
Molecular Mechanisms of RSV and Air Pollution Interaction: A Scoping Review

Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Protocol
2.1.1. Conceptual Framework
- a facilitated viral entry via an enhanced viral adhesion (as a result of expression/increased expression of the potential receptor/adhesion molecule on the cell surface), or facilitated entry due to the presence of a transporting canal or damaged natural protective barriers;
- an altered viral load (presumably an increased/prolonged viral replication) via either the virus gaining an unusual virulence or ability (per se) to replicate or a decreased antiviral defense of the host, or via a facilitated/increased viral release;
- an inappropriate host reaction, including but not limited to a prolonged or increased inflammatory reaction, hyperreactivity, or histopathological changes; may result in a more severe disease course, more damage to the host, and the presence of long-term sequelae.
2.1.2. Review Approach
2.1.3. Search Strategy
2.1.4. Eligibility Criteria
2.1.5. Extraction Methods
2.1.6. Critical Appraisal
2.1.7. Synthesis of the Results
3. Results
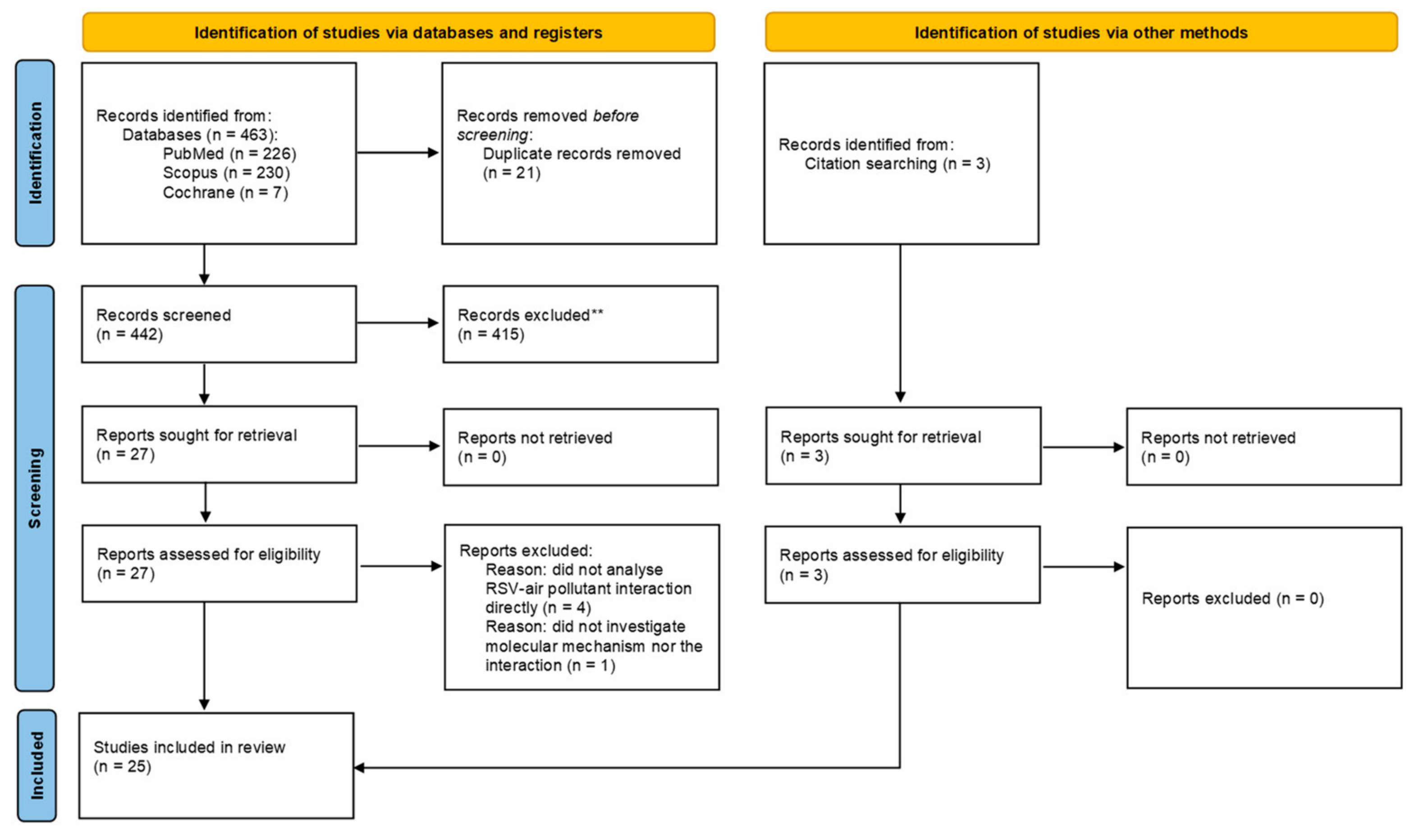
3.1. Search Results
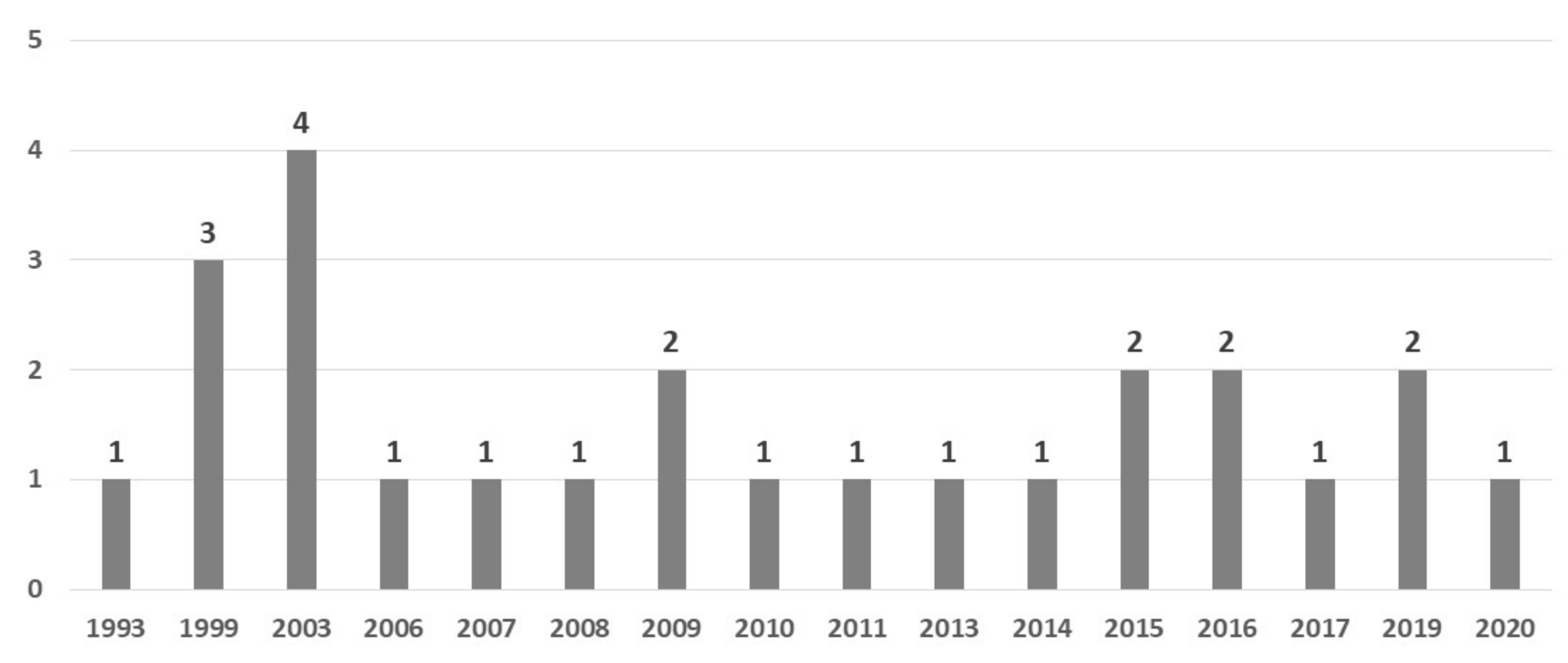
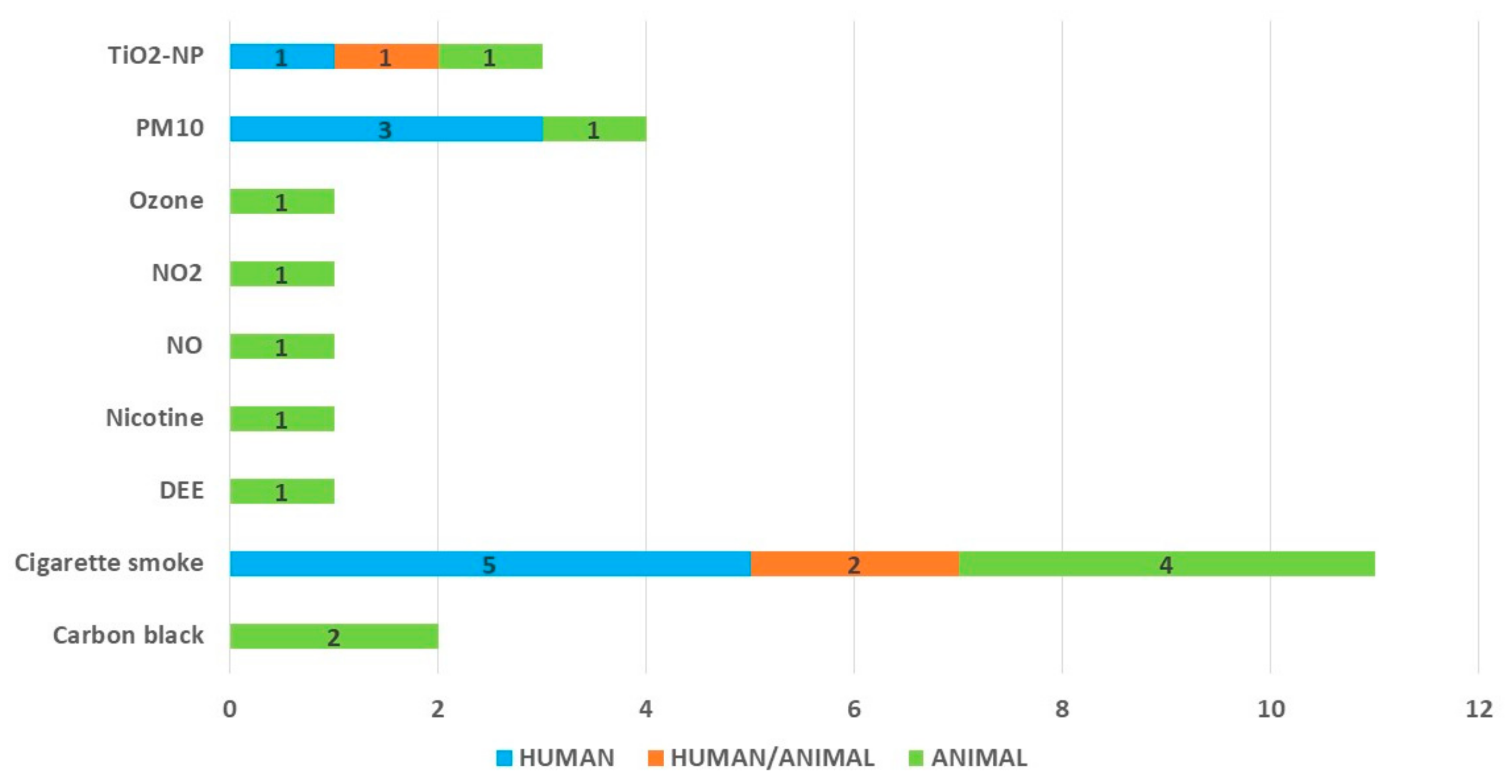
3.2. Study Characteristics
4. Molecular Mechanisms
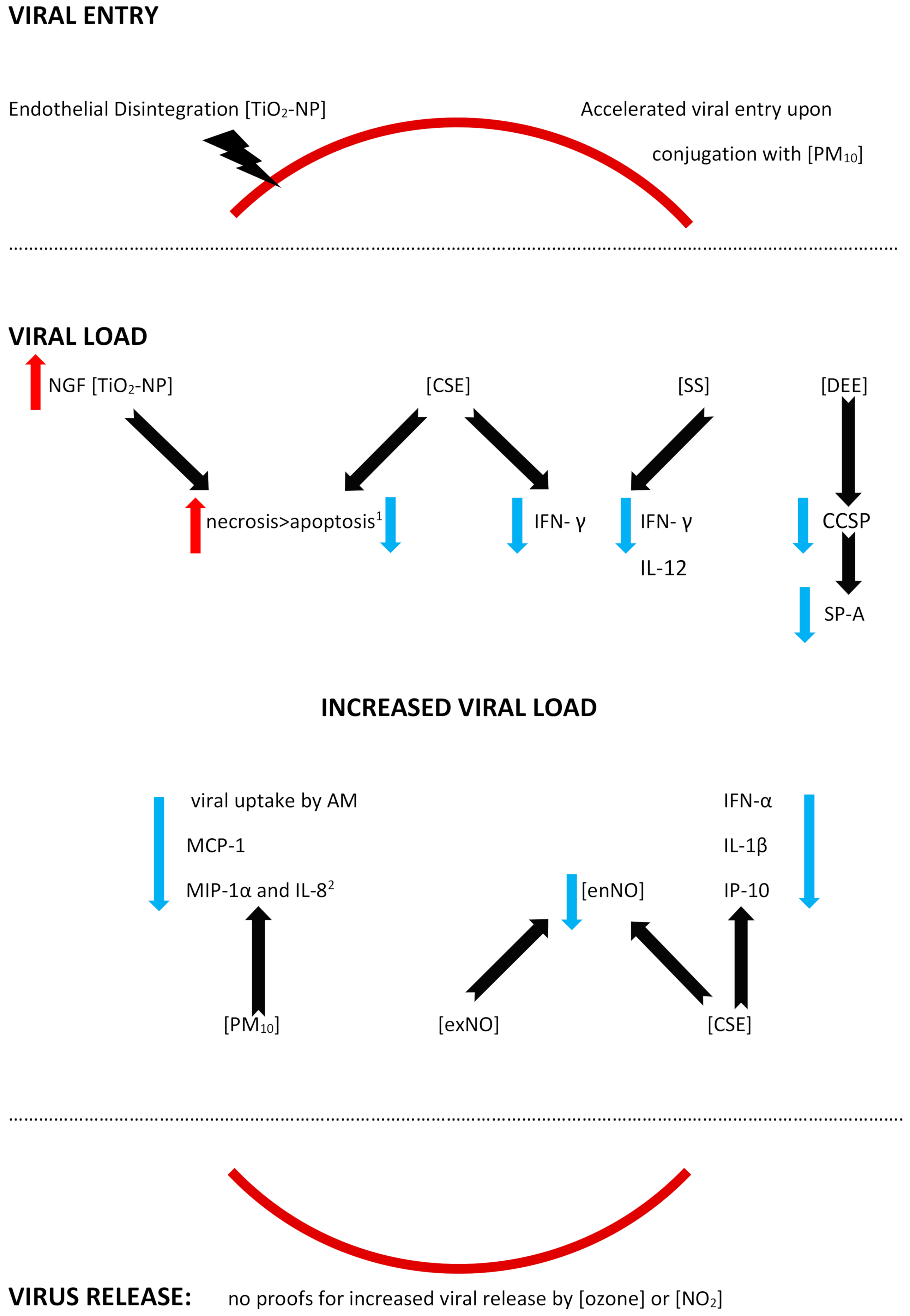
4.1. A Facilitated Viral Entry
4.1.1. Epithelial Barrier
4.1.2. A Facilitated Viral Entry
4.2. An Altered Viral Load
- (A)
- An Increased Viral Load
4.2.1. Autophagy, Decreased Apoptosis, and Enhanced Necrosis
4.2.2. Decreased Antiviral Defense
Epithelium
Alveolar Macrophages (AM)
Dendritic Cells
- (B)
- Viral load fluctuations
- (C)
- Virus release
4.3. An Inappropriate Host Reaction
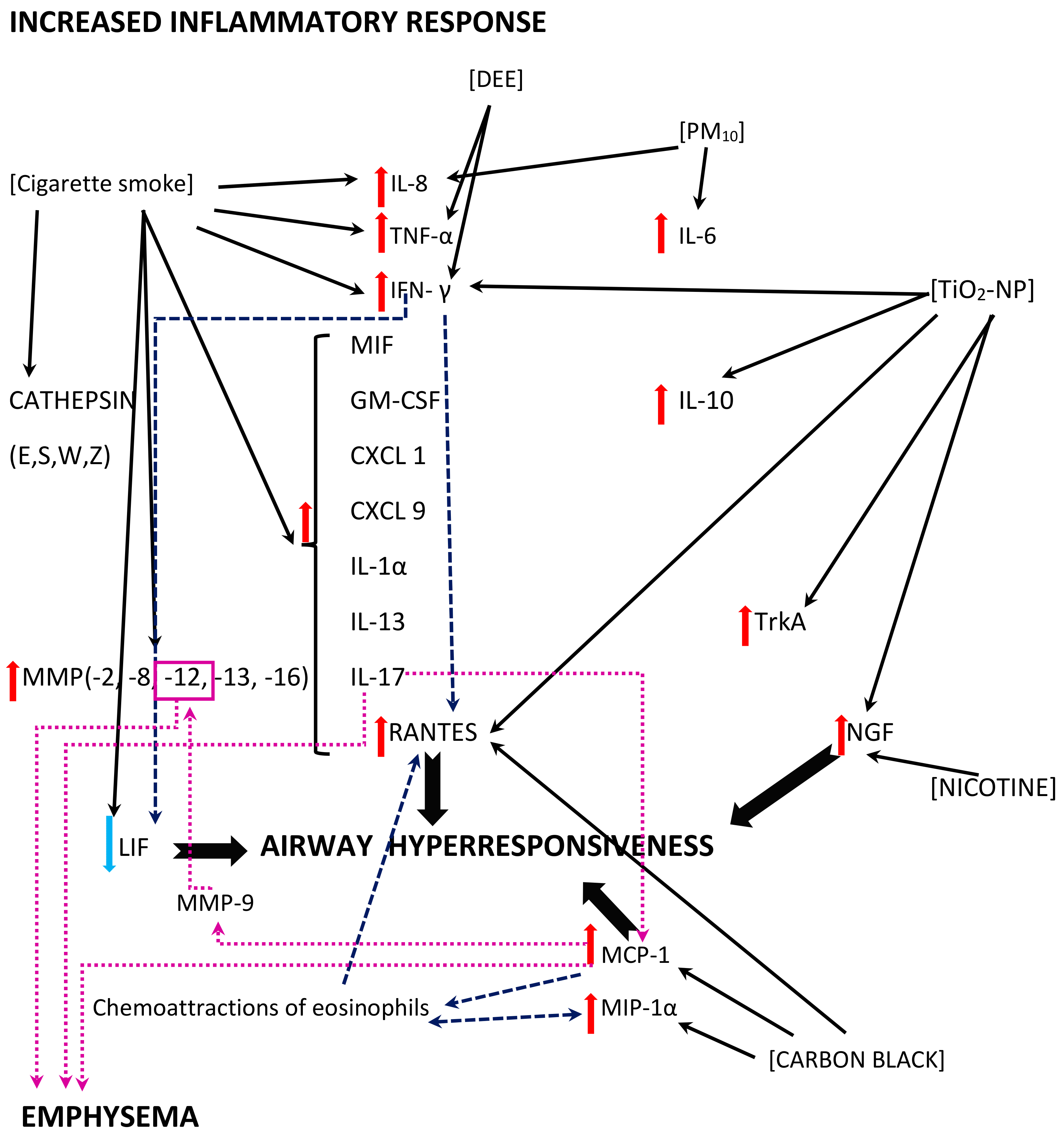
4.3.1. Inflammation
4.3.2. Airway Hyperresponsiveness (AHR)
4.3.3. Histopathological Changes
5. Discussion
- -
- air pollutants: the highest number of studies (although still not enough to identify and confirm the most important pathways in different models) that investigated the interaction between the RSV and cigarette smoke (or its derivatives); the data on the mechanisms of other pollutants is very scarce. While the studies on cigarette smoking and its relationship with the RSV are driven by the issue of COPD exacerbations due to infections (among which the RSV plays an important role), the influence of the other pollutants is hugely under-investigated. As for the six most significant air pollutants (according to the WHO: PM2.5, PM10, O₃, NO₂, SO₂ and CO) [1], there are only single studies;
- -
- the mechanisms facilitating an acute infection or worse disease course: the problem is of great clinical relevance; although the number of clinical studies underlying the relationship between the air pollutants and RSV morbidity is growing, the mechanisms of the interaction remain deeply unknown. We qualified the mechanisms underlying the increased morbidity together with a worse clinical course, since the studies on morbidity are performed mostly in hospital settings, and in fact present a problem of combined increased morbidity and severity, thus, common pathways might also be expected. An explanation of the mechanisms is of special meaning for short-term interventions, especially non-pharmacological ones, which might decrease RSV morbidity during periods of high air pollution;
- -
- the mechanisms of an inadequate response to the infection, resulting in a prolonged course of the acute infection, and probably related to long-term sequelae, such as the airway hyperreactivity following an RSV infection; here, a combination of air pollution and RSV infection might be particularly detrimental, and molecular mechanisms need to be well understood in order to take targeted actions, such as pharmacological interventions. In this regard, the scoping review identified some prospective targets for future considerations. Of interest, co-exposure to higher air pollution and RSV infection might play a role in long-term sequelae, and in this regard deserve more attention in clinical settings as well.
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Available online: https://www.who.int/news-room/questions-and-answers/item/who-global-air-quality-guidelines (accessed on 24 August 2022).
- Lelieveld, J.; Evans, J.S.; Fnais, M.; Giannadaki, D.; Pozzer, A. The Contribution of Outdoor Air Pollution Sources to Premature Mortality on a Global Scale. Nature 2015, 525, 367–371. [Google Scholar] [CrossRef]
- Bernstein, J.A.; Alexis, N.; Barnes, C.; Bernstein, I.L.; Nel, A.; Peden, D.; Diaz-Sanchez, D.; Tarlo, S.M.; Williams, B. Health Effects of Air Pollution. J. Allergy Clin. Immunol. 2004, 114, 1116–1123. [Google Scholar] [CrossRef] [PubMed]
- Bourdrel, T.; Bind, M.-A.; Béjot, Y.; Morel, O.; Argacha, J.-F. Cardiovascular Effects of Air Pollution. Arch. Cardiovasc. Dis. 2017, 110, 634–642. [Google Scholar] [CrossRef] [PubMed]
- IARC. Available online: https://monographs.iarc.who.int/list-of-classifications (accessed on 24 August 2022).
- Li, Y.; Wang, X.; Blau, D.M.; Caballero, M.T.; Feikin, D.R.; Gill, C.J.; Madhi, S.A.; Omer, S.B.; Simões, E.A.F.; Campbell, H.; et al. Global, Regional, and National Disease Burden Estimates of Acute Lower Respiratory Infections Due to Respiratory Syncytial Virus in Children Younger than 5 Years in 2019: A Systematic Analysis. Lancet 2022, 399, 2047–2064. [Google Scholar] [CrossRef]
