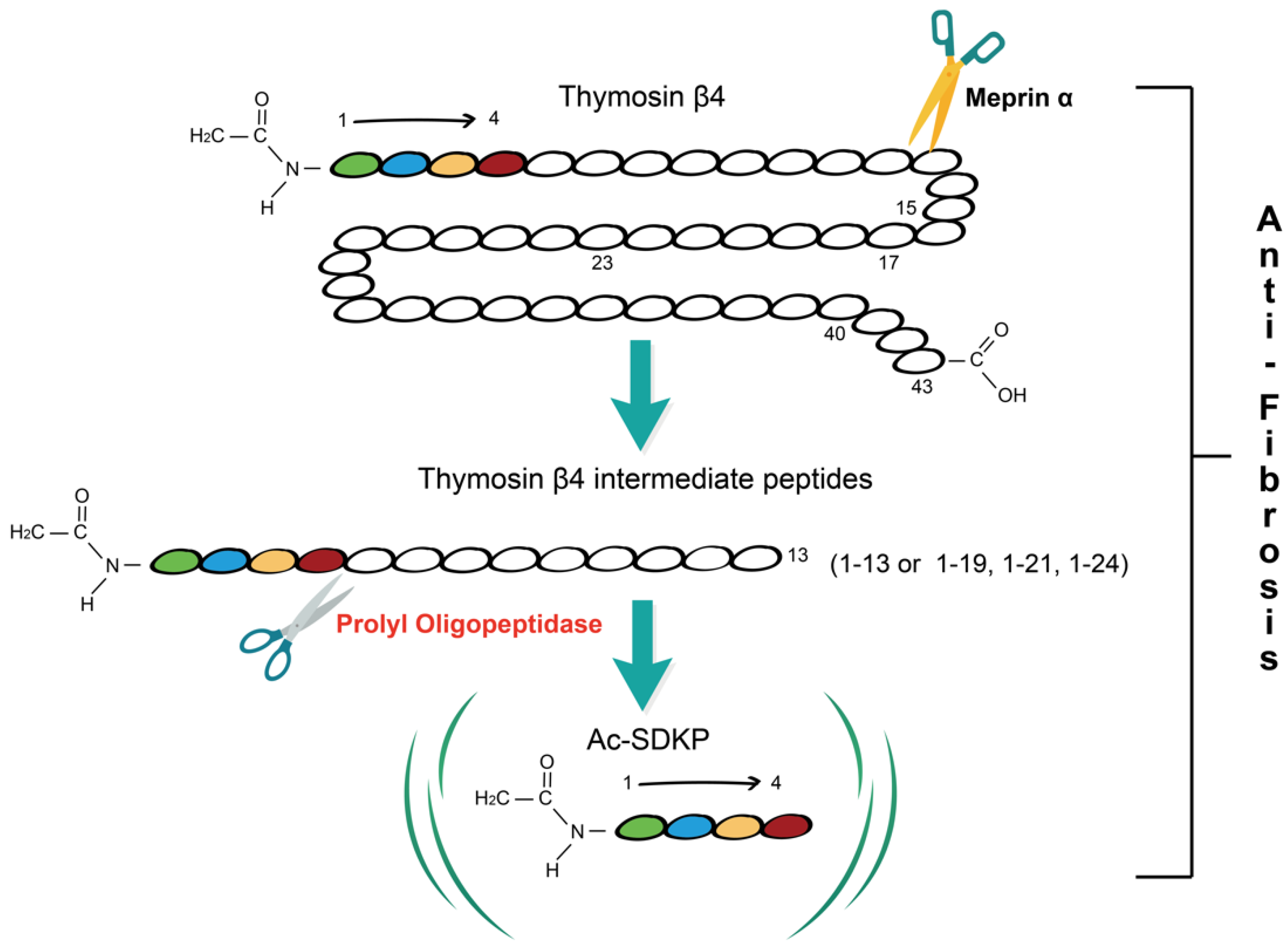
:1. Tβ4-POP-Ac-SDKP Axis
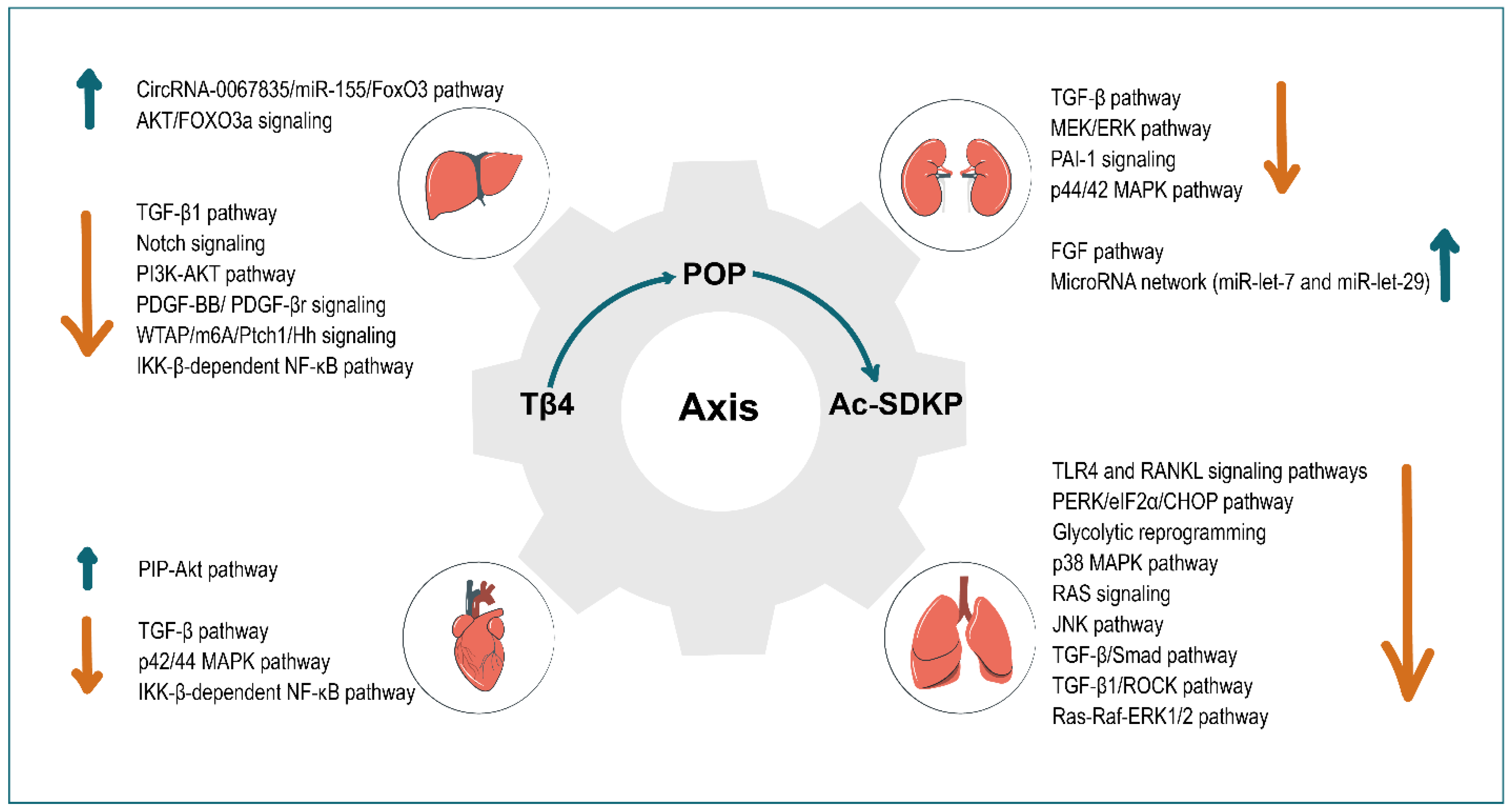
2. Tissue and Organ Fibrosis
3. Liver
4. Kidney
5. Heart
6. Lung
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Low, T.L.; Hu, S.K.; Goldstein, A.L. Complete amino acid sequence of bovine thymosin beta 4: A thymic hormone that induces terminal deoxynucleotidyl transferase activity in thymocyte populations. Proc. Natl. Acad. Sci. USA 1981, 78, 1162–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, Y.; Ye, Y.; Zuo, H.; Li, Y. Progress on the Function and Application of Thymosin β4. Front. Endocrinol. 2021, 12, 767785. [Google Scholar] [CrossRef] [PubMed]
- Smart, N.; Risebro, C.A.; Melville, A.A.D.; Moses, K.; Schwartz, R.J.; Chien, K.R.; Riley, P.R. Thymosin β4 induces adult epicardial progenitor mobilization and neovascularization. Nature 2007, 445, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Morris, D.; Chopp, M.; Zhang, L.; Lu, M.; Zhang, Z. Thymosin β4 improves functional neurological outcome in a rat model of embolic stroke. Neuroscience 2010, 169, 674–682. [Google Scholar] [CrossRef] [Green Version]
- Shah, R.; Reyes-Gordillo, K.; Cheng, Y.; Varatharajalu, R.; Ibrahim, J.; Lakshman, M.R. Thymosin β4 Prevents Oxidative Stress, Inflammation, and Fibrosis in Ethanol- and LPS-Induced Liver Injury in Mice. Oxidative Med. Cell. Longev. 2018, 2018, 9630175. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, A.L.; Hannappel, E.; Sosne, G.; Kleinman, H.K. Thymosin β4: A multi-functional regenerative peptide. Basic properties and clinical applications. Expert Opin. Biol. Ther. 2011, 12, 37–51. [Google Scholar] [CrossRef]
- Ehrlich, H.P.; Iii, S.W.H. Thymosin β4 enhances repair by organizing connective tissue and preventing the appearance of myofibroblasts. Ann. N. Y. Acad. Sci. 2010, 1194, 118–124. [Google Scholar] [CrossRef]
- Cavasin, M.A.; Rhaleb, N.-E.; Yang, X.-P.; Carretero, O.A. Prolyl Oligopeptidase Is Involved in Release of the Antifibrotic Peptide Ac-SDKP. Hypertension 2004, 43, 1140–1145. [Google Scholar] [CrossRef] [Green Version]
- Kumar, N.; Nakagawa, P.; Janic, B.; Romero, C.; Worou, M.E.; Monu, S.R.; Peterson, E.L.; Shaw, J.; Valeriote, F.; Ongeri, E.M.; et al. The anti-inflammatory peptide Ac-SDKP is released from thymosin-β4 by renal meprin-α and prolyl oligopeptidase. Am. J. Physiol.-Renal Physiol. 2016, 310, F1026–F1034. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-W.; Liu, B.-W.; Zhang, Y.-J.; Chen, Y.-W.; Dong, G.-F.; Ding, X.-D.; Xu, L.-M.; Pat, B.; Fan, J.-G.; Li, D.-G. Preservation of basal AcSDKP attenuates carbon tetrachloride-induced fibrosis in the rat liver. J. Hepatol. 2010, 53, 528–536. [Google Scholar] [CrossRef]
- Weber, L.W.D.; Boll, M.; Stampfl, A. Hepatotoxicity and Mechanism of Action of Haloalkanes: Carbon Tetrachloride as a Toxicological Model. Crit. Rev. Toxicol. 2003, 33, 105–136. [Google Scholar] [CrossRef] [PubMed]
- Cavasin, M.A.; Liao, T.-D.; Yang, X.-P.; Yang, J.J.; Carretero, O.A. Decreased Endogenous Levels of Ac-SDKP Promote Organ Fibrosis. Hypertension 2007, 50, 130–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azizi, M.; Rousseau, A.; Ezan, E.; Guyene, T.T.; Michelet, S.; Grognet, J.M.; Lenfant, M.; Corvol, P.; Ménard, J. Acute angiotensin-converting enzyme inhibition increases the plasma level of the natural stem cell regulator N-acetyl-seryl-aspartyl-lysyl-proline. J. Clin. Investig. 1996, 97, 839–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.-M.; Lawrence, F.; Kovacevic, M.; Bignon, J.; Papadimitriou, E.; Lallemand, J.-Y.; Katsoris, P.; Potier, P.; Fromes, Y.; Wdzieczak-Bakala, J. The tetrapeptide AcSDKP, an inhibitor of primitive hematopoietic cell proliferation, induces angiogenesis in vitro and in vivo. Blood 2003, 101, 3014–3020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenfant, M.; Wdzieczak-Bakala, J.; Guittet, E.; Prome, J.C.; Sotty, D.; Frindel, E. Inhibitor of hematopoietic pluripotent stem cell proliferation: Purification and determination of its structure. Proc. Natl. Acad. Sci. USA 1989, 86, 779–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Xu, L.-M.; Chen, Y.-W.; Ni, Q.-W.; Zhou, M.; Qu, C.-Y.; Zhang, Y. Antifibrotic effect of N-acetyl-seryl-aspartyl-lysyl-proline on bile duct ligation induced liver fibrosis in rats. World J. Gastroenterol. 2012, 18, 5283–5288. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Carretero, O.A.; Peterson, E.L.; Rhaleb, N.-E. Ac-SDKP inhibits transforming growth factor-β1-induced differentiation of human cardiac fibroblasts into myofibroblasts. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1357–H1364. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Yang, F.; Sun, Y.; Yuan, Y.; Cheng, H.; Wei, Z.; Li, S.; Cheng, T.; Brann, D.; Wang, R. A New Antifibrotic Target of Ac-SDKP: Inhibition of Myofibroblast Differentiation in Rat Lung with Silicosis. PLoS ONE 2012, 7, e40301. [Google Scholar] [CrossRef] [Green Version]
