
From Cyclo[18]carbon to the Novel Nanostructures—Theoretical Predictions


Abstract
:
1. Introduction
2. Results and Discussion
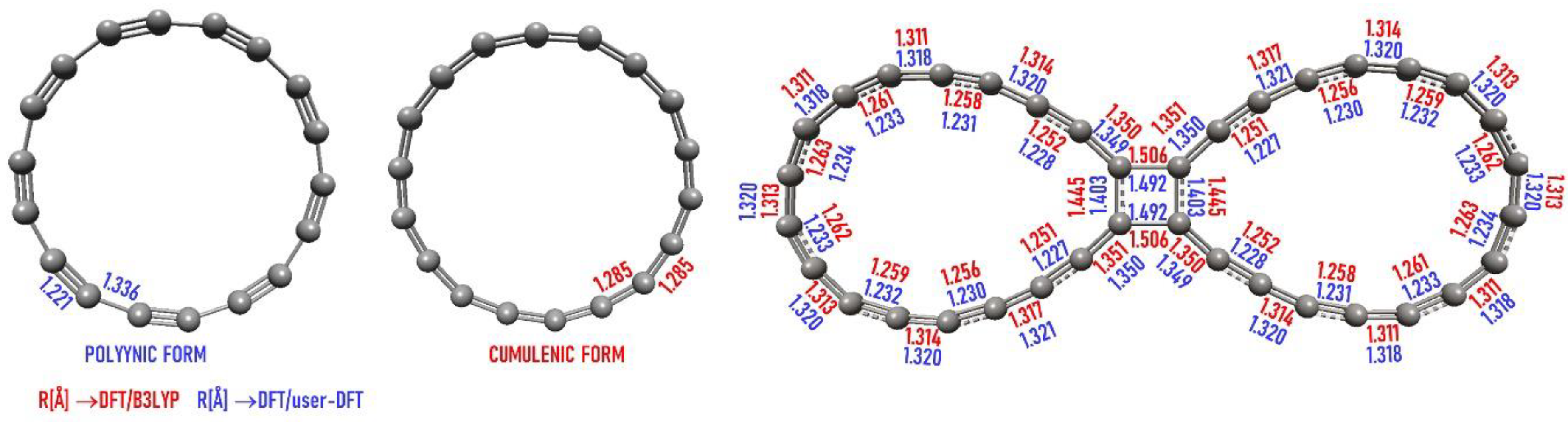
2.1. Polyynic versus Cumulenic
- in C36-p: 1.49 and 1.40 in the 4-membered ring and then 1.35 and 1.23, 1.32, 1.23, etc., in the circle
- in C36-c: 1.51 and 1.45 in the 4-membered ring and then 1.35 and 1.25, 1.32, 1.25, etc., in the circle
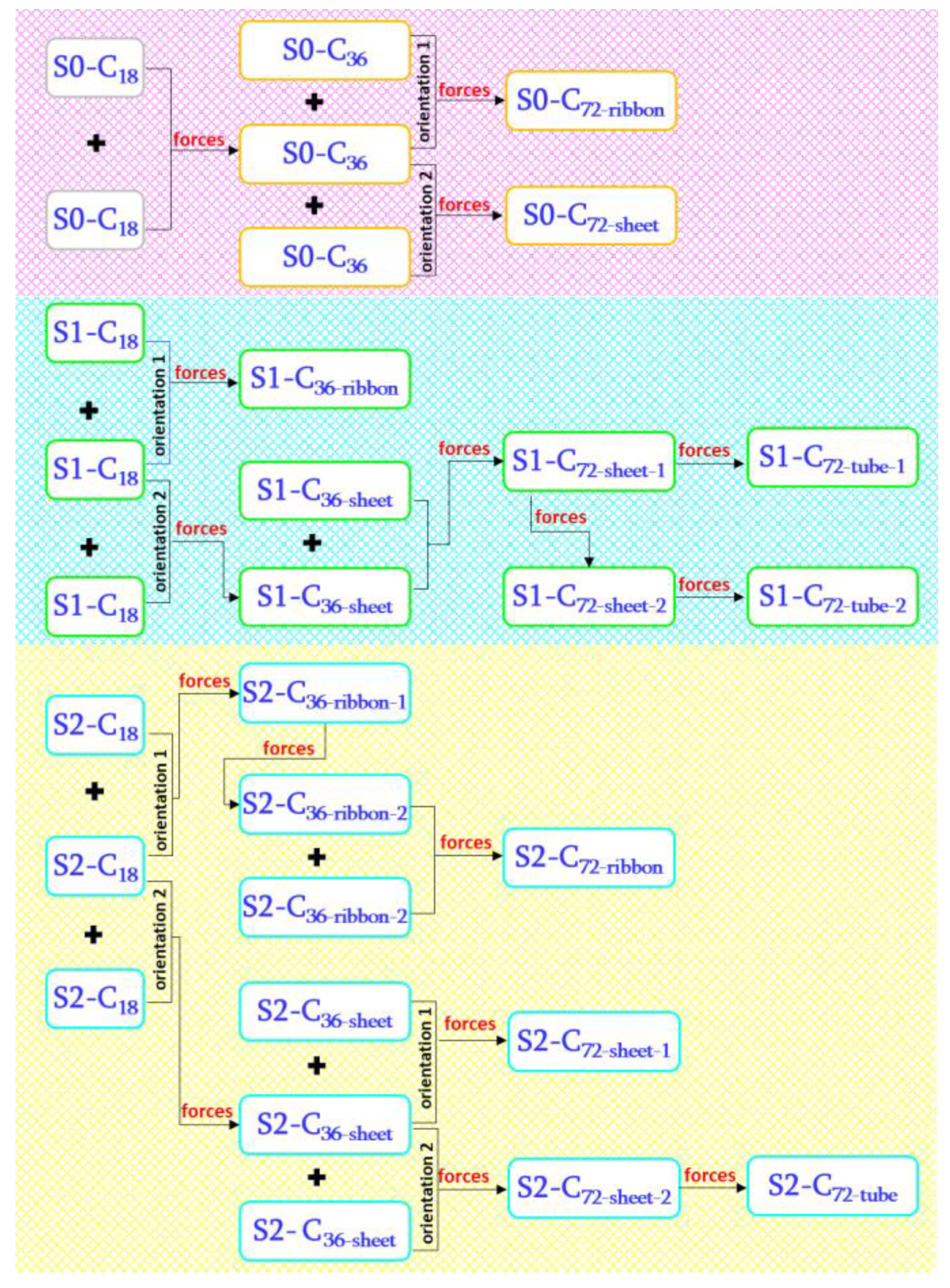
2.2. Running Virtual Reactions with External EGO Forces
2.3. Forming Squeezed S1C18 and S2C18 Molecules
2.4. Forming S0-C36 and S0-C72 Molecules
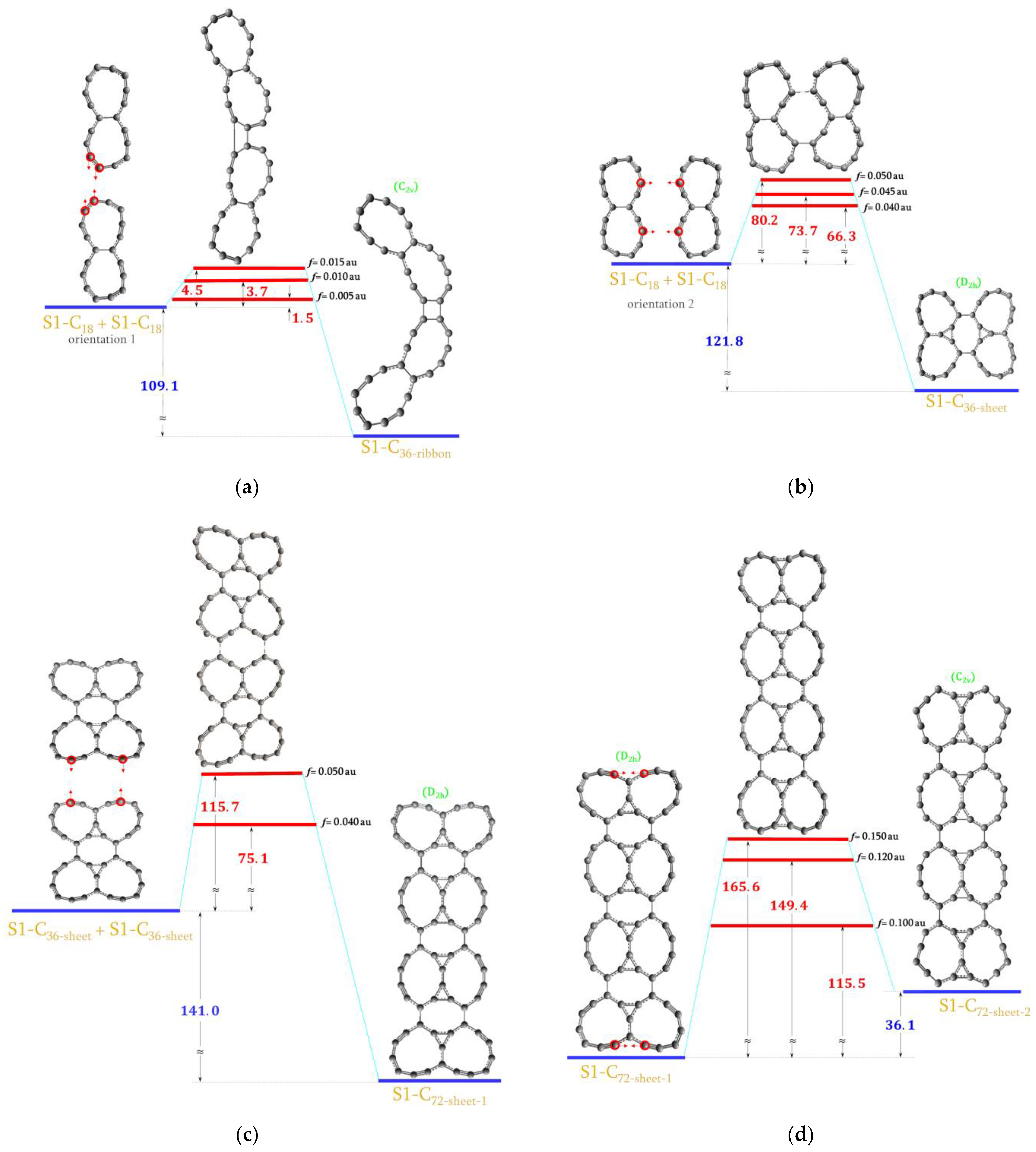
2.5. Forming S1-C36 and S1-C72 Molecules
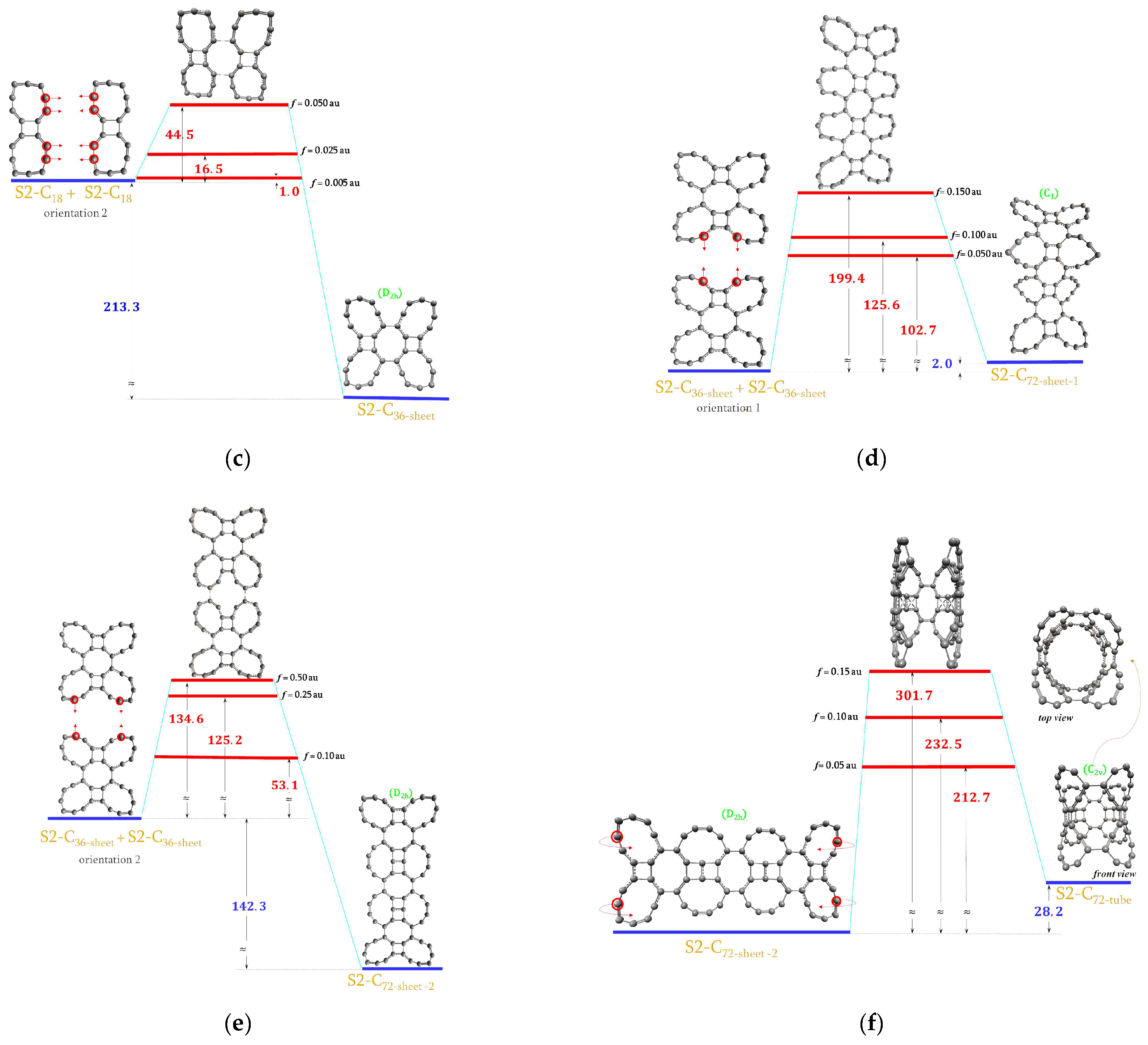
2.6. Forming S2-C36 and S2-C72 Molecules
2.7. Energy Requirement
2.8. HOMO–LUMO Gap
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Diederich, F.; Rubin, Y. Synthetic Approaches toward Molecular and Polymeric Carbon Allotropes. Angew. Chem. Int. Ed. Engl. 1992, 31, 1101–1123. [Google Scholar] [CrossRef]
- Diederich, F. Carbon scaffolding: Building acetylenic all-carbon and carbon-rich compounds. Nature 1994, 369, 199–207. [Google Scholar] [CrossRef]
- Diederich, F.; Kivala, M. All-Carbon Scaffolds by Rational Design. Adv. Mater. 2010, 22, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, K.; Scriven, L.M.; Schulz, F.; Gawel, P.; Gross, L.; Anderson, H.L. An sp-hybridized molecular carbon allotrope, cyclo [18]carbon. arXiv 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neiss, C.; Trushin, E.; Görling, A. The Nature of One-Dimensional Carbon: Polyynic versus Cumulenic. ChemPhysChem 2014, 15, 2497–2502. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Seal, P.; Chakrabarti, S. Explicit role of dynamical and nondynamical electron correlations on broken symmetry in C 4 N + 2 clusters. Phys. Rev. B 2006, 73, 245401. [Google Scholar] [CrossRef]
