
Evolution of an Iron-Detoxifying Protein: Eukaryotic and Rickettsia Frataxins Contain a Conserved Site Which Is Not Present in Their Bacterial Homologues
 , and
, and
Abstract
:1. Introduction
2. Results
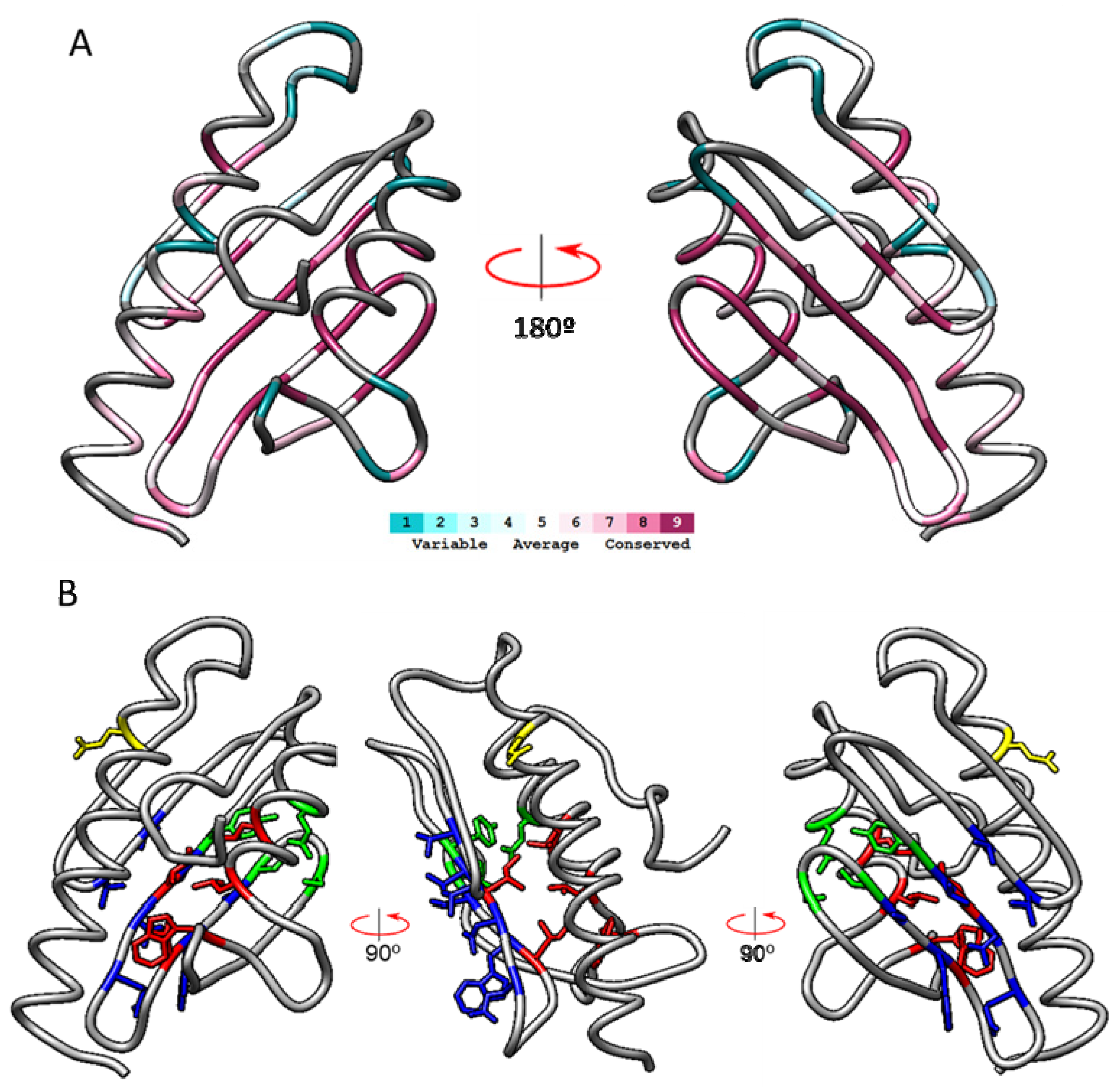
2.1. Analysis of Mature Frataxin by ConSurf
2.2. Coevolution of Amino Acids from Cluster 3
2.3. Cluster 3 in the Structures of S. cerevisiae Frataxin, E. coli CyaY and Rickettsia CyaY
2.4. Structural and Functional Effects of Cluster 3
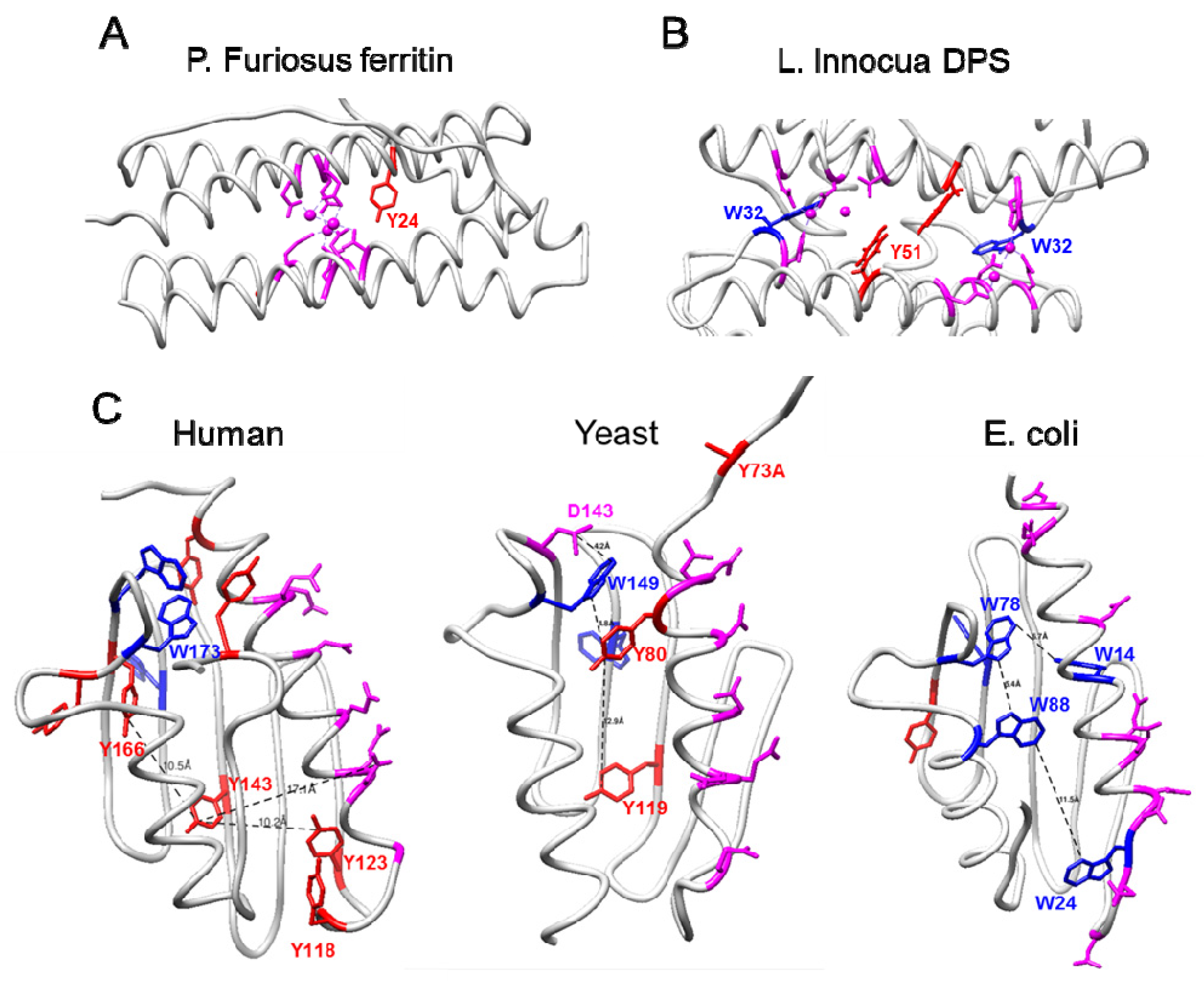
2.5. Similarities between Frataxins and Iron-Detoxifying Proteins
2.6. Variants in the Human Population
3. Discussion
4. Materials and Methods
4.1. Conservation Analysis
4.2. Protein Molecular Graphics and Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Campuzano, V.; Montermini, L.; Moltò, M.D.; Pianese, L.; Cossée, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s Ataxia: Autosomal Recessive Disease Caused by an Intronic GAA Triplet Repeat Expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef] [PubMed]
- Galea, C.A.; Huq, A.; Lockhart, P.J.; Tai, G.; Corben, L.A.; Yiu, E.M.; Gurrin, L.C.; Lynch, D.R.; Gelbard, S.; Durr, A.; et al. Compound heterozygousFXNmutations and clinical outcome in friedreich ataxia. Ann. Neurol. 2016, 79, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Cao, Y.; Dai, X.; Marelja, Z.; Zhou, D.; Mo, R.; Al-Mahdawi, S.; Pook, M.A.; Leimkühler, S.; Rouault, T.A.; et al. Novel Frataxin Isoforms May Contribute to the Pathological Mechanism of Friedreich Ataxia. PLoS ONE 2012, 7, e47847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrò, M.; Díaz-Nido, J. Effect of Mitochondrial and Cytosolic FXN Isoform Expression on Mitochondrial Dynamics and Metabolism. Int. J. Mol. Sci. 2020, 21, 8251. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Wang, Q.; Weng, L.; Hauser, L.A.; Strawser, C.J.; Mesaros, C.; Lynch, D.R.; Blair, I.A. Characterization of a new N-terminally acetylated extra-mitochondrial isoform of frataxin in human erythrocytes. Sci. Rep. 2018, 8, 17043. [Google Scholar] [CrossRef] [PubMed]
- Weng, L.; Laboureur, L.; Wang, Q.; Guo, L.; Xu, P.; Gottlieb, L.; Lynch, D.R.; Mesaros, C.; Blair, I.A. Extra-mitochondrial mouse frataxin and its implications for mouse models of Friedreich’s ataxia. Sci. Rep. 2020, 10, 15788. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, S.; Nair, M.; Politou, A.; Bayer, E.; Martin, S.; Temussi, P.; Pastore, A. The Factors Governing the Thermal Stability of Frataxin Orthologues: How To Increase a Protein’s Stability. Biochemistry 2004, 43, 6511–6518. [Google Scholar] [CrossRef] [PubMed]