- Omata, M.; Taniguchi, H.; Koya, D.; Kanasaki, K.; Sho, R.; Kato, Y.; Kojima, R.; Haneda, M.; Inomata, N. N-Acetyl-Seryl-Aspartyl-Lysyl-Proline Ameliorates the Progression of Renal Dysfunction and Fibrosis in WKY Rats with Established Anti–Glomerular Basement Membrane Nephritis. J. Am. Soc. Nephrol. 2006, 17, 674–685. [Google Scholar] [CrossRef] [Green Version]
- Ho, Y.Y.; Lagares, D.; Tager, A.M.; Kapoor, M. Fibrosis—A lethal component of systemic sclerosis. Nat. Rev. Rheumatol. 2014, 10, 390–402. [Google Scholar] [CrossRef]
- di Carlo, S.; Peduto, L. The perivascular origin of pathological fibroblasts. J. Clin. Investig. 2018, 128, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Fibrotic disease and the TH1/TH2 paradigm. Nat. Rev. Immunol. 2004, 4, 583–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound repair and regeneration. Nature 2008, 453, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: From mechanisms to medicines. Nature 2020, 587, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Borthwick, L.A.; Wynn, T.A.; Fisher, A.J. Cytokine mediated tissue fibrosis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2013, 1832, 1049–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gieseck, R.L.; Wilson, M.S.; Wynn, T.A. Type 2 immunity in tissue repair and fibrosis. Nat. Rev. Immunol. 2018, 18, 62–76. [Google Scholar] [CrossRef]
- Pakshir, P.; Hinz, B. The big five in fibrosis: Macrophages, myofibroblasts, matrix, mechanics, and miscommunication. Matrix Biol. 2018, 68–69, 81–93. [Google Scholar] [CrossRef]
- Gur, C.; Wang, S.Y.; Sheban, F.; Zada, M.; Li, B.; Kharouf, F.; Peleg, H.; Aamar, S.; Yalin, A.; Kirschenbaum, D.; et al. LGR5 expressing skin fibroblasts define a major cellular hub perturbed in scleroderma. Cell 2022, 185, 1373–1388.e1320. [Google Scholar] [CrossRef]
- Ruaro, B.; Soldano, S.; Smith, V.; Paolino, S.; Contini, P.; Montagna, P.; Pizzorni, C.; Casabella, A.; Tardito, S.; Sulli, A.; et al. Correlation between circulating fibrocytes and dermal thickness in limited cutaneous systemic sclerosis patients: A pilot study. Rheumatol. Int. 2019, 39, 1369–1376. [Google Scholar] [CrossRef]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiskirchen, R.; Weiskirchen, S.; Tacke, F. Organ and tissue fibrosis: Molecular signals, cellular mechanisms and translational implications. Mol. Asp. Med. 2018, 65, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Aydin, M.M.; Akcali, K.C. Liver fibrosis. Turk. J. Gastroenterol. 2018, 29, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-Y.; Ding, Z.-Y.; Zhou, Z.-Y.; Dai, S.-Z.; Zhang, J.; Li, H.; Du, Q.; Cai, Y.-Y.; Shang, Q.-L.; Luo, Y.-H.; et al. Multiparameter magnetic resonance imaging of liver fibrosis in a bile duct ligation mouse model. World J. Gastroenterol. 2021, 27, 8156–8165. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, Q.; Zhang, X.; Zheng, X.; Zhang, Q.; Hao, Z. Thymosin β4 suppresses CCl4 -induced murine hepatic fibrosis by down-regulating transforming growth factor β receptor-II. J. Gene Med. 2018, 20, e3043. [Google Scholar] [CrossRef]
- Kim, J.; Wang, S.; Hyun, J.; Choi, S.S.; Cha, H.; Ock, M.; Jung, Y. Hepatic Stellate Cells Express Thymosin Beta 4 in Chronically Damaged Liver. PLoS ONE 2015, 10, e0122758. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Hyun, J.; Wang, S.; Lee, C.; Lee, J.-W.; Moon, E.-Y.; Cha, H.; Diehl, A.M.; Jung, Y. Thymosin beta-4 regulates activation of hepatic stellate cells via hedgehog signaling. Sci. Rep. 2017, 7, 3815. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Gordillo, K.; Shah, R.; Arellanes-Robledo, J.; Rojkind, M.; Lakshman, M.R. Protective effects of thymosin β4 on carbon tetrachloride-induced acute hepatotoxicity in rats. Ann. N. Y. Acad. Sci. 2012, 1269, 61–68. [Google Scholar] [CrossRef]
- Li, X.; Wang, L.; Chen, C. Effects of exogenous thymosin β4 on carbon tetrachloride-induced liver injury and fibrosis. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Fu, W.-L.; Zhu, Y.; Wang, X.-G. Tβ4 suppresses lincRNA-p21-mediated hepatic apoptosis and fibrosis by inhibiting PI3K-AKT-NF-κB pathway. Gene 2020, 758, 144946. [Google Scholar] [CrossRef]