- Torelli, T.; Mitas, L. Electron Correlation in C4N+2 Carbon Rings: Aromatic versus Dimerized Structures. Phys. Rev. Lett. 2000, 85, 1702–1705. [Google Scholar] [CrossRef] [Green Version]
- Diederich, F.; Rubin, Y.; Knobler, C.B.; Whetten, R.L.; Schriver, K.E.; Houk, K.N.; Li, Y. All-Carbon Molecules: Evidence for the Generation of Cyclo[18]carbon from a Stable Organic Precursor. Science 1989, 245, 1088–1090. [Google Scholar] [CrossRef] [PubMed]
- Arulmozhiraja, S.; Ohno, T. CCSD calculations on C14, C18, and C22 carbon clusters. J. Chem. Phys. 2008, 128, 114301. [Google Scholar] [CrossRef] [PubMed]
- Ott, A.K.; Rechtsteiner, G.A.; Felix, C.; Hampe, O.; Jarrold, M.F.; Van Duyne, R.P.; Raghavachari, K. Raman spectra and calculated vibrational frequencies of size-selected C16, C18, and C20 clusters. J. Chem. Phys. 1998, 109, 9652–9655. [Google Scholar] [CrossRef]
- Rechtsteiner, G.A.; Felix, C.; Ott, A.K.; Hampe, O.; Van Duyne, R.P.; Jarrold, M.F.; Raghavachari, K. Raman and Fluorescence Spectra of Size-Selected, Matrix-Isolated C14 and C18 Neutral Carbon Clusters. J. Phys. Chem. A 2001, 105, 3029–3033. [Google Scholar] [CrossRef]
- Parasuk, V.; Almlof, J.; Feyereisen, M.W. The [18] all-carbon molecule: Cumulene or polyacetylene? J. Am. Chem. Soc. 1991, 113, 1049–1050. [Google Scholar] [CrossRef]
- Feyereisen, M.; Gutowski, M.; Simons, J.; Almlöf, J. Relative stabilities of fullerene, cumulene, and polyacetylene structures for Cn: N=18–60. J. Chem. Phys. 1992, 96, 2926–2932. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.O. Density functional study of carbon clusters C2n (2≤n≤16). I. Structure and bonding in the neutral clusters. J. Chem. Phys. 1999, 110, 5189–5200. [Google Scholar] [CrossRef] [Green Version]
- Boguslavskiy, A.E.; Ding, H.; Maier, J.P. Gas-phase electronic spectra of C18 and C22 rings. J. Chem. Phys. 2005, 123, 034305. [Google Scholar] [CrossRef]