- Simões, E.A.F. Respiratory Syncytial Virus Disease in Young Children and Older Adults in Europe: A Burden and Economic Perspective. J. Infect. Dis. 2022, 226 (Suppl. S1), S1–S9. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Lopardo, G.; Scarpellini, B.; Stein, R.T.; Ribeiro, D. Systematic Review on Respiratory Syncytial Virus Epidemiology in Adults and the Elderly in Latin America. Int. J. Infect. Dis. 2020, 90, 170–180. [Google Scholar] [CrossRef] [Green Version]
- Boattini, M.; Almeida, A.; Christaki, E.; Marques, T.M.; Tosatto, V.; Bianco, G.; Iannaccone, M.; Tsiolakkis, G.; Karagiannis, C.; Maikanti, P.; et al. Severity of RSV Infection in Southern European Elderly Patients during Two Consecutive Winter Seasons (2017–2018). J. Med. Virol. 2021, 93, 5152–5157. [Google Scholar] [CrossRef]
- Korsten, K.; Adriaenssens, N.; Coenen, S.; Butler, C.C.; Verheij, T.J.M.; Bont, L.J.; Wildenbeest, J.G.; Butler, C.; Nair, H.; Campbell, H.; et al. World Health Organization Influenza-Like Illness Underestimates the Burden of Respiratory Syncytial Virus Infection in Community-Dwelling Older Adults. J. Infect. Dis. 2022, 226 (Suppl. S1), S71–S78. [Google Scholar] [CrossRef]
- Mao, Z.; Li, X.; Korsten, K.; Bont, L.; Butler, C.; Wildenbeest, J.; Coenen, S.; Hens, N.; Bilcke, J.; Beutels, P.; et al. Economic Burden and Health-Related Quality of Life of Respiratory Syncytial Virus and Influenza Infection in European Community-Dwelling Older Adults. J. Infect. Dis. 2022, 226 (Suppl. S1), S87–S94. [Google Scholar] [CrossRef] [PubMed]
- Bowser, D.M.; Rowlands, K.R.; Hariharan, D.; Gervasio, R.M.; Buckley, L.; Halasa-Rappel, Y.; Glaser, E.L.; Nelson, C.B.; Shepard, D.S. Cost of Respiratory Syncytial Virus Infections in US Infants: Systematic Literature Review and Analysis. J. Infect. Dis. 2022, 226 (Suppl. S2), S225–S235. [Google Scholar] [CrossRef]
- Suh, M.; Movva, N.; Jiang, X.; Reichert, H.; Bylsma, L.C.; Fryzek, J.P.; Nelson, C.B. Respiratory Syncytial Virus Burden and Healthcare Utilization in United States Infants <1 Year of Age: Study of Nationally Representative Databases, 2011–2019. J. Infect. Dis. 2022, 226, S184–S194. [Google Scholar] [CrossRef] [PubMed]
- Mazur, N.I.; Terstappen, J.; Baral, R.; Bardají, A.; Beutels, P.; Buchholz, U.J.; Cohen, C.; Crowe, J.E.; Cutland, C.L.; Eckert, L.; et al. Respiratory Syncytial Virus Prevention within Reach: The Vaccine and Monoclonal Antibody Landscape. Lancet Infect. Dis. 2022. Epub ahead of print. [Google Scholar] [CrossRef]
- Vandini, S.; Corvaglia, L.; Alessandroni, R.; Aquilano, G.; Marsico, C.; Spinelli, M.; Lanari, M.; Faldella, G. Respiratory Syncytial Virus Infection in Infants and Correlation with Meteorological Factors and Air Pollutants. Ital. J. Pediatr. 2013, 39, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carugno, M.; Dentali, F.; Mathieu, G.; Fontanella, A.; Mariani, J.; Bordini, L.; Milani, G.P.; Consonni, D.; Bonzini, M.; Bollati, V.; et al. PM10 Exposure Is Associated with Increased Hospitalizations for Respiratory Syncytial Virus Bronchiolitis among Infants in Lombardy, Italy. Environ. Res. 2018, 166, 452–457. [Google Scholar] [CrossRef]
- Horne, B.D.; Joy, E.A.; Hofmann, M.G.; Gesteland, P.H.; Cannon, J.B.; Lefler, J.S.; Blagev, D.P.; Korgenski, E.K.; Torosyan, N.; Hansen, G.I.; et al. Short-Term Elevation of Fine Particulate Matter Air Pollution and Acute Lower Respiratory Infection. Am. J. Respir. Crit. Care Med. 2018, 198, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Karr, C.J.; Rudra, C.B.; Miller, K.A.; Gould, T.R.; Larson, T.; Sathyanarayana, S.; Koenig, J.Q. Infant Exposure to Fine Particulate Matter and Traffic and Risk of Hospitalization for RSV Bronchiolitis in a Region with Lower Ambient Air Pollution. Environ. Res. 2009, 109, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Nenna, R.; Evangelisti, M.; Frassanito, A.; Scagnolari, C.; Pierangeli, A.; Antonelli, G.; Nicolai, A.; Arima, S.; Moretti, C.; Papoff, P.; et al. Respiratory Syncytial Virus Bronchiolitis, Weather Conditions and Air Pollution in an Italian Urban Area: An Observational Study. Environ. Res. 2017, 158, 188–193. [Google Scholar] [CrossRef]
- Ye, Q.; Fu, J.-F.; Mao, J.-H.; Shang, S.-Q. Haze Is a Risk Factor Contributing to the Rapid Spread of Respiratory Syncytial Virus in Children. Environ. Sci. Pollut. Res. 2016, 23, 20178–20185. [Google Scholar] [CrossRef]
- Wrotek, A.; Badyda, A.; Czechowski, P.; Owczarek, T.; Dąbrowiecki, P.; Jackowska, T. Air Pollutants’ Concentrations Are Associated with Increased Number of RSV Hospitalizations in Polish Children. J. Clin. Med. 2021, 10, 3224. [Google Scholar] [CrossRef]
- Radhakrishnan, D.; Ouedraogo, A.; Shariff, S.Z.; McNally, J.D.; Benchimol, E.I.; Clemens, K.K. The Association between Climate, Geography and Respiratory Syncitial Virus Hospitalizations among Children in Ontario, Canada: A Population-Based Study. BMC Infect. Dis. 2020, 20, 157. [Google Scholar] [CrossRef]