- Zhu, Z.X.; Zhu, L.L.; Cheng, Z.; Zhao, X.K.; Liu, Y.M.; Fan, L.D.; Zou, G.L.; Ouyang, Q.Y.; Cheng, M.L. Cellular mechanism of Tbeta4 intervention in liver fibrosis by regulating NF-kappaB signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 1279–1290. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Ren, T.; Zhu, Z.; Cheng, M.; Mou, Q.; Mu, M.; Liu, Y.; Yao, Y.; Cheng, Y.; Zhang, B.; et al. Thymosin-β4 Mediates Hepatic Stellate Cell Activation by Interfering with CircRNA-0067835/miR-155/FoxO3 Signaling Pathway. Cell. Physiol. Biochem. 2018, 51, 1389–1398. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Gordillo, K.; Shah, R.; Popratiloff, A.; Fu, S.; Hindle, A.; Brody, F.; Rojkind, M. Thymosin-β4 (Tβ4) Blunts PDGF-Dependent Phosphorylation and Binding of AKT to Actin in Hepatic Stellate Cells. Am. J. Pathol. 2011, 178, 2100–2108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnaeva, E.; Nadezhda, A.; Hannappel, E.; Sjogren, M.H.; Rojkind, M. Thymosin beta4 Upregulates the Expression of Hepatocyte Growth Factor and Downregulates the Expression of PDGF-beta Receptor in Human Hepatic Stellate Cells. Ann. N. Y. Acad. Sci. 2007, 1112, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Knop, J.; App, C.; Huff, T.; Iavarone, F.; Castagnola, M.; Hannappel, E. Identification of PDGF-BB binding to thymosin β4by chemical cross-linking. Expert Opin. Biol. Ther. 2015, 15, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Li, X.; Wang, L. Thymosinβ4 alleviates cholestatic liver fibrosis in mice through downregulating PDGF/PDGFR and TGFβ/Smad pathways. Dig. Liver Dis. 2019, 52, 324–330. [Google Scholar] [CrossRef]
- Hong, Y.; Yao, Q.; Zheng, L. Thymosin β4 attenuates liver fibrosis via suppressing Notch signaling. Biochem. Biophys. Res. Commun. 2017, 493, 1396–1401. [Google Scholar] [CrossRef]
- Bataller, R.; Brenner, D. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Zhou, D.; Wang, J.; He, L.-N.; Li, B.-H.; Ding, Y.-N.; Chen, Y.-W.; Fan, J.-G. Prolyl oligopeptidase attenuates hepatic stellate cell activation through induction of Smad7 and PPAR-γ. Exp. Ther. Med. 2017, 13, 780–786. [Google Scholar] [CrossRef] [Green Version]
- Wei, A.; Zhao, F.; Hao, A.; Liu, B.; Liu, Z. N-acetyl-seryl-aspartyl-lysyl-proline (AcSDKP) mitigates the liver fibrosis via WTAP/m6A/Ptch1 axis through Hedgehog pathway. Gene 2021, 813, 146125. [Google Scholar] [CrossRef]
- Vasilopoulou, E.; Riley, P.R.; Long, D. Thymosin-β4: A key modifier of renal disease. Expert Opin. Biol. Ther. 2018, 18, 185–192. [Google Scholar] [CrossRef]
- Mizunuma, Y.; Kanasaki, K.; Nitta, K.; Nakamura, Y.; Ishigaki, Y.; Takagaki, Y.; Kitada, M.; Li, S.; Liu, H.; Li, J.; et al. CD-1db/db mice: A novel type 2 diabetic mouse model with progressive kidney fibrosis. J. Diabetes Investig. 2020, 11, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Pippin, J.W.; Brinkkoetter, P.T.; Cormack-Aboud, F.C.; Durvasula, R.V.; Hauser, P.V.; Kowalewska, J.; Krofft, R.D.; Logar, C.M.; Marshall, C.B.; Ohse, T.; et al. Inducible rodent models of acquired podocyte diseases. Am. J. Physiol.-Renal Physiol. 2009, 296, F213–F229. [Google Scholar] [CrossRef] [PubMed]
- Vasilopoulou, E.; Kolatsi-Joannou, M.; Lindenmeyer, M.T.; White, K.E.; Robson, M.G.; Cohen, C.D.; Sebire, N.J.; Riley, P.R.; Winyard, P.J.; Long, D.A. Loss of endogenous thymosin β4 accelerates glomerular disease. Kidney Int. 2016, 90, 1056–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwamoto, N.; Xano, H.J.; Yoshioka, T.; Shiraga, H.; Nitta, K.; Muraki, T.; Ito, K. Acetyl-seryl-aspartyl-lysyl-proline is a novel natural cell cycle regulator of renal cells. Life Sci. 2000, 66, 221–226. [Google Scholar] [CrossRef]
- Romero, C.A.; Kumar, N.; Nakagawa, P.; Worou, M.E.; Liao, T.-D.; Peterson, E.L.; Carretero, O.A. Renal release of N-acetyl-seryl-aspartyl-lysyl-proline is part of an antifibrotic peptidergic system in the kidney. Am. J. Physiol.-Renal Physiol. 2019, 316, F195–F203. [Google Scholar] [CrossRef] [Green Version]
- Zuo, Y.; Chun, B.; Potthoff, S.A.; Kazi, N.; Brolin, T.J.; Orhan, D.; Yang, H.-C.; Ma, L.-J.; Kon, V.; Myöhänen, T.; et al. Thymosin β4 and its degradation product, Ac-SDKP, are novel reparative factors in renal fibrosis. Kidney Int. 2013, 84, 1166–1175. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.J.; Shyr, Y.; Liang, X.; Ma, L.-J.; Donnert, E.M.; Roberts, J.D.; Zhang, X.; Kon, V.; Brown, N.J.; Caprioli, R.M.; et al. Proteomic Patterns and Prediction of Glomerulosclerosis and Its Mechanisms. J. Am. Soc. Nephrol. 2005, 16, 2967–2975. [Google Scholar] [CrossRef] [Green Version]
- Chan, G.C.W.; Yiu, W.H.; Wu, H.J.; Wong, D.W.L.; Lin, M.; Huang, X.R.; Lan, H.Y.; Tang, S.C.W. N-Acetyl-seryl-aspartyl-lysyl-proline Alleviates Renal Fibrosis Induced by Unilateral Ureteric Obstruction in BALB/C Mice. Mediat. Inflamm. 2015, 2015, 283123. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Shen, Y.; Yang, X.; Xie, Y.; Lin, X.; Zeng, W.; Zhao, Y.; Tian, M.; Zha, Y. Thymosin β4 alleviates renal fibrosis and tubular cell apoptosis through TGF-β pathway inhibition in UUO rat models. BMC Nephrol. 2017, 18, 314. [Google Scholar] [CrossRef]