- Pastore, A.; Puccio, H. Frataxin: A protein in search for a function. J. Neurochem. 2013, 126, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Gentry, L.E.; Thacker, M.A.; Doughty, R.; Timkovich, R.; Busenlehner, L.S. His86 from the N-Terminus of Frataxin Coordinates Iron and Is Required for Fe–S Cluster Synthesis. Biochemistry 2013, 52, 6085–6096. [Google Scholar] [CrossRef]
- Moreno-Cermeño, A.; Obis, E.; Bellí, G.; Cabiscol, E.; Ros, J.; Tamarit, J. Frataxin Depletion in Yeast Triggers Up-regulation of Iron Transport Systems before Affecting Iron-Sulfur Enzyme Activities. J. Biol. Chem. 2010, 285, 41653–41664. [Google Scholar] [CrossRef] [PubMed]
- Monnier, V.; Llorens, J.V.; Navarro, J.A. Impact of Drosophila Models in the Study and Treatment of Friedreich’s Ataxia. Int. J. Mol. Sci. 2018, 19, 1989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitnall, M.; Rahmanto, Y.S.; Huang, M.L.H.; Saletta, F.; Lok, H.C.; Gutierrez, L.; Lazaro, F.J.; Fleming, A.J.; St Pierre, T.; Mikhael, M.R.; et al. Identification of nonferritin mitochondrial iron deposits in a mouse model of Friedreich ataxia. Proc. Natl. Acad. Sci. USA 2012, 109, 20590–20595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patra, S.; Barondeau, D.P. Mechanism of activation of the human cysteine desulfurase complex by frataxin. Proc. Natl. Acad. Sci. USA 2019, 116, 19421–19430. [Google Scholar] [CrossRef] [Green Version]
- Maio, N.; Jain, A.; Rouault, T.A. Mammalian iron–sulfur cluster biogenesis: Recent insights into the roles of frataxin, acyl carrier protein and ATPase-mediated transfer to recipient proteins. Curr. Opin. Chem. Biol. 2020, 55, 34–44. [Google Scholar] [CrossRef]
- Tamarit, J.; Britti, E.; Delaspre, F.; Medina-Carbonero, M.; Sanz-Alcázar, A.; Cabiscol, E.; Ros, J. Crosstalk between nucleus and mitochondria in human disease: Mitochondrial iron and calcium homeostasis in Friedreich ataxia. IUBMB Life 2021, 73, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Cermeño, A.; Alsina, D.; Cabiscol, E.; Tamarit, J.; Ros, J. Metabolic remodeling in frataxin-deficient yeast is mediated by Cth2 and Adr1. Biochim. et Biophys. Acta 2013, 1833, 3326–3337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alsina, D.; Ros, J.; Tamarit, J. Nitric oxide prevents Aft1 activation and metabolic remodeling in frataxin-deficient yeast. Redox Biol. 2018, 14, 131–141. [Google Scholar] [CrossRef]