- Lanari, M.; Giovannini, M.; Giuffré, L.; Marini, A.; Rondini, G.; Rossi, G.A.; Merolla, R.; Zuccotti, G.V.; Salvioli, G.P. Prevalence of Respiratory Syncytial Virus Infection in Italian Infants Hospitalized for Acute Lower Respiratory Tract Infections, and Association between Respiratory Syncytial Virus Infection Risk Factors and Disease Severity. Pediatr. Pulmonol. 2002, 33, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, S.N.; Nguyen, T.N.T.; Vu, L.T.; Nguyen, T.D. Clinical Epidemiological Characteristics and Risk Factors for Severe Bronchiolitis Caused by Respiratory Syncytial Virus in Vietnamese Children. Int. J. Pediatr. 2021, 2021, 9704666. [Google Scholar] [CrossRef]
- Maedel, C.; Kainz, K.; Frischer, T.; Reinweber, M.; Zacharasiewicz, A. Increased Severity of Respiratory Syncytial Virus Airway Infection Due to Passive Smoke Exposure. Pediatr. Pulmonol. 2018, 53, 1299–1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomson, N.C. The Role of Smoking in Asthma and Chronic Obstructive Pulmonary Disease Overlap. Immunol. Allergy Clin. N. Am. 2022, 42, 615–630. [Google Scholar] [CrossRef] [PubMed]
- Sikkel, M.B.; Quint, J.K.; Mallia, P.; Wedzicha, J.A.; Johnston, S.L. Respiratory Syncytial Virus Persistence in Chronic Obstructive Pulmonary Disease. Pediatr. Infect. Dis. J. 2008, 27 (Suppl. S10), S63–S70. [Google Scholar] [CrossRef] [PubMed]
- Loaiza-Ceballos, M.C.; Marin-Palma, D.; Zapata, W.; Hernandez, J.C. Viral Respiratory Infections and Air Pollutants. Air Qual. Atmos. Health 2022, 15, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Harrod, K.S.; Jaramillo, R.J.; Rosenberger, C.L.; Wang, S.-Z.; Berger, J.A.; McDonald, J.D.; Reed, M.D. Increased Susceptibility to RSV Infection by Exposure to Inhaled Diesel Engine Emissions. Am. J. Respir. Cell Mol. Biol. 2003, 28, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Tricco, A.C.; Lillie, E.; Zarin, W.; O’Brien, K.K.; Colquhoun, H.; Levac, D.; Moher, D.; Peters, M.D.J.; Horsley, T.; Weeks, L.; et al. PRISMA Extension for Scoping Reviews (PRISMA-ScR): Checklist and Explanation. Ann. Intern. Med. 2018, 169, 467–473. [Google Scholar] [CrossRef] [Green Version]
- Munn, Z.; Peters, M.D.J.; Stern, C.; Tufanaru, C.; McArthur, A.; Aromataris, E. Systematic Review or Scoping Review? Guidance for Authors When Choosing between a Systematic or Scoping Review Approach. BMC Med. Res. Methodol. 2018, 18, 143. [Google Scholar] [CrossRef]
- Becker, S.; Soukup, J.M. Effect of Nitrogen Dioxide on Respiratory Viral Infection in Airway Epithelial Cells. Environ. Res. 1999, 81, 159–166. [Google Scholar] [CrossRef]
- Becker, S.; Soukup, J.M. Exposure to Urban Air Particulates Alters the Macrophage-Mediated Inflammatory Response to Respiratory Viral Infection. J. Toxicol. Environ. Health A 1999, 57, 445–457. [Google Scholar] [PubMed]
- Castro, S.M.; Kolli, D.; Guerrero-Plata, A.; Garofalo, R.P.; Casola, A. Cigarette Smoke Condensate Enhances Respiratory Syncytial Virus–Induced Chemokine Release by Modulating NF-kappa B and Interferon Regulatory Factor Activation. Toxicol. Sci. 2008, 106, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Castro, S.M.; Chakraborty, K.; Guerrero-Plata, A. Cigarette Smoke Suppresses TLR-7 Stimulation in Response to Virus Infection in Plasmacytoid Dendritic Cells. Toxicol. Vitr. 2011, 25, 1106–1113. [Google Scholar] [CrossRef]
- Chakraborty, S.; Castranova, V.; Perez, M.K.; Piedimonte, G. Nanoparticles Increase Human Bronchial Epithelial Cell Susceptibility to Respiratory Syncytial Virus Infection via Nerve Growth Factor-Induced Autophagy. Physiol. Rep. 2017, 5, e13344. [Google Scholar] [CrossRef]
- Cruz-Sanchez, T.M.; Haddrell, A.E.; Hackett, T.L.; Singhera, G.K.; Marchant, D.; Lekivetz, R.; Meredith, A.; Horne, D.; Knight, D.A.; van Eeden, S.F.; et al. Formation of a Stable Mimic of Ambient Particulate Matter Containing Viable Infectious Respiratory Syncytial Virus and Its Dry-Deposition Directly onto Cell Cultures. Anal. Chem. 2013, 85, 898–906. [Google Scholar] [CrossRef]
- Foronjy, R.F.; Dabo, A.J.; Taggart, C.C.; Weldon, S.; Geraghty, P. Respiratory Syncytial Virus Infections Enhance Cigarette Smoke Induced COPD in Mice. PLoS ONE 2014, 9, e90567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foronjy, R.F.; Ochieng, P.O.; Salathe, M.; Dabo, A.J.; Eden, E.; Baumlin, N.; Cummins, N.; Barik, S.; Campos, M.; Thorp, E.; et al. Protein Tyrosine Phosphatase 1B Negatively Regulates S100A9-Mediated Lung Damage during Respiratory Syncytial Virus Exacerbations. Mucosal Immunol. 2016, 9, 1317–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groskreutz, D.J.; Monick, M.M.; Babor, E.C.; Nyunoya, T.; Varga, S.M.; Look, D.C.; Hunninghake, G.W. Cigarette Smoke Alters Respiratory Syncytial Virus–Induced Apoptosis and Replication. Am. J. Respir. Cell Mol. Biol. 2009, 41, 189–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashiguchi, S.; Yoshida, H.; Akashi, T.; Komemoto, K.; Ueda, T.; Ikarashi, Y.; Miyauchi, A.; Konno, K.; Yamanaka, S.; Hirose, A.; et al. Titanium Dioxide Nanoparticles Exacerbate Pneumonia in Respiratory Syncytial Virus (RSV)-Infected Mice. Environ. Toxicol. Pharmacol. 2015, 39, 879–886. [Google Scholar] [CrossRef]
