
The Mutation Hotspots at UGT1A Locus May Be Associated with Gilbert’s Syndrome Affecting the Taiwanese Population
 , , and
, , and
Abstract
:1. Introduction
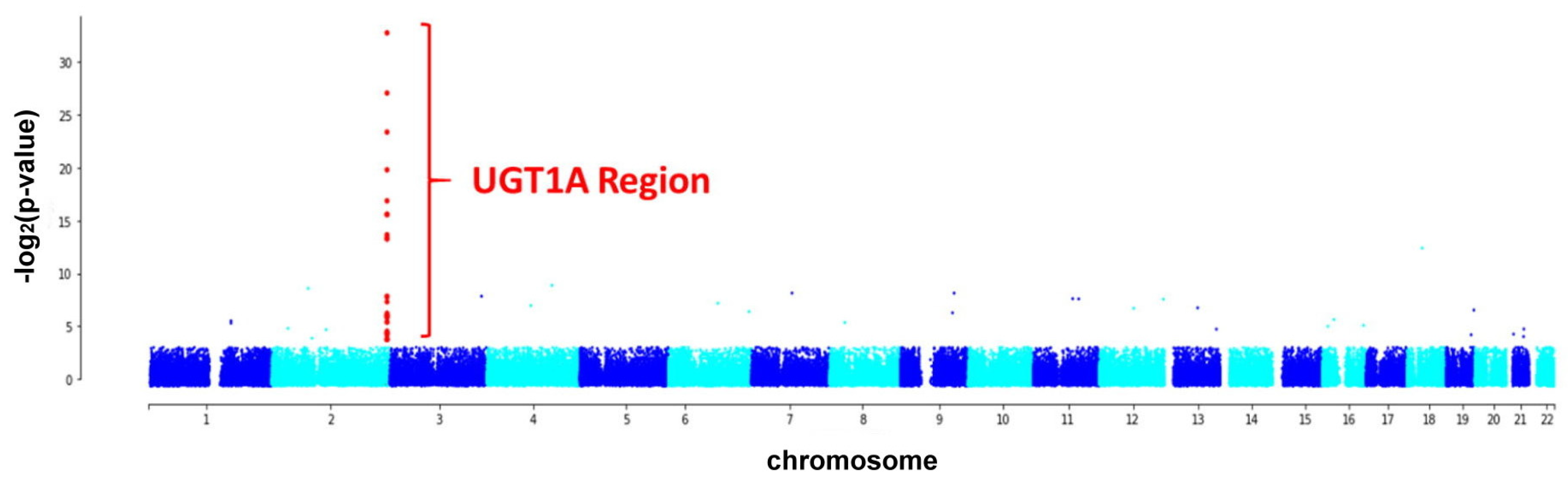
2. Results
2.1. Clinical Characteristics
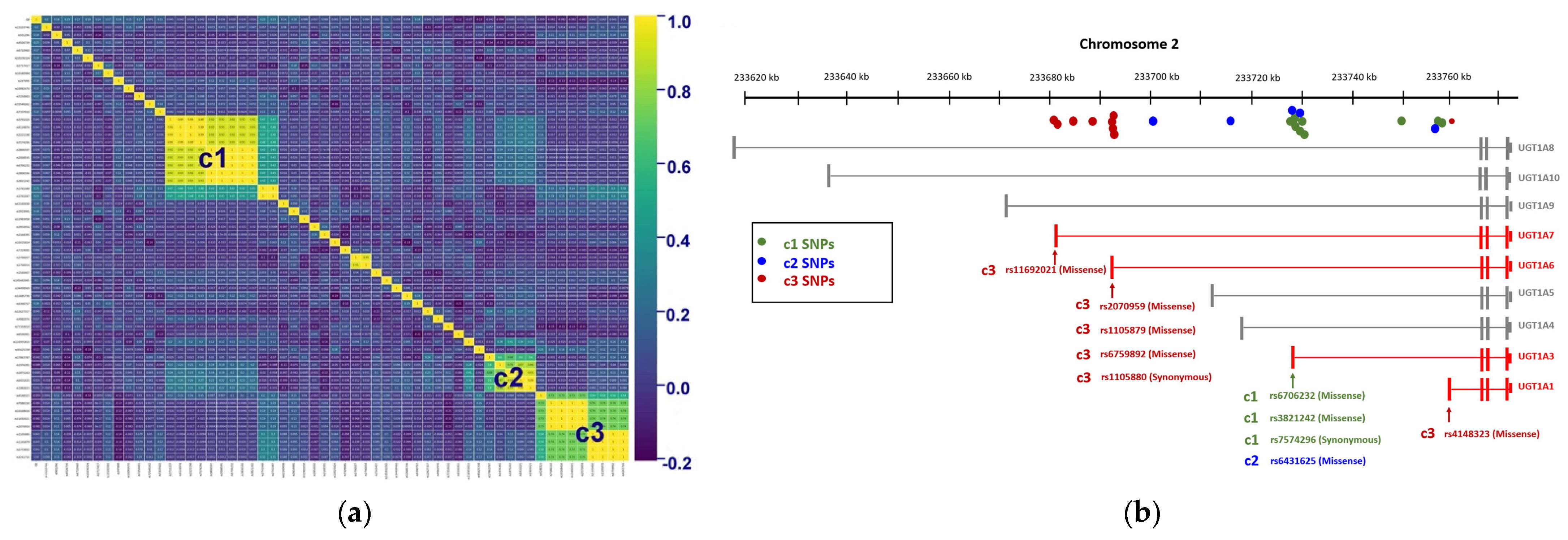
2.2. Genetic Variants Associated with GS
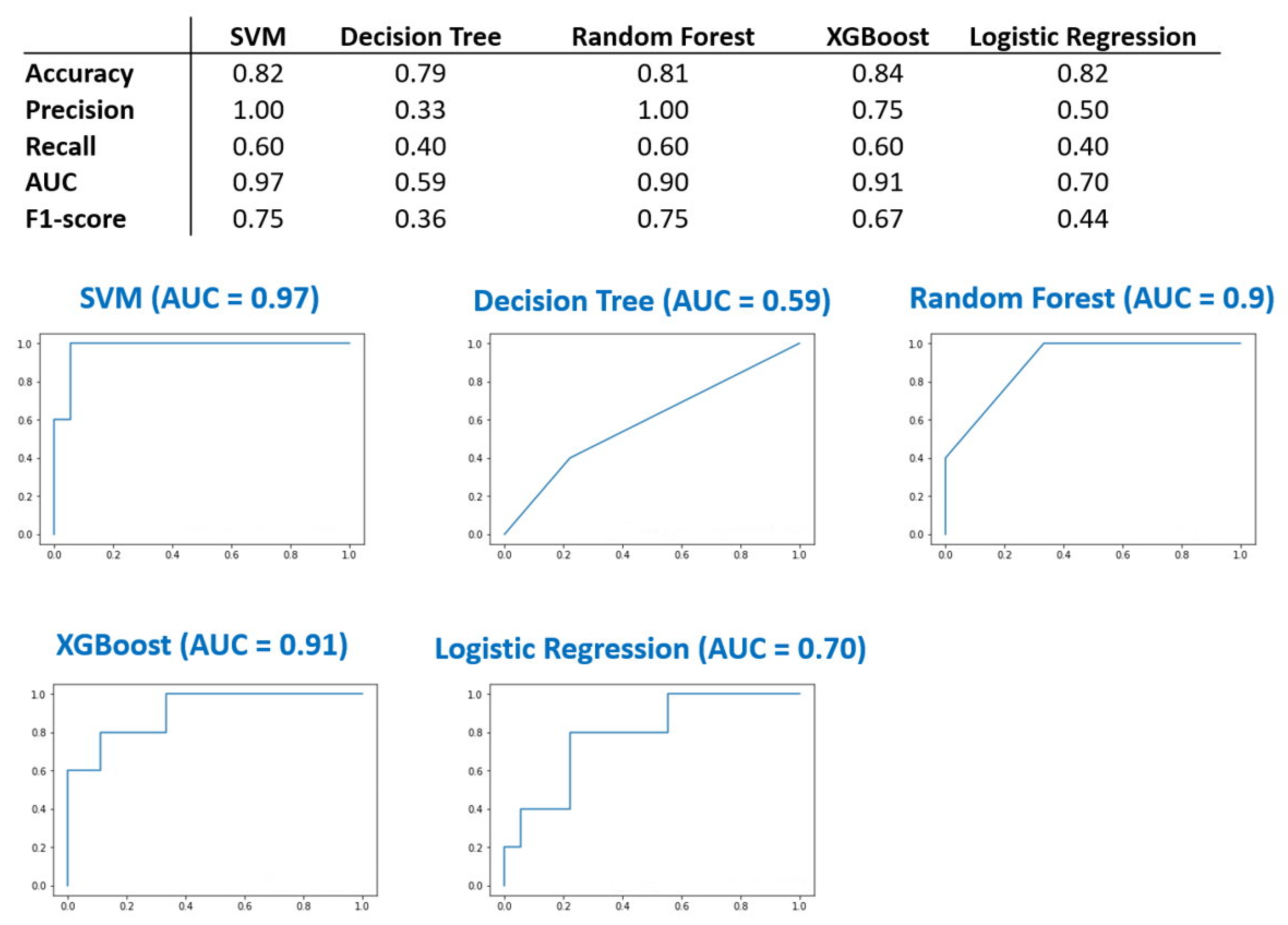
2.3. Machine Learning
2.4. Limitations
3. Discussion
4. Materials and Methods
4.1. Study Subjects
4.2. Clinical Assessment
4.3. DNA Extraction from White Blood Cells
4.4. Whole-Genome Single Nucleotide Polymorphism (SNP) Analysis
4.5. Correlation Heatmap and Machine Learning
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
The Animal Study Protocol
Clinical Trial Registration
Abbreviations
| GS | Gilbert’s syndrome |
| UGT | uridine diphosphoglucuronate glucuronosyltransferase |
| GOT | glutamic oxaloacetic transaminase |
| GPT | glutamic pyruvic transaminase |
| SNP | single nucleotide polymorphism |
| SVM | Support Vector Machine |
| ROC | receiver operating characteristic |
| AUC | area under the ROC curve. |
References
- Arias, I.M. Gilbert’s syndrome. Br. Med. J. 1968, 2, 702. [Google Scholar] [CrossRef] [PubMed]
- Erlinger, S.; Arias, I.M.; Dhumeaux, D. Inherited disorders of bilirubin transport and conjugation: New insights into molecular mechanisms and consequences. Gastroenterology 2014, 146, 1625–1638. [Google Scholar] [CrossRef] [PubMed]
- Vitek, L.; Schwertner, H.A. The heme catabolic pathway and its protective effects on oxidative stress-mediated diseases. Adv. Clin. Chem. 2007, 43, 1–57. [Google Scholar]