- Kanasaki, K.; Koya, D.; Sugimoto, T.; Isono, M.; Kashiwagi, A.; Haneda, M. N-Acetyl-Seryl-Aspartyl-Lysyl-Proline Inhibits TGF-β–Mediated Plasminogen Activator Inhibitor-1 Expression via Inhibition of Smad Pathway in Human Mesangial Cells. J. Am. Soc. Nephrol. 2003, 14, 863–872. [Google Scholar] [CrossRef] [Green Version]
- Xie, R.; Wang, M.; Liu, R.; Jia, X.; Mu, S. N-acetyl-seryl-aspartyl-lysyl-proline attenuates renal inflammation and tubulointerstitial fibrosis in rats. Int. J. Mol. Med. 2010, 26, 795–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, G.C.; Wu, H.; Chan, K.W.; Yiu, W.H.; Zou, A.; Huang, X.R.; Lan, H.Y.; Lai, K.N.; Tang, S.C.W. N-acetyl-seryl-aspartyl-lysyl-proline mediates the anti-fibrotic properties of captopril in unilateral ureteric obstructed BALB/C mice. Nephrology 2018, 23, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.P.; Goodwin, J.E.; Kanasaki, K.; Koya, D. Metabolic reprogramming by N-acetyl-seryl-aspartyl-lysyl-proline protects against diabetic kidney disease. J. Cereb. Blood Flow Metab. 2020, 177, 3691–3711. [Google Scholar] [CrossRef] [PubMed]
- Castoldi, G.; di Gioia, C.; Bombardi, C.; Preziuso, C.; Leopizzi, M.; Maestroni, S.; Corradi, B.; Zerbini, G.; Stella, A. Renal Antifibrotic Effect of N-Acetyl-Seryl-Aspartyl-Lysyl-Proline in Diabetic Rats. Am. J. Nephrol. 2013, 37, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Nagai, T.; Nitta, K.; Kanasaki, M.; Koya, D.; Kanasaki, K. The biological significance of angiotensin-converting enzyme inhibition to combat kidney fibrosis. Clin. Exp. Nephrol. 2014, 19, 65–74. [Google Scholar] [CrossRef]
- Li, J.; Liu, H.; Srivastava, S.P.; Hu, Q.; Gao, R.; Li, S.; Kitada, M.; Wu, G.; Koya, D.; Kanasaki, K. Endothelial FGFR1 (Fibroblast Growth Factor Receptor 1) Deficiency Contributes Differential Fibrogenic Effects in Kidney and Heart of Diabetic Mice. Hypertension 2020, 76, 1935–1944. [Google Scholar] [CrossRef]
- Gao, R.; Kanasaki, K.; Li, J.; Kitada, M.; Okazaki, T.; Koya, D. βklotho is essential for the anti-endothelial mesenchymal transition effects of N-acetyl-seryl-aspartyl-lysyl-proline. FEBS Open Bio 2019, 9, 1029–1038. [Google Scholar] [CrossRef] [Green Version]
- Nitta, K.; Shi, S.; Nagai, T.; Kanasaki, M.; Kitada, M.; Srivastava, S.P.; Haneda, M.; Kanasaki, K.; Koya, D. Oral Administration of N-Acetyl-seryl-aspartyl-lysyl-proline Ameliorates Kidney Disease in Both Type 1 and Type 2 Diabetic Mice via a Therapeutic Regimen. BioMed Res. Int. 2016, 2016, 9172157. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.P.; Shi, S.; Kanasaki, M.; Nagai, T.; Kitada, M.; He, J.; Nakamura, Y.; Ishigaki, Y.; Kanasaki, K.; Koya, D. Effect of Antifibrotic MicroRNAs Crosstalk on the Action of N-acetyl-seryl-aspartyl-lysyl-proline in Diabetes-related Kidney Fibrosis. Sci. Rep. 2016, 6, 29884. [Google Scholar] [CrossRef]
- Nagai, T.; Kanasaki, M.; Srivastava, S.P.; Nakamura, Y.; Ishigaki, Y.; Kitada, M.; Shi, S.; Kanasaki, K.; Koya, D. N-acetyl-seryl-aspartyl-lysyl-proline Inhibits Diabetes-Associated Kidney Fibrosis and Endothelial-Mesenchymal Transition. BioMed Res. Int. 2014, 2014, 696475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, S.P.; Goodwin, J.E.; Kanasaki, K.; Koya, D. Inhibition of Angiotensin-Converting Enzyme Ameliorates Renal Fibrosis by Mitigating DPP-4 Level and Restoring Antifibrotic MicroRNAs. Genes 2020, 11, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savoia, C.; Schiffrin, E.L. Inflammation in hypertension. Curr. Opin. Intern. Med. 2006, 5, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Zoccali, C. Endothelial Dysfunction and the Kidney: Emerging Risk Factors for Renal Insufficiency and Cardiovascular Outcomes in Essential Hypertension. J. Am. Soc. Nephrol. 2006, 17, S61–S63. [Google Scholar] [CrossRef] [Green Version]
- Zoccali, C.; Mallamaci, F.; Tripepi, G. Inflammation and Atherosclerosis in End-Stage Renal Disease. Blood Purif. 2003, 21, 29–36. [Google Scholar] [CrossRef]
- Kelly, D.; Zhang, Y.; Cox, A.; Gilbert, R. Combination therapy with tranilast and angiotensin-converting enzyme inhibition provides additional renoprotection in the remnant kidney model. Kidney Int. 2006, 69, 1954–1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Lu, Y.; Hu, X.; Li, H.; Li, X.; Xiao, C.; Meng, T.; Peng, L.; Gan, L.; Zhou, Q.; et al. Interleukin-22 exacerbates angiotensin II-induced hypertensive renal injury. Int. Immunopharmacol. 2022, 109, 108840. [Google Scholar] [CrossRef]