- Hirota, J.A.; Marchant, D.J.; Singhera, G.K.; Moheimani, F.; Dorscheid, D.R.; Carlsten, C.; Sin, D.; Knight, D. Urban Particulate Matter Increases Human Airway Epithelial Cell IL-1β Secretion Following Scratch Wounding and H1N1 Influenza A Exposure In Vitro. Exp. Lung Res. 2015, 41, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Hobson, L.; Everard, M.L. Persistent of Respiratory Syncytial Virus in Human Dendritic Cells and Influence of Nitric Oxide. Clin. Exp. Immunol. 2008, 151, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Ivanciuc, T.; Sbrana, E.; Casola, A.; Garofalo, R.P. Cystathionine γ-Lyase Deficiency Enhances Airway Reactivity and Viral-Induced Disease in Mice Exposed to Side-Stream Tobacco Smoke. Pediatr. Res. 2019, 86, 39–46. [Google Scholar] [CrossRef]
- Kaan, P.M.; Hegele, R.G. Interaction between Respiratory Syncytial Virus and Particulate Matter in Guinea Pig Alveolar Macrophages. Am. J. Respir. Cell Mol. Biol. 2003, 28, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.L.; Trasti, F.S.; Mangum, J.B.; Everitt, J.I. Effect of Preexposure to Ultrafine Carbon Black on Respiratory Syncytial Virus Infection in Mice. Toxicol. Sci. 2003, 72, 331–338. [Google Scholar] [CrossRef] [Green Version]
- Lambert, A.L.; Mangum, J.B.; Delorme, M.P.; Everitt, J.I. Ultrafine Carbon Black Particles Enhance Respiratory Syncytial Virus-Induced Airway Reactivity, Pulmonary Inflammation, and Chemokine Expression. Toxicol. Sci. 2003, 72, 339–346. [Google Scholar] [CrossRef] [Green Version]
- Mebratu, Y.A.; Smith, K.R.; Agga, G.E.; Tesfaigzi, Y. Inflammation and Emphysema in Cigarette Smoke-Exposed Mice When Instilled with Poly (I:C) or Infected with Influenza A or Respiratory Syncytial Viruses. Respir. Res. 2016, 17, 75. [Google Scholar] [CrossRef] [Green Version]
- Modestou, M.A.; Manzel, L.J.; El-Mahdy, S.; Look, D.C. Inhibition of IFN-γ-Dependent Antiviral Airway Epithelial Defense by Cigarette Smoke. Respir. Res. 2010, 11, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phaybouth, V.; Wang, S.-Z.; Hutt, J.A.; McDonald, J.D.; Harrod, K.S.; Barrett, E.G. Cigarette Smoke Suppresses Th1 Cytokine Production and Increases RSV Expression in a Neonatal Model. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2006, 290, L222–L231. [Google Scholar] [CrossRef]
- Poon, J.; Campos, M.A.; Foronjy, R.F.; Nath, S.; Gupta, G.; Railwah, C.; Dabo, A.J.; Baumlin, N.; Salathe, M.A.; Geraghty, P. Cigarette Smoke Exposure Reduces Leukemia Inhibitory Factor Levels During Respiratory Syncytial Viral Infection. Int. J. Chron. Obstruct. Pulmon. Dis. 2019, 14, 1305–1315. [Google Scholar] [CrossRef] [Green Version]
- Smallcombe, C.C.; Harford, T.J.; Linfield, D.T.; Lechuga, S.; Bokun, V.; Piedimonte, G.; Rezaee, F. Titanium Dioxide Nanoparticles Exaggerate Respiratory Syncytial Virus-Induced Airway Epithelial Barrier Dysfunction. Am. J. Physiol. Cell. Mol. Physiol. 2020, 319, L481–L496. [Google Scholar] [CrossRef]
- Raza, M.W.; Essery, S.D.; Weir, D.M.; Ogilvie, M.M.; Elton, R.A.; Blackwell, C.C. Infection with Respiratory Syncytial Virus and Water-Soluble Components of Cigarette Smoke Alter Production of Tumour Necrosis Factor α and Nitric Oxide by Human Blood Monocytes. FEMS Immunol. Med. Microbiol. 1999, 24, 387–394. [Google Scholar] [CrossRef]
- Soukup, J.; Koren, H.; Becker, S. Ozone Effect on Respiratory Syncytial Virus Infectivity and Cytokine Production by Human Alveolar Macrophages. Environ. Res. 1993, 60, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Urrego, F.; Scuri, M.; Auais, A.; Mohtasham, L.; Piedimonte, G. Combined Effects of Chronic Nicotine and Acute Virus Exposure on Neurotrophin Expression in Rat Lung. Pediatr. Pulmonol. 2009, 44, 1075–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.L.; Wang, Y.Y.; Yang, Z.H.; Huang, D.; Weng, H.; Seng, X.T. Methodological Quality (Risk of Bias) Assessment Tools for Primary and Secondary Medical Studies: What Are They and Which Is Better? Mil. Med. Res. 2020, 7, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooijmans, C.R.; Rovers, M.M.; de Vries, R.B.M.; Leenaars, M.; Ritskes-Hoitinga, M.; Langendam, M.W. SYRCLE’s Risk of Bias Tool for Animal Studies. BMC Med. Res. Methodol. 2014, 14, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, A.M.; Sargeant, J.M. Critical Appraisal of Studies Using Laboratory Animal Models. ILAR J. 2014, 55, 405–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tufanaru, C.; Aromataris, E.; Campbell, J.; Hopp, L. Systematic Reviews of Effectiveness. In JBI Manual for Evidence Synthesis; Aromataris E, Ed.; The Joanna Briggs Institute: Adelaide, Australia, 2020. [Google Scholar]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 Statement: An Updated Guideline for Reporting Systematic Reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]
- Rezaee, F.; Harford, T.J.; Linfield, D.T.; Altawallbeh, G.; Midura, R.J.; Ivanov, A.I.; Piedimonte, G. cAMP-Dependent Activation of Protein Kinase a Attenuates Respiratory Syncytial Virus-Induced Human Airway Epithelial Barrier Disruption. PLoS ONE 2017, 12, e0181876. [Google Scholar] [CrossRef] [Green Version]
- Ambalavanan, N.; Stanishevsky, A.; Bulger, A.; Halloran, B.; Steele, C.; Vohra, Y.; Matalon, S. Titanium Oxide Nanoparticle Instillation Induces Inflammation and Inhibits Lung Development in Mice. Am. J. Physiol. Cell. Mol. Physiol. 2013, 304, L152–L161. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, N.; Naya, M.; Endoh, S.; Maru, J.; Yamamoto, K.; Nakanishi, J. Comparative Pulmonary Toxicity Study of Nano-TiO(2) Particles of Different Sizes and Agglomerations in Rats: Different Short- and Long-Term Post-instillation Results. Toxicology 2009, 264, 110–118. [Google Scholar] [CrossRef]