- Ghosh, S.S.; Sappal, B.S.; Kalpana, G.V.; Lee, S.W.; Chowdhury, J.R.; Chowdhury, N.R. Homodimerization of human bilirubin-uridine-diphosphoglucuronate glucuronosyltransferase-1 (UGT1A1) and its functional implications. J. Biol. Chem. 2001, 276, 42108–42115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fevery, J. Bilirubin in clinical practice: A review. Liver Int. 2008, 28, 592–605. [Google Scholar] [CrossRef]
- Tiribelli, C.; Ostrow, J.D. Intestinal flora and bilirubin. J. Hepatol. 2005, 42, 170–172. [Google Scholar] [CrossRef]
- Gollan, J.L.; Dallinger, K.J.; Billing, B.H. Excretion of conjugated bilirubin in the isolated perfused rat kidney. Clin. Sci. Mol. Med. 1978, 54, 381–389. [Google Scholar] [CrossRef] [Green Version]
- Hoilat, G.J.; John, S. Bilirubinuria. In BTI-StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Rodrigues, C.; Vieira, E.; Santos, R.; de Carvalho, J.; Santos-Silva, A.; Costa, E.; Bronze-da-Rocha, E. Impact of UGT1A1 gene variants on total bilirubin levels in Gilbert syndrome patients and in healthy subjects. Blood Cells Mol. Dis. 2012, 48, 166–172. [Google Scholar] [CrossRef]
- Shi, X.; Aronson, S.; Khan, A.S.; Bosma, P.J. A novel UGT1A1 gene mutation causing severe unconjugated hyperbilirubinemia: A case report. BMC Pediatr. 2019, 19, 173. [Google Scholar] [CrossRef]
- Agrawal, S.K.; Kumar, P.; Rathi, R.; Sharma, N.; Das, R.; Prasad, R.; Narang, A. UGT1A1 gene polymorphisms in North Indian neonates presenting with unconjugated hyperbilirubinemia. Pediatr. Res. 2009, 65, 675–680. [Google Scholar] [CrossRef] [Green Version]
- Ostanek, B.; Furlan, D.; Mavec, T.; Lukac-Bajalo, J. UGT1A1(TA)n promoter polymorphism--a new case of a (TA)8 allele in Caucasians. Blood Cells Mol. Dis. 2007, 38, 78–82. [Google Scholar] [CrossRef]