- Fowler, P.W.; Mizoguchi, N.; Bean, D.E.; Havenith, R.W.A. Double Aromaticity and Ring Currents in All-Carbon Rings. Chem. Eur. J. 2009, 15, 6964–6972. [Google Scholar] [CrossRef]
- Hutter, J.; Luethi, H.P.; Diederich, F. Structures and vibrational frequencies of the carbon molecules C2-C18 calculated by density functional theory. J. Am. Chem. Soc. 1994, 116, 750–756. [Google Scholar] [CrossRef]
- Remya, K.; Suresh, C.H. Carbon rings: A DFT study on geometry, aromaticity, intermolecular carbon–carbon interactions and stability. RSC Adv. 2016, 6, 44261–44271. [Google Scholar] [CrossRef]
- Murphy, V.L.; Farfan, C.; Kahr, B. Chiroptical structure-property relations in cyclo[18]carbon and its in silico hydrogenation products. Chirality 2018, 30, 325–331. [Google Scholar] [CrossRef]
- Stasyuk, A.J.; Stasyuk, O.A.; Solŕ, M.; Voityuk, A.A. Cyclo[18]carbon: The smallest all-carbon electron acceptor. Chem. Commun. 2020, 56, 352–355. [Google Scholar] [CrossRef]
- Baryshnikov, G.V.; Valiev, R.R.; Kuklin, A.V.; Sundholm, D.; Ĺgren, H. Cyclo[18]carbon: Insight into Electronic Structure, Aromaticity, and Surface Coupling. J. Phys. Chem. Lett. 2019, 10, 6701–6705. [Google Scholar] [CrossRef]
- Plattner, D.A.; Houk, K.N. C18 Is a Polyyne. J. Am. Chem. Soc. 1995, 117, 4405–4406. [Google Scholar] [CrossRef]
- Fang, S.; Hu, Y.H. Cyclo[18]carbon as an ultra-elastic molecular O-ring with unique mechanical properties. Carbon 2021, 171, 96–103. [Google Scholar] [CrossRef]
- Altass, H.M.; Morad, M.; Khder, A.E.-R.S.; Mannaa, M.A.; Jassas, R.S.; Alsimaree, A.A.; Ahmed, S.A.; Salama, R.S. Enhanced Catalytic Activity for CO Oxidation by Highly Active Pd Nanoparticles Supported on Reduced Graphene Oxide /Copper Metal Organic Framework. J. Taiwan Inst. Chem. Eng. 2021, 128, 194–208. [Google Scholar] [CrossRef]