- Morimoto, Y.; Kobayashi, N.; Shinohara, N.; Myojo, T.; Tanaka, I.; Nakanishi, J. Hazard Assessments of Manufactured Nanomaterials. J. Occup. Health 2010, 52, 325–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, S.; Swanson, M.; Lieberman, A.; Reed, M.; Yue, Z.; Lindell, D.M.; Lukacs, N.W. Autophagy-Mediated Dendritic Cell Activation Is Essential for Innate Cytokine Production and APC Function with Respiratory Syncytial Virus Responses. J. Immunol. 2011, 187, 3953–3961. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, A.; Tanaka, N.; Tamai, K.; Kyuuma, M.; Ishikawa, Y.; Sato, H.; Yoshimori, T.; Saito, S.; Sugamura, K. Survival of Parvovirus B19-Infected Cells by Cellular Autophagy. Virology 2006, 349, 254–263. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.-C.; Chang, C.-L.; Wang, P.-S.; Tsai, Y.; Liu, H.-S. Enterovirus 71-Induced Autophagy Detected In Vitro and In Vivo Promotes Viral Replication. J. Med. Virol. 2009, 81, 1241–1252. [Google Scholar] [CrossRef]
- Heaton, N.S.; Randall, G. Dengue Virus and Autophagy. Viruses 2011, 3, 1332–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosso, P.; Moreno, S.; Fracassi, A.; Rocco, M.L.; Aloe, L. Nerve Growth Factor and Autophagy: Effect of Nasal Anti-NGF-Antibodies Administration on Ambra1 and Beclin-1 Expression in Rat Brain. Growth Factors 2015, 33, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Franco, M.L.; Melero, C.; Sarasola, E.; Acebo, P.; Luque, A.; Calatayud-Baselga, I.; García-Barcina, M.; Vilar, M. Mutations in TrkA Causing Congenital Insensitivity to Pain with Anhidrosis (CIPA) Induce Misfolding, Aggregation, and Mutation-dependent Neurodegeneration by Dysfunction of the Autophagic Flux. J. Biol. Chem. 2016, 291, 21363–21374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.-G.; Li, H.; Huang, Y.; Li, R.; Wang, X.-F.; Yu, L.-X.; Guang, X.-Q.; Li, L.; Zhang, H.-Y.; Zhao, Y.-Z.; et al. Nerve Growth Factor-Induced Akt/mTOR Activation Protects the Ischemic Heart via Restoring Autophagic Flux and Attenuating Ubiquitinated Protein Accumulation. Oncotarget 2017, 8, 5400–5413. [Google Scholar] [CrossRef] [Green Version]
- Othumpangat, S.; Gibson, L.; Samsell, L.; Piedimonte, G. NGF Is an Essential Survival Factor for Bronchial Epithelial Cells during Respiratory Syncytial Virus Infection. PLoS ONE 2009, 4, e6444. [Google Scholar] [CrossRef]
- Cuff, S.; Ruby, J. Evasion of Apoptosis by DNA Viruses. Immunol. Cell Biol. 1996, 74, 527–537. [Google Scholar] [CrossRef]
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity 2019, 50, 907–923. [Google Scholar] [CrossRef] [PubMed]
- Bach, E.A.; Aguet, M.; Schreiber, R.D. The IFN γ Receptor: A Paradigm for Cytokine Receptor Signaling. Annu. Rev. Immunol. 1997, 15, 563–591. [Google Scholar] [CrossRef]
- Greenlund, A.C.; Morales, M.O.; Viviano, B.L.; Yan, H.; Krolewski, J.; Schreiber, R.D. Stat Recruitment by Tyrosine-Phosphorylated Cytokine Receptors: An Ordered Reversible Affinity-Driven Process. Immunity 1995, 2, 677–687. [Google Scholar] [CrossRef] [Green Version]
- Widdicombe, J.G.; Pack, R.J. The Clara Cell. Eur. J. Respir. Dis. 1982, 63, 202–220. [Google Scholar] [PubMed]
- Wang, S.-Z.; Rosenberger, C.L.; Bao, Y.-X.; Stark, J.M.; Harrod, K. Clara Cell Secretory Protein Modulates Lung Inflammatory and Immune Responses to Respiratory Syncytial Virus Infection. J. Immunol. 2003, 171, 1051–1060. [Google Scholar] [CrossRef] [Green Version]
- Miyata, R.; van Eeden, S.F. The Innate and Adaptive Immune Response Induced by Alveolar Macrophages Exposed to Ambient Particulate Matter. Toxicol. Appl. Pharmacol. 2011, 257, 209–226. [Google Scholar] [CrossRef]
- Panuska, J.R.; Cirino, N.M.; Midulla, F.; Despot, J.E.; McFadden, E.R.; Huang, Y.T. Productive Infection of Isolated Human Alveolar Macrophages by Respiratory Syncytial Virus. J. Clin. Investig. 1990, 86, 113–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franke, G.; Freihorst, J.; Steinmüller, C.; Verhagen, W.; Hockertz, S.; Lohmann-Matthes, M.-L. Interaction of Alveolar Macrophages and Respiratory Syncytial Virus. J. Immunol. Methods 1994, 174, 173–184. [Google Scholar] [CrossRef]
- Dakhama, A.; Kaan, P.M.; Hegele, R.G. Permissiveness of Guinea Pig Alveolar Macrophage Subpopulations to Acute Respiratory Syncytial Virus Infection In Vitro. Chest 1998, 114, 1681–1688. [Google Scholar] [CrossRef] [Green Version]
- Giuffrida, M.J.; Valero, N.; Mosquera, J.; De Mon, M.A.; Chacín, B.; Espina, L.M.; Gotera, J.; Bermudez, J.; Mavarez, A. Increased Cytokine/Chemokines in Serum from Asthmatic and Non-asthmatic Patients with Viral Respiratory Infection. Influ. Other Respir. Viruses 2014, 8, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Cella, M.; Jarrossay, D.; Facchetti, F.; Alebardi, O.; Nakajima, H.; Lanzavecchia, A.; Colonna, M. Plasmacytoid Monocytes Migrate to Inflamed Lymph Nodes and Produce Large Amounts of Type I Interferon. Nat. Med. 1999, 5, 919–923. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald-Bocarsly, P.; Dai, J.; Singh, S. Plasmacytoid Dendritic Cells and Type I IFN: 50 Years of Convergent History. Cytokine Growth Factor Rev. 2008, 19, 3–19. [Google Scholar] [CrossRef] [Green Version]