- Raijmakers, M.T.; Jansen, P.L.; Steegers, E.A.; Peters, W.H. Association of human liver bilirubin UDP-glucuronyltransferase activity with a polymorphism in the promoter region of the UGT1A1 gene. J. Hepatol. 2000, 33, 348–351. [Google Scholar] [CrossRef]
- Fargo, M.V.; Grogan, S.P.; Saguil, A. Evaluation of Jaundice in Adults. Am. Fam. Physician 2017, 95, 164–168. [Google Scholar] [PubMed]
- Gil, J.; Sasiadek, M.M. Gilbert syndrome: The UGT1A1*28 promoter polymorphism as a biomarker of multifactorial diseases and drug metabolism. Biomark. Med. 2012, 6, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Bosma, P.J.; Chowdhury, J.R.; Bakker, C.; Gantla, S.; de Boer, A.; Oostra, B.A.; Lindhout, D.; Tytgat, G.N.; Jansen, P.L.; Oude Elferink, R.P.; et al. The genetic basis of the reduced expression of bilirubin UDP-glucuronosyltransferase 1 in Gilbert’s syndrome. N. Engl. J. Med. 1995, 333, 1171–1175. [Google Scholar] [CrossRef]
- Bosma, P.; Chowdhury, J.R.; Jansen, P.H. Genetic inheritance of Gilbert’s syndrome. Lancet 1995, 346, 314–315. [Google Scholar] [CrossRef]
- Teng, H.C.; Huang, M.J.; Tang, K.S.; Yang, S.S.; Tseng, C.S.; Huang, C.S. Combined UGT1A1 and UGT1A7 variant alleles are associated with increased risk of Gilbert’s syndrome in Taiwanese adults. Clin. Genet. 2007, 72, 321–328. [Google Scholar] [CrossRef]
- Wei, C.Y.; Yang, J.H.; Yeh, E.C.; Tsai, M.F.; Kao, H.J.; Lo, C.Z.; Chang, L.P.; Lin, W.J.; Hsieh, F.J.; Belsare, S.; et al. Genetic profiles of 103,106 individuals in the Taiwan Biobank provide insights into the health and history of Han Chinese. NPJ Genom. Med. 2021, 6, 10. [Google Scholar] [CrossRef]