- Park, S.; Gakh, O.; Mooney, S.M.; Isaya, G. The Ferroxidase Activity of Yeast Frataxin. J. Biol. Chem. 2002, 277, 38589–38595. [Google Scholar] [CrossRef] [Green Version]
- Bou-Abdallah, F.; Adinolfi, S.; Pastore, A.; Laue, T.M.; Chasteen, N.D. Iron Binding and Oxidation Kinetics in Frataxin CyaY of Escherichia coli. J. Mol. Biol. 2004, 341, 605–615. [Google Scholar] [CrossRef]
- Bedekovics, T.; Gajdos, G.B.; Kispal, G.; Isaya, G. Partial conservation of functions between eukaryotic frataxin and the Escherichia coli frataxin homolog CyaY. FEMS Yeast Res. 2007, 7, 1276–1284. [Google Scholar] [CrossRef]
- Panchenko, A.R.; Kondrashov, F.; Bryant, S. Prediction of functional sites by analysis of sequence and structure conservation. Protein Sci. 2004, 13, 884–892. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wu, M. An integrated phylogenomic approach toward pinpointing the origin of mitochondria. Sci. Rep. 2015, 5, srep07949. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Peng, Z.; Zhang, Y.; Yang, J. COACH-D: Improved protein–ligand binding sites prediction with refined ligand-binding poses through molecular docking. Nucleic Acids Res. 2018, 46, W438–W442. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, A.J.M.; Holliday, G.; Furnham, N.; Tyzack, J.; Ferris, K.; Thornton, J. Mechanism and Catalytic Site Atlas (M-CSA): A database of enzyme reaction mechanisms and active sites. Nucleic Acids Res. 2017, 46, D618–D623. [Google Scholar] [CrossRef]
- Handing, K.B.; Niedzialkowska, E.; Shabalin, I.G.; Kuhn, M.L.; Zheng, H.; Minor, W. Characterizing metal-binding sites in proteins with X-ray crystallography. Nat. Protoc. 2018, 13, 1062–1090. [Google Scholar] [CrossRef]
- Ebrahimi, K.H.; Hagedoorn, P.-L.; Hagen, W.R. A Conserved Tyrosine in Ferritin Is a Molecular Capacitor. ChemBioChem 2013, 14, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Bellapadrona, G.; Ardini, M.; Ceci, P.; Stefanini, S.; Chiancone, E. Dps proteins prevent Fenton-mediated oxidative damage by trapping hydroxyl radicals within the protein shell. Free Radic. Biol. Med. 2010, 48, 292–297. [Google Scholar] [CrossRef]