- Siegal, F.P.; Kadowaki, N.; Shodell, M.; Fitzgerald-Bocarsly, P.A.; Shah, K.; Ho, S.; Antonenko, S.; Liu, Y.-J. The Nature of the Principal Type 1 Interferon-Producing Cells in Human Blood. Science 1999, 284, 1835–1837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domachowske, J.B.; Ali-Ahmad, D.; Bonville, C.A.; Rosenberg, H.F. Replication of Respiratory Syncytial Virus Is Inhibited in Target Cells Generating Nitric Oxide In Situ. Front. Biosci. 2003, 8, a48–a53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, J.M.; Khan, A.M.; Chiappetta, C.L.; Xue, H.; Alcorn, J.L.; Colasurdo, G.N. Immune and Functional Role of Nitric Oxide in a Mouse Model of Respiratory Syncytial Virus Infection. J. Infect. Dis. 2005, 191, 387–395. [Google Scholar] [CrossRef]
- Colasurdo, G.N.; Fullmer, J.J.; Elidemir, O.; Atkins, C.; Khan, A.M.; Stark, J.M. Respiratory Syncytial Virus Infection in a Murine Model of Cystic Fibrosis. J. Med. Virol. 2006, 78, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Mestan, J.; Digel, W.; Mittnacht, S.; Hillen, H.; Blohm, D.; Möller, A.; Jacobsen, H.; Kirchner, H. Antiviral Effects of Recombinant Tumour Necrosis Factor In Vitro. Nature 1986, 323, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Neville, L.F.; Mathiak, G.; Bagasra, O. The Immunobiology of Interferon-γ Inducible Protein 10 kD (IP-10): A Novel, Pleiotropic Member of the C-X-C Chemokine Superfamily. Cytokine Growth Factor Rev. 1997, 8, 207–219. [Google Scholar] [CrossRef]
- Roe, M.F.E.; Bloxham, D.M.; Cowburn, A.S.; O’Donnell, D.R. Changes in Helper Lymphocyte Chemokine Receptor Expression and Elevation of IP-10 During Acute Respiratory Syncytial Virus Infection in Infants. Pediatr. Allergy Immunol. 2011, 22, 229–234. [Google Scholar] [CrossRef]
- Xu, H.; An, H.; Hou, J.; Han, C.; Wang, P.; Yu, Y.; Cao, X. Phosphatase PTP1B Negatively Regulates MyD88- and TRIF-Dependent Proinflammatory Cytokine and Type I Interferon Production in TLR-Triggered Macrophages. Mol. Immunol. 2008, 45, 3545–3552. [Google Scholar] [CrossRef]
- Medgyesi, D.; Hobeika, E.; Biesen, R.; Kollert, F.; Taddeo, A.; Voll, R.E.; Hiepe, F.; Reth, M. The Protein Tyrosine Phosphatase PTP1B is a Negative Regulator of CD40 and BAFF-R Signaling and Controls B Cell Autoimmunity. J. Exp. Med. 2014, 211, 427–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, S.-Y.; Segovia, J.A.; Chang, T.-H.; Morris, I.R.; Berton, M.T.; Tessier, P.A.; Tardif, M.R.; Cesaro, A.; Bose, S. DAMP Molecule S100A9 Acts as a Molecular Pattern to Enhance Inflammation during Influenza A Virus Infection: Role of DDX21-TRIF-TLR4-MyD88 Pathway. PLoS Pathog. 2014, 10, e1003848. [Google Scholar] [CrossRef]
- Brunwasser, S.M.; Snyder, B.; Driscoll, A.J.; Fell, D.B.; Savitz, D.A.; Feikin, D.R.; Skidmore, B.; Bhat, N.; Bont, L.J.; Dupont, W.D.; et al. Assessing the Strength of Evidence for a Causal Effect of Respiratory Syncytial Virus Lower Respiratory Tract Infections on Subsequent Wheezing Illness: A Systematic Review and Meta-Analysis. Lancet Respir. Med. 2020, 8, 795–806. [Google Scholar] [CrossRef]
- Olszewska-Pazdrak, B.; Casola, A.; Saito, T.; Alam, R.; Crowe, S.E.; Mei, F.; Ogra, P.L.; Garofalo, R.P. Cell-Specific Expression of RANTES, MCP-1, and MIP-1α by Lower Airway Epithelial Cells and Eosinophils Infected with Respiratory Syncytial Virus. J. Virol. 1998, 72, 4756–4764. [Google Scholar] [CrossRef] [Green Version]
- Rajajendram, R.; Tham, C.L.; Akhtar, M.N.; Sulaiman, M.R.; Israf, D.A. Inhibition of Epithelial CC-Family Chemokine Synthesis by the Synthetic Chalcone DMPF-1 via Disruption of NF-κB Nuclear Translocation and Suppression of Experimental Asthma in Mice. Mediat. Inflamm. 2015, 2015, 176926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, X.-H.; Pang, L.-L.; Yang, J.; Jin, Y. Comparison of Immune Response to Human Rhinovirus C and Respiratory Syncytial Virus in Highly Differentiated Human Airway Epithelial Cells. Virol. J. 2022, 19, 81. [Google Scholar] [CrossRef] [PubMed]
- Piedimonte, G. Contribution of Neuroimmune Mechanisms to Airway Inflammation and Remodeling during and after Respiratory Syncytial Virus Infection. Pediatr. Infect. Dis. J. 2003, 22 (Suppl. S2), S66–S75. [Google Scholar] [CrossRef] [PubMed]
- Zang, N.; Li, S.; Li, W.; Xie, X.; Ren, L.; Long, X.; Xie, J.; Deng, Y.; Fu, Z.; Xu, F.; et al. Resveratrol Suppresses Persistent Airway Inflammation and Hyperresponsivess Might Partially via Nerve Growth Factor in Respiratory Syncytial Virus-Infected Mice. Int. Immunopharmacol. 2015, 28, 121–128. [Google Scholar] [CrossRef]
- Foronjy, R.F.; Dabo, A.J.; Cummins, N.; Geraghty, P. Leukemia Inhibitory Factor Protects the Lung during Respiratory Syncytial Viral Infection. BMC Immunol. 2014, 15, 41. [Google Scholar] [CrossRef] [Green Version]
- Ulich, T.R.; Fann, M.J.; Patterson, P.H.; Williams, J.H.; Samal, B.; Del Castillo, J.; Yin, S.; Guo, K.; Remick, D.G. Intratracheal Injection of LPS and Cytokines. V. LPS Induces Expression of LIF and LIF Inhibits Acute Inflammation. Am. J. Physiol. Content 1994, 267 (4 Pt 1), L442–L446. [Google Scholar] [CrossRef]