- Wagner, K.H.; Shiels, R.G.; Lang, C.A.; Seyed Khoei, N.; Bulmer, A.C. Diagnostic criteria and contributors to Gilbert’s syndrome. Crit. Rev. Clin. Lab. Sci. 2018, 55, 129–139. [Google Scholar] [CrossRef] [Green Version]
- Urawa, N.; Kobayashi, Y.; Araki, J.; Sugimoto, R.; Iwasa, M.; Kaito, M.; Adachi, Y. Linkage disequilibrium of UGT1A1*6 and UGT1A1*28 in relation to UGT1A6 and UGT1A7 polymorphisms. Oncol. Rep. 2006, 16, 801–806. [Google Scholar] [CrossRef]
- Ramirez, J.; Mirkov, S.; House, L.K.; Ratain, M.J. Glucuronidation of OTS167 in Humans Is Catalyzed by UDP-Glucuronosyltransferases UGT1A1, UGT1A3, UGT1A8, and UGT1A10. Drug Metab. Dispos. 2015, 43, 928–935. [Google Scholar] [CrossRef] [PubMed]
- Peters, W.H.; te Morsche, R.H.; Roelofs, H.M. Combined polymorphisms in UDP-glucuronosyltransferases 1A1 and 1A6: Implications for patients with Gilbert’s syndrome. J. Hepatol. 2003, 38, 3–8. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Healthy Control n = 180 | Gilbert’s Syndrome n = 45 | p-Value | |
|---|---|---|---|
| Age, years | 60.45 ± 14.90 | 67.49 ± 13.99 | 0.005 |
| Female, n (%) | 68 (37.8%) | 17 (37.8%) | 1 |
| T_Bilirubin 1 (mg/dL) | 0.63 ± 0.24 | 1.73 ± 0.30 | <0.001 |
| D_Bilirubin 2 (mg/dL) | 0.22 ± 0.08 | 0.22 ± 0.06 | 0.719 |
| D_Bilirubin/T_Bilirubin | 37.82 ± 12.63 | 12.78 ± 3.08 | <0.001 |
| Anemia, n (%) | 11 (6.1%) | 2 (4.4%) | 0.668 |
| WBC 3 (×103/μL) | 6.11 ± 2.64 | 6.17 ± 1.95 | 0.885 |
| RBC 4 (×103/μL) | 4.8 ± 0.60 | 4.86 ± 0.49 | 0.513 |
| Hemoglobin (g/dL) | 14.08 ± 1.42 | 14.3 ± 1.69 | 0.356 |
| Hematocrit (%) | 41.91 ± 3.69 | 42.16 ± 4.07 | 0.689 |