- Liao, T.-D.; Yang, X.-P.; D’Ambrosio, M.; Zhang, Y.; Rhaleb, N.-E.; Carretero, O.A. N-Acetyl-seryl-aspartyl-lysyl-proline Attenuates Renal Injury and Dysfunction in Hypertensive Rats With Reduced Renal Mass: Council for high blood pressure research. Hypertension 2010, 55, 459–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhaleb, N.-E.; Pokharel, S.; Sharma, U.; Carretero, O.A. Renal protective effects of N-acetyl-Ser-Asp-Lys-Pro in deoxycorticosterone acetate–salt hypertensive mice. J. Hypertens. 2011, 29, 330–338. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Zhao, Q.; Kong, W. Extracellular matrix remodeling and cardiac fibrosis. Matrix Biol. 2018, 68–69, 490–506. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2020, 117, 1450–1488. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Asp. Med. 2018, 65, 70–99. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Liao, T.-D.; Romero, C.A.; Maheshwari, M.; Peterson, E.L.; Carretero, O.A. Thymosin β4 Deficiency Exacerbates Renal and Cardiac Injury in Angiotensin-II–Induced Hypertension. Hypertension 2018, 71, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Xu, J.; Yang, X.-P.; Dai, X.; Peterson, E.L.; Carretero, O.A.; Rhaleb, N.-E. Thymosin-β4 prevents cardiac rupture and improves cardiac function in mice with myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H741–H751. [Google Scholar] [CrossRef] [Green Version]
- Evans, M.A.; Smart, N.; Dubé, K.N.; Bollini, S.; Clark, J.E.; Evans, H.G.; Taams, L.S.; Richardson, R.; Lévesque, M.; Martin, P.; et al. Thymosin β4-sulfoxide attenuates inflammatory cell infiltration and promotes cardiac wound healing. Nat. Commun. 2013, 4, 2081. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Gupta, S. Thymosin Beta 4 Prevents Oxidative Stress by Targeting Antioxidant and Anti-Apoptotic Genes in Cardiac Fibroblasts. PLoS ONE 2011, 6, e26912. [Google Scholar] [CrossRef]
- Yan, B.; Singla, R.D.; Abdelli, L.S.; Singal, P.K.; Singla, D.K. Regulation of PTEN/Akt Pathway Enhances Cardiomyogenesis and Attenuates Adverse Left Ventricular Remodeling following Thymosin β4 Overexpressing Embryonic Stem Cell Transplantation in the Infarcted Heart. PLoS ONE 2013, 8, e75580. [Google Scholar] [CrossRef] [Green Version]
- Sopko, N.; Qin, Y.; Finan, A.; Dadabayev, A.; Chigurupati, S.; Qin, J.; Penn, M.S.; Gupta, S. Significance of Thymosin β4 and Implication of PINCH-1-ILK-α-Parvin (PIP) Complex in Human Dilated Cardiomyopathy. PLoS ONE 2011, 6, e20184. [Google Scholar] [CrossRef] [Green Version]
- Stark, C.; Taimen, P.; Savunen, T.; Koskenvuo, J. Pegylated and liposomal doxorubicin is associated with high mortality and causes limited cardiotoxicity in mice. BMC Res. Notes 2018, 11, 148. [Google Scholar] [CrossRef]
- Cingolani, O.H.; Yang, X.-P.; Liu, Y.-H.; Villanueva, M.; Rhaleb, N.-E.; Carretero, O.A. Reduction of Cardiac Fibrosis Decreases Systolic Performance Without Affecting Diastolic Function in Hypertensive Rats. Hypertension 2004, 43, 1067–1073. [Google Scholar] [CrossRef]
- Rhaleb, N.-E.; Peng, H.; Harding, P.; Tayeh, M.; LaPointe, M.C.; Carretero, O.A. Effect of N-Acetyl-seryl-aspartyl-lysyl-proline on DNA and Collagen Synthesis in Rat Cardiac Fibroblasts. Hypertension 2001, 37, 827–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Yang, X.-P.; Liu, Y.-H.; Xu, J.; Cingolani, O.; Rhaleb, N.-E.; Carretero, O.A. Ac-SDKP Reverses Inflammation and Fibrosis in Rats With Heart Failure After Myocardial Infarction. Hypertension 2004, 43, 229–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, U.C.; Sonkawade, S.D.; Baird, A.; Chen, M.; Xu, S.; Sexton, S.; Singh, A.K.; Groman, A.; Turowski, S.G.; Spernyak, J.A.; et al. Effects of a novel peptide Ac-SDKP in radiation-induced coronary endothelial damage and resting myocardial blood flow. Cardio-Oncology 2018, 4, 8. [Google Scholar] [CrossRef] [Green Version]
- Sharma, U.C.; Sonkawade, S.D.; Spernyak, J.A.; Sexton, S.; Nguyen, J.; Dahal, S.; Attwood, K.M.; Singh, A.K.; van Berlo, J.H.; Pokharel, S. A Small Peptide Ac-SDKP Inhibits Radiation-Induced Cardiomyopathy. Circ. Heart Fail. 2018, 11, e004867. [Google Scholar] [CrossRef]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-β signaling in fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Carretero, O.A.; Brigstock, D.R.; Oja-Tebbe, N.; Rhaleb, N.-E. Ac-SDKP Reverses Cardiac Fibrosis in Rats with Renovascular Hypertension. Hypertension 2003, 42, 1164–1170. [Google Scholar] [CrossRef] [Green Version]
- Rasoul, S.; Carretero, O.A.; Peng, H.; Cavasin, M.A.; Zhuo, J.; Sánchez-Mendoza, A.; Brigstock, D.R.; Rhaleb, N.-E. Antifibrotic effect of Ac-SDKP and angiotensin-converting enzyme inhibition in hypertension. J. Hypertens. 2004, 22, 593–603. [Google Scholar] [CrossRef]