- Wang, J.; Chen, Q.; Corne, J.; Zhu, Z.; Lee, C.G.; Bhandari, V.; Homer, R.; Elias, J.A. Pulmonary Expression of Leukemia Inhibitory Factor Induces B Cell Hyperplasia and Confers Protection in Hyperoxia. J. Biol. Chem. 2003, 278, 31226–31232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinton, L.J.; Mizgerd, J.; Hilliard, K.L.; Jones, M.; Kwon, C.Y.; Allen, E. Leukemia Inhibitory Factor Signaling Is Required for Lung Protection during Pneumonia. J. Immunol. 2012, 188, 6300–6308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamohara, H.; Sakamoto, K.; Ishiko, T.; Masuda, Y.; Abe, T.; Ogawa, M. Leukemia Inhibitory Factor Induces Apoptosis and Proliferation of Human Carcinoma Cells through Different Oncogene Pathways. Int. J. Cancer 1997, 72, 687–695. [Google Scholar] [CrossRef]
- Schere-Levy, C.; Buggiano, V.; Quaglino, A.; Gattelli, A.; Cirio, M.C.; Piazzon, I.; Vanzulli, S.; Kordon, E.C. Leukemia Inhibitory Factor Induces Apoptosis of the Mammary Epithelial Cells and Participates in Mouse Mammary Gland Involution. Exp. Cell Res. 2003, 282, 35–47. [Google Scholar] [CrossRef]
- Furue, M.; Okamoto, T.; Hayashi, Y.; Okochi, H.; Fujimoto, M.; Myoishi, Y.; Abe, T.; Ohnuma, K.; Sato, G.H.; Asashima, M.; et al. Leukemia Inhibitory Factor as an Anti-apoptotic Mitogen for Pluripotent Mouse Embryonic Stem Cells in a Serum-Free Medium without Feeder Cells. Vitr. Cell. Dev. Biol. Anim. 2005, 41, 19–28. [Google Scholar] [CrossRef]
- Lu, Y.; Fukuyama, S.; Yoshida, R.; Kobayashi, T.; Saeki, K.; Shiraishi, H.; Yoshimura, A.; Takaesu, G. Loss of SOCS3 Gene Expression Converts STAT3 Function from Anti-apoptotic to Pro-apoptotic. J. Biol. Chem. 2006, 281, 36683–36690. [Google Scholar] [CrossRef] [Green Version]
- Fayon, M.; Rebola, M.; Berger, P.; Daburon, S.; Ousova, O.; Lavrand, F.; Moukaïla, B.; Pujol, W.; Taupin, J.L.; Labbé, A.; et al. Increased Secretion of Leukemia Inhibitory Factor by Immature Airway Smooth Muscle Cells Enhances Intracellular Signaling and Airway Contractility. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2006, 291, L244–L251. [Google Scholar] [CrossRef]
- Arici, A.; Engin, O.; Attar, E.; Olive, D.L. Modulation of Leukemia Inhibitory Factor Gene Expression and Protein Biosynthesis in Human Endometrium. J. Clin. Endocrinol. Metab. 1995, 80, 1908–1915. [Google Scholar]
- Carlson, C.D.; Bai, Y.; Miller Jonakait, G.; Hart, R.P. Interleukin-1 β Increases Leukemia Inhibitory Factor mRNA Levels through Transient Stimulation of Transcription Rate. Glia 1996, 18, 141–151. [Google Scholar] [CrossRef]
- Trojanek, J.B.; Cobos-Correa, A.; Diemer, S.; Kormann, M.; Schubert, S.C.; Zhou-Suckow, Z.; Agrawal, R.; Duerr, J.; Wagner, C.J.; Schatterny, J.; et al. Airway Mucus Obstruction Triggers Macrophage Activation and Matrix Metalloproteinase 12–Dependent Emphysema. Am. J. Respir. Cell Mol. Biol. 2014, 51, 709–720. [Google Scholar] [CrossRef]
- Chen, K.; Pociask, D.A.; McAleer, J.P.; Chan, Y.R.; Alcorn, J.F.; Kreindler, J.L.; Keyser, M.R.; Shapiro, S.D.; Houghton, A.M.; Kolls, J.K.; et al. IL-17RA Is Required for CCL2 Expression, Macrophage Recruitment, and Emphysema in Response to Cigarette Smoke. PLoS ONE 2011, 6, e20333. [Google Scholar] [CrossRef] [PubMed]
- Makino, A.; Shibata, T.; Nagayasu, M.; Hosoya, I.; Nishimura, T.; Nakano, C.; Nagata, K.; Ito, T.; Takahashi, Y.; Nakamura, S. RSV Infection-Elicited High MMP-12–Producing Macrophages Exacerbate Allergic Airway Inflammation with Neutrophil Infiltration. iScience 2021, 24, 103201. [Google Scholar] [CrossRef] [PubMed]
- Lagente, V.; Le Quement, C.; Boichot, E. Macrophage Metalloelastase (MMP-12) as a Target for Inflammatory Respiratory Diseases. Expert Opin. Ther. Targets 2009, 13, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Marchant, D.J.; Bellac, C.L.; Moraes, T.J.; Wadsworth, S.J.; Dufour, A.; Butler, G.S.; Bilawchuk, L.M.; Hendry, R.G.; Robertson, A.G.; Cheung, C.T.; et al. A New Transcriptional Role for Matrix Metalloproteinase-12 in Antiviral Immunity. Nat. Med. 2014, 20, 493–502. [Google Scholar] [CrossRef]
- Churg, A.; Wang, R.D.; Tai, H.; Wang, X.; Xie, C.; Dai, J.; Shapiro, S.D.; Wright, J.L. Macrophage Metalloelastase Mediates Acute Cigarette Smoke–induced Inflammation via Tumor Necrosis Factor-α Release. Am. J. Respir. Crit. Care Med. 2003, 167, 1083–1089. [Google Scholar] [CrossRef]
- Churg, A.; Wang, R.D.; Tai, H.; Wang, X.; Xie, C.; Wright, J.L. Tumor Necrosis Factor-α Drives 70% of Cigarette Smoke–induced Emphysema in the Mouse. Am. J. Respir. Crit. Care Med. 2004, 170, 492–498. [Google Scholar] [CrossRef]
- Vlahos, R.; Bozinovski, S.; Chan, S.P.J.; Ivanov, S.; Lindén, A.; Hamilton, J.A.; Anderson, G.P. Neutralizing Granulocyte/Macrophage Colony–Stimulating Factor Inhibits Cigarette Smoke–induced Lung Inflammation. Am. J. Respir. Crit. Care Med. 2010, 182, 34–40. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inclusion Criteria | Exclusion Criteria |
|---|---|
| Original study | Review article |
| Study performed either on cellular, animal, or human models | Analysis on the molecular mechanism(s) of air pollution only |
| Analysis of the interaction between air pollutants and RSV | Analysis on the molecular mechanism(s) of RSV only |
| Investigation on molecular mechanism(s), i.e., altered immune system response, including cytokine/chemokine production and/or release, receptor stimulation, gene expression, histological study | Lack of a potential pathomechanism analysis |
| Only prenatal exposure to RSV or air pollution | |