| MCV 5 (fL) | 88.04 ± 7.48 | 87.2 ± 8.14 | 0.504 |
| MCH 6 (pg) | 29.58 ± 2.91 | 29.57 ± 3.34 | 0.993 |
| MCHC 7 (g/dl) | 33.55 ± 1.03 | 33.86 ± 1.22 | 0.092 |
| RDW 8 (%) | 13.39 ± 1.03 | 13.58 ± 1.63 | 0.33 |
| Platelets (×103/μL) | 244.28 ± 62.13 | 244.18 ± 62.05 | 0.992 |
| Transferrin (ng/mL) | 245.36 ± 34.75 | 235.14 ± 35.12 | 0.08 |
| Clustering | SNP | Chromosome | Position (bp) | p-Value | Location |
|---|---|---|---|---|---|
| c1 | rs3755319 | 2 | 233,758,936 | 0.003031869 | UGT1A region: Intronic Variant |
| rs4124874 | 2 | 233,757,013 | 0.005113795 | UGT1A region: Intronic Variant | |
| rs2221198 | 2 | 233,749,977 | 0.00333 | UGT1A region: Intronic Variant | |
| rs7574296 | 2 | 233,729,603 | 0.002707358 | UGT1A3: Synonymous Variant | |
| rs3806597 | 2 | 233,728,923 | 0.026915457 | UGT1A region: Intronic Variant | |
| rs2008595 | 2 | 233,728,546 | 0.026915457 | UGT1A region: Intronic Variant | |
| rs6706232 | 2 | 233,729,207 | 0.0152489 | UGT1A3: Missense Variant | |
| rs3806596 | 2 | 233,729,061 | 0.015291563 | UGT1A region: Intronic Variant | |
| rs3821242 | 2 | 233,729,157 | 0.012727593 | UGT1A3: Missense Variant | |
| c2 | rs17863787 | 2 | 233,702,448 | 0.002450485 | UGT1A region: Intronic Variant |
| rs1976391 | 2 | 233,757,337 | 6.66 × 10−15 | UGT1A region: Intronic Variant | |
| rs1875263 | 2 | 233,716,976 | 0.000759234 | UGT1A region: Intronic Variant | |
| rs6431625 | 2 | 233,729,266 | 1.99 × 10−12 | UGT1A3: Missense Variant | |
| rs1983023 | 2 | 233,728,376 | 8.00 × 10−11 | UGT1A region: Intronic Variant | |
| c3 | rs4148323 | 2 | 233,760,498 | 0.00000151 | UGT1A1: Missense Variant |