- Alshorifi, F.T.; Alswat, A.A.; Salama, R.S. Gold-selenide quantum dots supported onto cesium ferrite nanocomposites for the efficient degradation of rhodamine B. Heliyon 2022, 8, e09652. [Google Scholar] [CrossRef]
- Wolinski, K.; Baker, J. Theoretical predictions of enforced structural changes in molecules. Mol. Phys. 2009, 107, 2403–2417. [Google Scholar] [CrossRef] [Green Version]
- Wolinski, K.; Brzyska, A. Theoretical studies of the pyranose ring under mechanical stress. Carbohydr. Res. 2018, 470, 64–72. [Google Scholar] [CrossRef]
- Baker, J.; Wolinski, K. Isomerization of stilbene using enforced geometry optimization. J. Comput. Chem. 2011, 32, 43–53. [Google Scholar] [CrossRef]
- Baker, J.; Wolinski, K. Kinetically stable high-energy isomers of C14H12 and C12H10N2 derived from cis-stilbene and cis-azobenzene. J. Mol. Model. 2011, 17, 1335–1342. [Google Scholar] [CrossRef] [Green Version]
- Brzyska, A.; Płaziński, W.; Woliński, K. Force-induced structural changes in non-sulfated carrageenan based oligosaccharides—A theoretical study. Soft Matter 2018, 14, 6264–6277. [Google Scholar] [CrossRef]
- Brzyska, A.; Woliński, K. Simulation of the conformational flexibility of the mycodextran under external forces. Biopolymers 2020, 111, e23357. [Google Scholar] [CrossRef] [PubMed]
- Cybulska, J.; Brzyska, A.; Zdunek, A.; Woliński, K. Simulation of Force Spectroscopy Experiments on Galacturonic Acid Oligomers. PLoS ONE 2014, 9, e107896. [Google Scholar] [CrossRef] [PubMed]
- Brzyska, A.; Woliński, K. Enforced conformational changes in the structural units of glycosaminoglycan (non-sulfated heparin-based oligosaccharides). RSC Adv. 2014, 4, 36640–36648. [Google Scholar] [CrossRef]
- Brzyska, A.; Korycki, P.; Woliński, K. The carbohydrate glycosylphosphatidylinositol anchor chain under mechanical stress. Carbohydr. Res. 2022, in press. [Google Scholar] [CrossRef]