- Bogatyreva, N.; Finkelstein, A.; Galzitskaya, O.V. Trend of amino acid composition of proteins of different taxa. J. Bioinform. Comput. Biol. 2006, 4, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Stephenson, J.D.; Sillitoe, I.; Orengo, C.A.; Thornton, J. VarSite: Disease variants and protein structure. Protein Sci. 2019, 29, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Gakh, O.; Iv, D.Y.S.; Ranatunga, W.K.; Isaya, G. Missense Mutations Linked to Friedreich Ataxia Have Different but Synergistic Effects on Mitochondrial Frataxin Isoforms. J. Biol. Chem. 2013, 288, 4116–4127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavadini, P.; Adamec, J.; Taroni, F.; Gakh, O.; Isaya, G. Two-step Processing of Human Frataxin by Mitochondrial Processing Peptidase. J. Biol. Chem. 2000, 275, 41469–41475. [Google Scholar] [CrossRef] [Green Version]
- Shan, Y.; Napoli, E.; Cortopassi, G. Mitochondrial frataxin interacts with ISD11 of the NFS1/ISCU complex and multiple mitochondrial chaperones. Hum. Mol. Genet. 2007, 16, 929–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pace, C.; Horn, G.; Hebert, E.J.; Bechert, J.; Shaw, K.; Urbanikova, L.; Scholtz, J.; Sevcik, J. Tyrosine hydrogen bonds make a large contribution to protein stability. J. Mol. Biol. 2001, 312, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Moosmann, B.; Behl, C. Cytoprotective antioxidant function of tyrosine and tryptophan residues in transmembrane proteins. JBIC J. Biol. Inorg. Chem. 2000, 267, 5687–5692. [Google Scholar] [CrossRef] [Green Version]
- Foury, F.; Pastore, A.; Trincal, M. Acidic residues of yeast frataxin have an essential role in Fe–S cluster assembly. EMBO Rep. 2006, 8, 194–199. [Google Scholar] [CrossRef] [Green Version]
- Bridwell-Rabb, J.; Iannuzzi, C.; Pastore, A.; Barondeau, D.P. Effector Role Reversal during Evolution: The Case of Frataxin in Fe–S Cluster Biosynthesis. Biochemistry 2012, 51, 2506–2514. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SEQUENCE | Y143 | S158 | S161 | E189 | Organism |
|---|---|---|---|---|---|
| Input Sequence (HUMAN) | yes | yes | yes | yes | Homo sapiens |
| FRDA_BOVIN | yes | yes | yes | yes | Bos taurus |
| FRDA_MOUSE | yes | yes | yes | yes | Mus musculus |
| FRDA_RAT | yes | G | yes | yes | Rattus norvegicus |
| FRDA_DROME | yes | yes | yes | yes | Drosophila melanogaster |
| FRDA_CAEEL | yes | yes | yes | yes | Caenorhabditis elegans |
| FRDA_ARATH | yes | yes | yes | yes | Arabidopsis thaliana |
| FRDA_SCHPO | yes | yes | yes | yes | Schizosaccharomyces pombe |