- González, G.E.; Rhaleb, N.-E.; Nakagawa, P.; Liao, T.-D.; Liu, Y.; Leung, P.; Dai, X.; Yang, X.-P.; Carretero, O.A. N-acetyl-seryl-aspartyl-lysyl-proline reduces cardiac collagen cross-linking and inflammation in angiotensin II-induced hypertensive rats. Clin. Sci. 2013, 126, 85–94. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-X.; Rhaleb, N.-E.; Yang, X.-P.; Liao, T.-D.; D’Ambrosio, M.A.; Carretero, O.A. Prevention of aortic fibrosis by N-acetyl-seryl-aspartyl-lysyl-proline in angiotensin II-induced hypertension. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1253–H1261. [Google Scholar] [CrossRef] [Green Version]
- Castoldi, G.; Di Gioia, C.R.T.; Bombardi, C.; Perego, C.; Perego, L.; Mancini, M.; Leopizzi, M.; Corradi, B.; Perlini, S.; Zerbini, G.; et al. Prevention of myocardial fibrosis by N-acetyl-seryl-aspartyl-lysyl-proline in diabetic rats. Clin. Sci. 2010, 118, 211–220. [Google Scholar] [CrossRef]
- Pokharel, S.; van Geel, P.P.; Sharma, U.C.; Cleutjens, J.P.; Bohnemeier, H.; Tian, X.-L.; Schunkert, H.; Crijns, H.J.; Paul, M.; Pinto, Y.M. Increased Myocardial Collagen Content in Transgenic Rats Overexpressing Cardiac Angiotensin-Converting Enzyme Is Related to Enhanced Breakdown of N-Acetyl-ser-asp-lys-pro and Increased Phosphorylation of Smad2/3. Circulation 2004, 110, 3129–3135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.-H.; D’Ambrosio, M.; Liao, T.-D.; Peng, H.; Rhaleb, N.-E.; Sharma, U.; André, S.; Gabius, H.-J.; Carretero, O.A. N-acetyl-seryl-aspartyl-lysyl-proline prevents cardiac remodeling and dysfunction induced by galectin-3, a mammalian adhesion/growth-regulatory lectin. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H404–H412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pokharel, S.; Rasoul, S.; Roks, A.J.; van Leeuwen, R.E.; van Luyn, M.J.; Deelman, L.E.; Smits, J.F.; Carretero, O.; van Gilst, W.H.; Pinto, Y.M. N-Acetyl-ser-asp-lys-pro Inhibits Phosphorylation of Smad2 in Cardiac Fibroblasts. Hypertension 2002, 40, 155–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, H.; Carretero, O.A.; Liao, T.-D.; Peterson, E.L.; Rhaleb, N.-E. Role of N-Acetyl-Seryl-Aspartyl-Lysyl-Proline in the Antifibrotic and Anti-Inflammatory Effects of the Angiotensin-Converting Enzyme Inhibitor Captopril in Hypertension. Hypertension 2007, 49, 695–703. [Google Scholar] [CrossRef]
- Rhaleb, N.-E.; Pokharel, S.; Sharma, U.C.; Peng, H.; Peterson, E.; Harding, P.; Yang, X.-P.; Carretero, O.A. N-acetyl-Ser-Asp-Lys-Pro inhibits interleukin-1β-mediated matrix metalloproteinase activation in cardiac fibroblasts. Pflügers Arch.-Eur. J. Physiol. 2013, 465, 1487–1495. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Yang, X.-P.; Janic, B.; Rhaleb, N.-E.; Harding, P.; Nakagawa, P.; Peterson, E.L.; Carretero, O.A. Ac-SDKP suppresses TNF-α-induced ICAM-1 expression in endothelial cells via inhibition of IκB kinase and NF-κB activation. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H1176–H1183. [Google Scholar] [CrossRef] [Green Version]
- Thannickal, V.J.; Toews, G.B.; White, E.S.; Iii, J.P.L.; Martinez, F.J. Mechanisms of Pulmonary Fibrosis. Annu. Rev. Med. 2004, 55, 395–417. [Google Scholar] [CrossRef]
- Tian, Z.; Yao, N.; Wang, F.; Ruan, L. Thymosin β4 Suppresses LPS-Induced Murine Lung Fibrosis by Attenuating Oxidative Injury and Alleviating Inflammation. Inflammation 2021, 45, 59–73. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Richeldi, L.; Thomson, C.C.; Inoue, Y.; Johkoh, T.; Kreuter, M.; Lynch, D.A.; Maher, T.M.; Martinez, F.J.; et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2022, 205, e18–e47. [Google Scholar] [CrossRef]
- Ruaro, B.; Matucci Cerinic, M.; Salton, F.; Baratella, E.; Confalonieri, M.; Hughes, M. Editorial: Pulmonary fibrosis: One manifestation, various diseases. Front. Pharmacol. 2022, 13. [Google Scholar] [CrossRef]
- Li, S.; Li, Y.; Zhang, Y.; Li, S.; Zhang, M.; Jin, F.; Wei, Z.; Yang, Y.; Gao, X.; Mao, N.; et al. N-Acetyl-Seryl-Asparyl-Lysyl-Proline regulates lung renin angiotensin system to inhibit epithelial–mesenchymal transition in silicotic mice. Toxicol. Appl. Pharmacol. 2020, 408, 115255. [Google Scholar] [CrossRef] [PubMed]