- Luo, Y.; Ren, C.; Xu, Y.; Yu, J.; Wang, S.; Sun, M. A first principles investigation on the structural, mechanical, electronic, and catalytic properties of biphenylene. Sci. Rep. 2021, 11, 19008. [Google Scholar] [CrossRef]
- Xie, Y.; Chen, L.; Xu, J.; Liu, W. Effective regulation of the electronic properties of a biphenylene network by hydrogenation and halogenation. RSC Adv. 2022, 12, 20088–20095. [Google Scholar] [CrossRef]
- Németh, P.; McColl, K.; Garvie, L.A.J.; Salzmann, C.G.; Murri, M.; McMillan, P.F. Complex nanostructures in diamond. Nat. Mater. 2020, 19, 1126–1131. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wu, C.; Pan, Q.; Jin, Y.; Lyu, R.; Martinez, V.; Huang, S.; Wu, J.; Wayment, L.J.; Clark, N.A.; et al. Synthesis of γ-graphyne using dynamic covalent chemistry. Nat. Synth. 2022, 1, 449–454. [Google Scholar] [CrossRef]
- Pavliček, N.; Gawel, P.; Kohn, D.R.; Majzik, Z.; Xiong, Y.; Meyer, G.; Anderson, H.L.; Gross, L. Polyyne formation via skeletal rearrangement induced by atomic manipulation. Nat. Chem. 2018, 10, 853–858. [Google Scholar] [CrossRef]
- Scriven, L.M.; Kaiser, K.; Schulz, F.; Sterling, A.J.; Woltering, S.L.; Gawel, P.; Christensen, K.E.; Anderson, H.L.; Gross, L. Synthesis of Cyclo[18]carbon via Debromination of C18 Br6. J. Am. Chem. Soc. 2020, 142, 12921–12924. [Google Scholar] [CrossRef] [PubMed]
- Wolinski, K. Exploring Potential Energy Surface with External Forces. J. Chem. Theory Comput. 2018, 14, 6306–6316. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Harabuchi, Y.; Takagi, M.; Taketsugu, T.; Morokuma, K. Artificial Force Induced Reaction (AFIR) Method for Exploring Quantum Chemical Potential Energy Surfaces. Chem. Rec. 2016, 16, 2232–2248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatanaka, M.; Yoshimura, T.; Maeda, S. Artificial Force-Induced Reaction Method for Systematic Elucidation of Mechanism and Selectivity in Organometallic Reactions. In New Directions in the Modeling of Organometallic Reactions; Topics in Organometallic Chemistry; Lledós, A., Ujaque, G., Eds.; Springer International Publishing: Cham, Switzerland, 2020; Volume 67, pp. 57–80. ISBN 978-3-030-56995-2. [Google Scholar]