| rs7586110 | 2 | 233,681,881 | 0.000000195 | UGT1A region: Intronic Variant | |
| rs10168416 | 2 | 233,688,441 | 0.000000195 | UGT1A region: Intronic Variant | |
| rs11692021 | 2 | 233,682,559 | 5.39 × 10−8 | UGT1A7: Missense Variant | |
| rs2070959 | 2 | 233,693,545 | 0.000000195 | UGT1A6: Missense Variant | |
| rs1105880 | 2 | 233,693,319 | 0.00000193 | UGT1A6: Synonymous Variant | |
| rs1105879 | 2 | 233,693,556 | 0.00000193 | UGT1A6: Missense Variant | |
| rs6759892 | 2 | 233,693,023 | 0.00000193 | UGT1A6: Missense Variant | |
| rs4261716 | 2 | 233,684,471 | 0.00000133 | UGT1A region: Intronic Variant |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, P.W.-C.; Liao, P.-C.; Kao, Y.-H.; Lin, X.-Y.; Chien, R.-N.; Yeh, C.-T.; Lai, C.-C.; Shyu, Y.-C.; Lin, C.-L. The Mutation Hotspots at UGT1A Locus May Be Associated with Gilbert’s Syndrome Affecting the Taiwanese Population. Int. J. Mol. Sci. 2022, 23, 12709. https://doi.org/10.3390/ijms232012709
Hsu PW-C, Liao P-C, Kao Y-H, Lin X-Y, Chien R-N, Yeh C-T, Lai C-C, Shyu Y-C, Lin C-L. The Mutation Hotspots at UGT1A Locus May Be Associated with Gilbert’s Syndrome Affecting the Taiwanese Population. International Journal of Molecular Sciences. 2022; 23(20):12709. https://doi.org/10.3390/ijms232012709
Chicago/Turabian StyleHsu, Paul Wei-Che, Po-Cheng Liao, Yu-Hsiang Kao, Xin-Yu Lin, Rong-Nan Chien, Chau-Ting Yeh, Chi-Chun Lai, Yu-Chiau Shyu, and Chih-Lang Lin. 2022. "The Mutation Hotspots at UGT1A Locus May Be Associated with Gilbert’s Syndrome Affecting the Taiwanese Population" International Journal of Molecular Sciences 23, no. 20: 12709. https://doi.org/10.3390/ijms232012709