- Conte, E.; Fagone, E.; Gili, E.; Fruciano, M.; Iemmolo, M.; Pistorio, M.P.; Impellizzeri, D.; Cordaro, M.; Cuzzocrea, S.; Vancheri, C. Preventive and therapeutic effects of thymosin β4 N-terminal fragment Ac-SDKP in the bleomycin model of pulmonary fibrosis. Oncotarget 2016, 7, 33841–33854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conte, E.; Genovese, T.; Gili, E.; Esposito, E.; Iemmolo, M.; Fruciano, M.; Fagone, E.; Pistorio, M.P.; Crimi, N.; Cuzzocrea, S.; et al. Thymosin β4 protects C57BL/6 mice from bleomycin-induced damage in the lung. Eur. J. Clin. Investig. 2012, 43, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Conte, E.; Iemmolo, M.; Fruciano, M.; Fagone, E.; Gili, E.; Genovese, T.; Esposito, E.; Cuzzocrea, S.; Vancheri, C. Effects of thymosin β4 and its N-terminal fragment Ac-SDKP on TGF-β-treated human lung fibroblasts and in the mouse model of bleomycin-induced lung fibrosis. Expert Opin. Biol. Ther. 2015, 15, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Conte, E.; Iemmolo, M.; Fagone, E.; Gili, E.; Fruciano, M.; Genovese, T.; Esposito, E.; Cuzzocrea, S.; Vancheri, C. Thymosin β4 reduces IL-17-producing cells and IL-17 expression, and protects lungs from damage in bleomycin-treated mice. Immunobiology 2014, 219, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Xiao, H.D.; Xu, J.; Ong, F.S.; Kwon, M.; Roman, J.; Gal, A.; Bernstein, K.E.; Fuchs, S. Angiotensin-Converting Enzyme N-Terminal Inactivation Alleviates Bleomycin-Induced Lung Injury. Am. J. Pathol. 2010, 177, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.-B.; Zhang, L.-J.; Li, Q. Anti-fibrotic effect of N-acetyl-seryl-aspartyl-lysyl-proline in lung of rat with silicosis. Zhonghua lao dong wei sheng zhi ye bing za zhi = Chin. J. Ind. Hyg. Occup. Dis. 2008, 26, 401–405. [Google Scholar]
- Kasai, H.; Allen, J.T.; Mason, R.M.; Kamimura, T.; Zhang, Z. TGF-β1 induces human alveolar epithelial to mesenchymal cell transition (EMT). Respir. Res. 2005, 6, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.-F.; Chiang, W.-C.; Lai, C.-F.; Chang, F.-C.; Chen, Y.-T.; Chou, Y.-H.; Wu, T.-H.; Linn, G.R.; Ling, H.; Wu, K.-D.; et al. Transforming Growth Factor β-1 Stimulates Profibrotic Epithelial Signaling to Activate Pericyte-Myofibroblast Transition in Obstructive Kidney Fibrosis. Am. J. Pathol. 2012, 182, 118–131. [Google Scholar] [CrossRef] [Green Version]
- Deng, H.; Xu, H.; Zhang, X.; Sun, Y.; Wang, R.; Brann, D.; Yang, F. Protective effect of Ac-SDKP on alveolar epithelial cells through inhibition of EMT via TGF-β1/ROCK1 pathway in silicosis in rat. Toxicol. Appl. Pharmacol. 2016, 294, 1–10. [Google Scholar] [CrossRef]
- Yuan, Y.; Yang, F.; Xu, H.; Yu, W.-Y.; Sun, Y.; Deng, H.-J.; Ma, W.-D.; Wei, Z.-Q.; Wang, R.-M. Effect of N-acetyl-seryl-aspartyl-lysyl-proline on differentiation from pulmonary fibroblast to myofibroblast mediated by Rho-associated coiled-coil forming protein kinase pathway. Zhonghua lao dong wei sheng zhi ye bing za zhi = Chin. J. Ind. Hyg. Occup. Dis. 2013, 31, 654–660. [Google Scholar]
- Deng, H.; Yang, F.; Xu, H.; Sun, Y.; Xue, X.; Du, S.; Wang, X.; Li, S.; Liu, Y.; Wang, R. Ac-SDKP suppresses epithelial–mesenchymal transition in A549 cells via HSP27 signaling. Exp. Mol. Pathol. 2014, 97, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Yao, S.S.; Gong, H.B.; Zhu, L.Y.; Miao, Z.Y.; Deng, H.J. Regulatory effect of Ac-SDKP on phosphorylated heat shock protein 27/SNAI1 pathway in silicotic rats. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi 2022, 40, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, V.; Ntsekhe, M.; Sturrock, E. Investigating the antifibrotic potential of N-acetyl seryl-aspartyl-lysyl-proline sequence peptides. Clin. Exp. Pharmacol. Physiol. 2021, 48, 1558–1565. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yang, F.; Yan, J.; Li, Q.; Wei, Z.; Feng, H.; Wang, R.; Zhang, L.; Zhang, X. New anti-fibrotic mechanisms of n-acetyl-seryl-aspartyl-lysyl-proline in silicon dioxide-induced silicosis. Life Sci. 2010, 87, 232–239. [Google Scholar] [CrossRef]
- Wei, Z.-Q.; Yu, W.-Y.; Feng, H.-L.; Ma, W.-D.; Li, Z.-G.; Xu, H.; Wang, R.-M.; Yang, F. Regulating effect of N-acetyl-seryl-aspartyl-lysyl-proline on activation of c-jun N-terminal kinase pathway in rats with silicosis. Zhonghua lao dong wei sheng zhi ye bing za zhi = Chin. J. Ind. Hyg. Occup. Dis. 2013, 31, 335–340. [Google Scholar]
- Wei, Z.; Sun, Y.; Cheng, H.; Ma, W.; Xu, H.; Li, Q.; Zhang, L.; Wang, R.; Yang, F. Anti-fibrotic role of AcSDKP through inhibition of P38MAPK pathway activity mediated transforming growth beta receptors in rat with silicosis. Zhonghua lao dong wei sheng zhi ye bing za zhi = Chin. J. Ind. Hyg. Occup. Dis. 2014, 32, 340–347. [Google Scholar]
- Li, Q.; Yang, F.; Zhang, L.-J.; Yan, J.-B.; Chen, P.; Li, D.-D.; Wu, K.-F. Antifibrotic effects of N-acetyl-seryl-aspartyl-lysyl-proline mediated by regulation of transforming growth factor beta and connective tissue growth factor expression on rats with silicosis. Zhonghua lao dong wei sheng zhi ye bing za zhi = Chin. J. Ind. Hyg. Occup. Dis. 2009, 27, 390–394. [Google Scholar]