- Wolinski, K.; Baker, J. Geometry optimization in the presence of external forces: A theoretical model for enforced structural changes in molecules. Mol. Phys. 2010, 108, 1845–1856. [Google Scholar] [CrossRef]
- Brzyska, A.; Woliński, K. Isomerization and Decomposition of 2-Methylfuran with External Forces. J. Chem. Inf. Modeling 2019, 59, 3454–3463. [Google Scholar] [CrossRef]
- Zhan, C.-G.; Nichols, J.A.; Dixon, D.A. Ionization Potential, Electron Affinity, Electronegativity, Hardness, and Electron Excitation Energy: Molecular Properties from Density Functional Theory Orbital Energies. J. Phys. Chem. A 2003, 107, 4184–4195. [Google Scholar] [CrossRef] [Green Version]
- Perepichka, D.F.; Bryce, M.R. Molecules with Exceptionally Small HOMO-LUMO Gaps. Angew. Chem. Int. Ed. 2005, 44, 5370–5373. [Google Scholar] [CrossRef] [PubMed]
- Berlin, A.; Zotti, G.; Zecchin, S.; Schiavon, G.; Vercelli, B.; Zanelli, A. New Low-Gap Polymers from 3,4-Ethylenedioxythiophene-Bis-Substituted Electron-Poor Thiophenes. The Roles of Thiophene, Donor−Acceptor Alternation, and Copolymerization in Intrinsic Conductivity. Chem. Mater. 2004, 16, 3667–3676. [Google Scholar] [CrossRef]
- Karaush-Karmazin, N.N.; Baryshnikov, G.V.; Kuklin, A.V.; Saykova, D.I.; Ĺgren, H.; Minaev, B.F. Impact of molecular and packing structure on the charge-transport properties of hetero[8]circulenes. J. Mater. Chem. C 2021, 9, 1451–1466. [Google Scholar] [CrossRef]
- Xu, Y.; Wu, W. High-efficiency switching effect and negative differential conductance in cyclo[18]carbon–graphene nanoribbon junction. J. Appl. Phys. 2020, 128, 194303. [Google Scholar] [CrossRef]