| FRDA_DICDI | yes | yes | yes | yes | Dictyostelium discoideum |
| FRDA_YEAST | yes | yes | yes | yes | Saccharomyces cerevisiae |
| CYAY_RICAE | yes | yes | yes | yes | Rickettsia africae |
| CYAY_RICFE | yes | yes | yes | yes | Rickettsia felis |
| FRDA_ENCCU | yes | yes | T | yes | Encephalitozoon cuniculi |
| CYAY_NEIMB | I | A | G | A | Neisseria meningitidis serogroup B |
| CYAY_JANMA | I | A | G | M | Janthinobacterium sp. |
| CYAY_RICBR | yes | yes | yes | yes | Rickettsia bellii |
| CYAY_RICAH | yes | yes | yes | yes | Rickettsia akari |
| CYAY_AZOVD | L | A | G | V | Azotobacter vinelandii |
| CYAY_RICTY | yes | yes | yes | yes | Rickettsia typhi |
| CYAY_PSEPG | L | D | G | I | Pseudomonas putida |
| CYAY_TOLAT | I | T | N | A | Tolumonas auensis |
| CYAY_SALPK | I | T | yes | A | Salmonella paratyphi A |
| CYAY_PSEPF | L | A | G | I | Pseudomonas fluorescens |
| CYAY_THISH | L | yes | G | - | Thioalkalivibrio sulfidiphilus |
| CYAY_SALA4 | I | T | G | A | Salmonella agona |
| CYAY_AERS4 | V | T | N | A | Aeromonas salmonicida |
| CYAY_ECO81 | I | T | G | A | Escherichia coli O81 |
| CYAY_EDWI9 | I | T | G | - | Edwardsiella ictaluri |
| CYAY_ALISL | I | yes | G | yes | Aliivibrio salmonicida |
| CYAY_CHLT3 | yes | E | N | L | Chloroherpeton thalassium |
| CYAY_PSEF5 | V | A | G | I | Pseudomonas fluorescens |
| CYAY_VIBCH | I | yes | G | yes | Vibrio cholerae serotype O1 |
| CYAY_PSEU2 | L | A | G | F | Pseudomonas syringae pv. syringae |
| CYAY_PSESM | L | A | G | M | Pseudomonas syringae pv. tomato |
| CYAY_VIBPA | I | yes | G | yes | Vibrio parahaemolyticus serotype O3:K6 |
| CYAY_VIBVU | I | yes | G | yes | Vibrio vulnificus |
| CYAY_ALIFM | I | yes | G | yes | Aliivibrio fischeri |
| CYAY_PSEPW | L | D | G | I | Pseudomonas putida |
| CYAY_VIBTL | I | yes | G | yes | Vibrio atlanticus |
| CYAY_PSEFS | L | A | G | L | Pseudomonas fluorescens |
| CYAY_PHOLL | I | T | G | A | Photorhabdus laumondii subsp. laumondii |
| CYAY_HERAR | I | T | G | - | Herminiimonas arsenicoxydans |
| CYAY_CROS8 | I | T | G | A | Cronobacter sakazakii |
| CYAY_SALNS | I | T | G | A | Salmonella newport |
| CYAY_PSEAE | L | A | G | - | Pseudomonas aeruginosa |
| CYAY_ENT38 | I | T | G | A | Enterobacter sp. |
| CYAY_DICCH | I | T | G | A | Dickeya chrysanthemi |
| Cluster | Position | Variant GnomAD | Freq v3.1.1 | Freq v2.1.1 | Pathological Variant |
|---|---|---|---|---|---|
| C1 | I145 | No | No | ||