- Tian, J.R.; Yang, F.; Li, D.D.; Zhang, L.J.; Wei, Z.Q.; Feng, H.L.; Li, Z.G.; Wang, R.M. Effect of N-acetyl-seryl-aspartyl-lysyl-proline on regulation of expression of ras-raf-ERK1/2 signal transduction pathway in lung of rats with silicosis. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi 2010, 28, 760–765. [Google Scholar]
- Xiaojun, W.; Yan, L.; Hong, X.; Xianghong, Z.; Shifeng, L.; Dingjie, X.; Xuemin, G.; Lijuan, Z.; Bonan, Z.; Zhongqiu, W.; et al. Acetylated α-Tubulin Regulated by N-Acetyl-Seryl-Aspartyl-Lysyl-Proline(Ac-SDKP) Exerts the Anti-fibrotic Effect in Rat Lung Fibrosis Induced by Silica. Sci. Rep. 2016, 6, 32257. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Gao, X.; Xu, D.; Wang, X.; Liu, Y.; Zhang, L.; Deng, H.; Wei, Z.; Tian, J.; Xu, H.; et al. Inhibition effect of N-acetyl-seryl-aspartyl-lysyl-proline on myofibroblast differentiation by regulating acetylated tubulin α in silicotic rat model. Zhonghua lao dong wei sheng zhi ye bing za zhi = Chin. J. Ind. Hyg. Occup. Dis. 2015, 33, 816–821. [Google Scholar]
- Shifeng, L.; Hong, X.; Xue, Y.; Siyu, N.; Qiaodan, Z.; Dingjie, X.; Lijuan, Z.; Zhongqiu, W.; Xuemin, G.; Wenchen, C.; et al. Ac-SDKP increases α-TAT 1 and promotes the apoptosis in lung fibroblasts and epithelial cells double-stimulated with TGF-β1 and silica. Toxicol. Appl. Pharmacol. 2019, 369, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Budinger, G.R.S. Angiotensin II and pulmonary fibrosis, a new twist on an old story. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2011, 301, L267–L268. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Li, T.; Zhou, G.-S.; Chen, Y.; Yu, C.-H.; Pang, M.-X.; Li, W.; Li, Y.; Zhang, W.-Y.; Li, X. The Angiotensin-Converting Enzyme 2/Angiotensin (1–7)/Mas Axis Protects Against Lung Fibroblast Migration and Lung Fibrosis by Inhibiting the NOX4-Derived ROS-Mediated RhoA/Rho Kinase Pathway. Antioxidants Redox Signal. 2015, 22, 241–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenoy, V.; Ferreira, A.J.; Qi, Y.; Fraga-Silva, R.A.; Díez-Freire, C.; Dooies, A.; Jun, J.Y.; Sriramula, S.; Mariappan, N.; Pourang, D.; et al. The Angiotensin-Converting Enzyme 2/Angiogenesis-(1–7)/Mas Axis Confers Cardiopulmonary Protection against Lung Fibrosis and Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2010, 182, 1065–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Xu, H.; Zhang, B.; Tao, T.; Liu, Y.; Xu, D.; Cai, W.; Wei, Z.; Li, S.; Zhang, H.; et al. Interaction of N-acetyl-seryl-aspartyl-lysyl-proline with the angiotensin-converting enzyme 2–angiotensin-(1–7)–Mas axis attenuates pulmonary fibrosis in silicotic rats. Exp. Physiol. 2019, 104, 1562–1574. [Google Scholar] [CrossRef]
- Zhang, B.; Xu, H.; Zhang, Y.; Yi, X.; Zhang, G.; Zhang, X.; Xu, D.; Gao, X.; Li, S.; Zhu, Y.; et al. Targeting the RAS axis alleviates silicotic fibrosis and Ang II-induced myofibroblast differentiation via inhibition of the hedgehog signaling pathway. Toxicol. Lett. 2019, 313, 30–41. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, F.; Liu, Y.; Peng, H.; Geng, Y.; Li, S.; Xu, H.; Zhu, L.; Yang, X.; Brann, D. Influence of the interaction between Ac-SDKP and Ang II on the pathogenesis and development of silicotic fibrosis. Mol. Med. Rep. 2018, 17, 7467–7476. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Xu, D.; Li, Q.; Yang, Y.; Xu, H.; Wei, Z.; Wang, R.; Zhang, W.; Liu, Y.; Geng, Y.; et al. N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) attenuates silicotic fibrosis by suppressing apoptosis of alveolar type II epithelial cells via mediation of endoplasmic reticulum stress. Toxicol. Appl. Pharmacol. 2018, 350, 1–10. [Google Scholar] [CrossRef]
- Jin, F.; Geng, F.; Xu, D.; Li, Y.; Li, T.; Yang, X.; Liu, S.; Zhang, H.; Wei, Z.; Li, S.; et al. Ac-SDKP Attenuates Activation of Lung Macrophages and Bone Osteoclasts in Rats Exposed to Silica by Inhibition of TLR4 and RANKL Signaling Pathways. J. Inflamm. Res. 2021, 14, 1647–1660. [Google Scholar] [CrossRef]
- Mao, N.; Yang, H.; Yin, J.; Li, Y.; Jin, F.; Li, T.; Yang, X.; Sun, Y.; Liu, H.; Xu, H.; et al. Glycolytic Reprogramming in Silica-Induced Lung Macrophages and Silicosis Reversed by Ac-SDKP Treatment. Int. J. Mol. Sci. 2021, 22, 10063. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.; Jia, W.; Zhang, C. The Role of Tβ4-POP-Ac-SDKP Axis in Organ Fibrosis. Int. J. Mol. Sci. 2022, 23, 13282. https://doi.org/10.3390/ijms232113282
Wang W, Jia W, Zhang C. The Role of Tβ4-POP-Ac-SDKP Axis in Organ Fibrosis. International Journal of Molecular Sciences. 2022; 23(21):13282. https://doi.org/10.3390/ijms232113282
Chicago/Turabian StyleWang, Wei, Wenning Jia, and Chunping Zhang. 2022. "The Role of Tβ4-POP-Ac-SDKP Axis in Organ Fibrosis" International Journal of Molecular Sciences 23, no. 21: 13282. https://doi.org/10.3390/ijms232113282