- Sajid, H.; Ayub, K.; Mahmood, T. Sensing behaviour of monocyclic C18 and B9N9 analogues toward chemical warfare agents (CWAs); quantum chemical approach. Surf. Interfaces 2022, 30, 101912. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef] [Green Version]
- Baker, J.; Wolinski, K.; Malagoli, M.; Kinghorn, D.; Wolinski, P.; Magyarfalvi, G.; Saebo, S.; Janowski, T.; Pulay, P. Quantum Chemistry in Parallel with PQS. J. Comput. Chem. 2009, 30, 317–335. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.W.; Shenderova, O.A.; Harrison, J.A.; Stuart, S.J.; Ni, B.; Sinnott, S.B. A second-generation reactive empirical bond order (REBO) potential energy expression for hydrocarbons. J. Phys. Condens. Matter 2002, 14, 783–802. [Google Scholar] [CrossRef] [Green Version]
- Stuart, S.J.; Tutein, A.B.; Harrison, J.A. A reactive potential for hydrocarbons with intermolecular interactions. J. Chem. Phys. 2000, 112, 6472–6486. [Google Scholar] [CrossRef] [Green Version]
- Jagusiak, A.; Goclon, J.; Panczyk, T. Adsorption of Evans blue and Congo red on carbon nanotubes and its influence on the fracture parameters of defective and functionalized carbon nanotubes studied using computational methods. Appl. Surf. Sci. 2021, 539, 148236. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | EH | EL | ΔEH-L | IP | EA | μ | η | s | ω |
|---|---|---|---|---|---|---|---|---|---|
| C18 | −6.313 | −3.184 | 3.129 | 6.313 | 3.184 | 4.748 | 1.565 | 0.639 | 0.258 |
| S0-C36 | −5.552 | −3.981 | 1.572 | 5.552 | 3.981 | 4.767 | 0.786 | 1.272 | 0.065 |
| S0-C72-sheet | −5.348 | −4.575 | 0.773 | 5.348 | 4.575 | 4.962 | 0.387 | 2.586 | 0.015 |
| S0-C72-ribbon | −5.338 | −4.344 | 0.994 | 5.338 | 4.344 | 4.841 | 0.497 | 2.013 | 0.025 |
| S1-C18 | −6.093 | −3.119 | 2.974 | 6.093 | 3.119 | 4.606 | 1.487 | 0.672 | 0.240 |
| S1-C36-ribbon | −5.580 | −3.650 | 1.931 | 5.580 | 3.650 | 4.615 | 0.965 | 1.036 | 0.101 |