| I154 | F | 0.742 | 0.872 | Yes | |
| V | 0.742 | 9.41 | No | ||
| W173 | No | Yes | |||
| L182 | No | Yes | |||
| L186 | No | Yes | |||
| C2 | V131 | No | No | ||
| T133 | A | 0.742 | 1.922 | No | |
| V144 | No | No | |||
| N146 | No | Yes | |||
| Q148 | No | Yes | |||
| W155 | No | Yes | |||
| S157 | No | No | |||
| C3 | Y143 | No | No | ||
| S158 | P | 0.742 | 4.703 | No | |
| S161 | R | 0.742 | No | ||
| E189 | No | No | |||
| C4 | E111 | No | No | ||
| Acidic Ridge | D112 | Y | 0.961 | No | |
| H | 0.742 | No | |||
| E100 | A | 9.65 | 24.505 | No |
| Residue | Mutation | rs ID | ConSurf Score (35ID) | ConSurf Score (20ID) | Conservation Cluster |
|---|---|---|---|---|---|
| LEU106 | S | rs104894105 | 7 | 7 | - |
| ASP122 | Y | rs142157346 | 1 | 6 | - |
| GLY130 | V | rs104894107 | 6 | 8 | - |
| ASN146 | K | rs146818694 | 9 | 9 | Cluster 2 |
| GLN148 | R | rs140472905 | 9 | 9 | Cluster 2 |
| ILE154 | F | rs104894106 | 9 | 9 | Cluster 1 |
| TRP155 | R | rs138471431 | 9 | 9 | Cluster 2 |
| LEU156 | P | rs143340609 | 6 | 7 | - |
| ARG165 | C | rs138034837 | 7 | 9 | Cluster 2 |
| TRP173 | G | rs56214919 | 9 | 9 | Cluster 1 |
| LEU182 | F | rs139616452 | 9 | 8 | Cluster 1 |
| H | rs149335881 | ||||
| HIS183 | R | rs144610605 | 3 | 1 | - |
| LEU186 | R | rs148443992 | 9 | 9 | Cluster 1 |
| LEU198 | R | rs144104124 | 5 | 5 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alves, R.; Pazos-Gil, M.; Medina-Carbonero, M.; Sanz-Alcázar, A.; Delaspre, F.; Tamarit, J. Evolution of an Iron-Detoxifying Protein: Eukaryotic and Rickettsia Frataxins Contain a Conserved Site Which Is Not Present in Their Bacterial Homologues. Int. J. Mol. Sci. 2022, 23, 13151. https://doi.org/10.3390/ijms232113151
Alves R, Pazos-Gil M, Medina-Carbonero M, Sanz-Alcázar A, Delaspre F, Tamarit J. Evolution of an Iron-Detoxifying Protein: Eukaryotic and Rickettsia Frataxins Contain a Conserved Site Which Is Not Present in Their Bacterial Homologues. International Journal of Molecular Sciences. 2022; 23(21):13151. https://doi.org/10.3390/ijms232113151
Chicago/Turabian StyleAlves, Rui, Maria Pazos-Gil, Marta Medina-Carbonero, Arabela Sanz-Alcázar, Fabien Delaspre, and Jordi Tamarit. 2022. "Evolution of an Iron-Detoxifying Protein: Eukaryotic and Rickettsia Frataxins Contain a Conserved Site Which Is Not Present in Their Bacterial Homologues" International Journal of Molecular Sciences 23, no. 21: 13151. https://doi.org/10.3390/ijms232113151