| S1-C36-sheet | −5.450 | −3.754 | 1.696 | 5.450 | 3.754 | 4.602 | 0.848 | 1.179 | 0.078 |
| S1-C72-sheet−1 | −4.960 | −4.263 | 0.696 | 4.960 | 4.263 | 4.612 | 0.348 | 2.873 | 0.013 |
| S1-C72-sheet−2 | −5.476 | −4.550 | 0.926 | 5.476 | 4.550 | 5.013 | 0.463 | 2.160 | 0.021 |
| S1-C72-tube−1 | −4.843 | −3.730 | 1.113 | 4.843 | 3.730 | 4.286 | 0.556 | 1.797 | 0.036 |
| S1-C72-tube−2 | −5.024 | −4.191 | 0.833 | 5.024 | 4.191 | 4.607 | 0.416 | 2.402 | 0.019 |
| S2-C18 | −6.198 | −5.809 | 0.389 | 6.198 | 5.809 | 6.003 | 0.194 | 5.143 | 0.003 |
| S2-C36-ribbon−1 | −6.381 | −4.739 | 1.642 | 6.381 | 4.739 | 5.560 | 0.821 | 1.218 | 0.061 |
| S2-C36-ribbon−2 | −6.180 | −5.458 | 0.722 | 6.180 | 5.458 | 5.819 | 0.361 | 2.768 | 0.011 |
| S2-C36-sheet | −5.663 | −3.802 | 1.861 | 5.663 | 3.802 | 4.733 | 0.930 | 1.075 | 0.091 |
| S2-C72-ribbon | −5.857 | −5.420 | 0.437 | 5.857 | 5.420 | 5.638 | 0.219 | 4.574 | 0.004 |
| S2-C72-sheet1 | −5.724 | −4.605 | 1.119 | 5.724 | 4.605 | 5.164 | 0.560 | 1.787 | 0.030 |
| S2-C72-sheet2 | −5.272 | −3.865 | 1.407 | 5.272 | 3.865 | 4.569 | 0.704 | 1.421 | 0.054 |
| S2-C72-tube | −5.318 | −3.806 | 1.512 | 5.318 | 3.806 | 4.562 | 0.756 | 1.323 | 0.063 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brzyska, A.; Panczyk, T.; Wolinski, K. From Cyclo[18]carbon to the Novel Nanostructures—Theoretical Predictions. Int. J. Mol. Sci. 2022, 23, 12960. https://doi.org/10.3390/ijms232112960
Brzyska A, Panczyk T, Wolinski K. From Cyclo[18]carbon to the Novel Nanostructures—Theoretical Predictions. International Journal of Molecular Sciences. 2022; 23(21):12960. https://doi.org/10.3390/ijms232112960
Chicago/Turabian StyleBrzyska, Agnieszka, Tomasz Panczyk, and Krzysztof Wolinski. 2022. "From Cyclo[18]carbon to the Novel Nanostructures—Theoretical Predictions" International Journal of Molecular Sciences 23, no. 21: 12960. https://doi.org/10.3390/ijms232112960