| Lack of a possibility to obtain a full-text article |
| Author | Air Pollutant | Material/Subjects | Country |
|---|---|---|---|
| Becker and Soukup, 1999 [32] | NO2 (different concentrations: 0.5, 1.0, and 1.5 ppm) | Human bronchial epithelial cell line BEAS-2B | USA |
| Becker and Soukup, 1999 “Exposure to urban air particulates alters…” [33] | PM10 (EHC-93) | human bronchial epithelial cell line BEAS-2B subclone S6 and human alveolar macrophage from BAL from volunteers | USA |
| Castro, 2008 [34] | Cigarette smoke condensate | A549, human alveolar type II–like epithelial cells, and 293, a human embryonic kidney epithelial cell line | USA |
| Castro, 2011 [35] | cigarette smoke extract | Human plasmacytoid dendritic cells | USA |
| Chakraborty, 2017 [36] | TiO2-NP | Human primary bronchial epithelial cells) | USA |
| Cruz-Sanchez, 2013 [37] | Mimics of ambient particulate matter (PM10) | human epithelial-2 (HEp-2) cells 1HAEo-cells- normal human airway epithelial cells transformed with Simian virus 40 | Canada |
| Foronjy, 2014 [38] | Cigarette smoke | C57BL/6J mice (repeated RSV exposition- 6 times) | USA |
| Foronjy, 2016 [39] | Cigarette smoke | lung BALF from age-matched healthy control subjects, smokers, and subjects with COPD; Ptp1b (Ptpn1 gene) knockout (-/-) mice; human primary small airway epithelial and mouse bone marrow derived macrophages | USA |
| Groskreutz, 2009 [40] | Cigarette smoke extract | Primary human tracheobronchial epithelial | USA |
| Harrod, 2003 [29] | Diesel engine emissions | C57Bl/6 mice | USA |
| Hashiguchi [41] | TiO2-NP | BALB/c mice | Japan |
| Hirota, 2015 [42] | PM10 (EHC93) | human airway epithelial cell line (HBEC-6KT) | Canada |
| Hobson and Everard, 2007 [43] | Nitric oxide | human monocyte-derived dendritic cells (DCs) | UK |
| Ivanciuc 2019 [44] | side-stream tobacco smoke | cystathionine γ-lyase enzyme (CSE)- deficient and wild-type mice | USA |
| Kaan and Hegele, 2003 [45] | PM10 (EHC-93) | Guinea pig alveolar macrophages | Canada |
| Lambert, 2003 “Effect of Preexposure to Ultrafine Carbon Black…” [46] | Preexposure to ultrafine carbon black | BALB/c mice | USA |
| Lambert, 2003 “Ultrafine Carbon Black Particles Enhance…” [47] | Ultrafine carbon black after RSV infection | BALB/c mice | USA |
| Mebratu, 2016 [48] | Cigarette smoke | C57BL/6 mice | USA |
| Modestou, 2010 [49] | Cigarette smoke extract | Human trachea and bronchial samples Primary human tracheobronchial epithelial cells | USA |
| Phaybouth, 2006 [50] | side-stream cigarette smoke | Newborn BALB/cmice (RSV infection twice) | USA |
| Poon, 2019 [51] | Cigarette smoke-mice COPD patients Smokers | Mice exposed to cigarette smoke; BALF from healthy never smokers, smokers, and COPD patients; Human bronchial epithelial (HBE) | USA |
| Smallcombe, 2020 [52] | Titanium dioxide nanoparticles | Immortalized human bronchial epithelial cells; C57BL/6 mice | USA |
| Raza, 1999 [53] | water-soluble cigarette smoke extract (CSE), nicotine, cotinine | monocytes of the blood from donors | UK |
| Soukup, Koren, and Becker, 1993 [54] | ozone | Human alveolar macrophages | USA |
| Urrego, 2009 [55] | Nicotine exposure | Rats | USA |
| JBI Critical Appraisal Checklist For Quasi-Experimental Studies | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Articles | Model | D1 | D2 | D3 | D4 | D5 | D6 | D7 | D8 | D9 |
| Becker and Soukup, 1999 [32] | H |  | | | | | | | | |
| Becker and Soukup, 1999 “Exposure to urban air particulates alters…” [33] | H | | | | | | | | | |
| Castro, 2008 [34] | H | | | | | | | | | |
| Castro, 2011 [35] | H | | | | |  | | | | |
| Chakraborty, 2017 [36] | H | | | | | | | | | |
| Cruz-Sanchez, 2013 [37] | H | | | | | | | | | |
| Foronjy 2014 [38] A | A | | | | | | | | | |
| Foronjy, 2016 [39] B | H/A | | | | | | | | | |
| Groskreutz, 2009 [40] | H | | | | | | | | | |
| Harrod, 2003 [29] C | A | | | | | | | | | |
| Hashiguchi [41] D | A | | | | | | | | | |
| Hirota, 2015 [42] | H | | | | | | | | | |
| Hobson and Everard, 2007 [43] | H | | | | | | | | | |
| Ivanciuc, 2019 [44] E | A | | | | | | | | | |
| Kaan and Hegele, 2003 [45] F | A | | | | | | | | | |
| Lambert, 2003 “Effect of Preexposure…” [46] C | A | | | | |  | | | | |
| Lambert, 2003 “Ultrafine Carbon Black …” [47] C | A | | | | | | | | | |
| Mebratu, 2016 [48] C | A | | | | | | | | | |
| Modestou, 2010 [49] | H | | | | | | | | | |
| Phaybouth, 2006 [50] G | A | | | | | | | | | |
| Poon, 2019 [51] H | H/A | | | | | | | | | |
| Smallcombe, 2020 [52] I | H/A | | | | | | | | | |
| Raza, 1999 [53] | H | | | | | | | | | |
| Soukup, Koren, and Becker, 1993 [54] | H | | | | | | | | | |
| Urrego, 2009 [55] C | A | | | | | | | | | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wrotek, A.; Jackowska, T. Molecular Mechanisms of RSV and Air Pollution Interaction: A Scoping Review. Int. J. Mol. Sci. 2022, 23, 12704. https://doi.org/10.3390/ijms232012704
Wrotek A, Jackowska T. Molecular Mechanisms of RSV and Air Pollution Interaction: A Scoping Review. International Journal of Molecular Sciences. 2022; 23(20):12704. https://doi.org/10.3390/ijms232012704
Chicago/Turabian StyleWrotek, August, and Teresa Jackowska. 2022. "Molecular Mechanisms of RSV and Air Pollution Interaction: A Scoping Review" International Journal of Molecular Sciences 23, no. 20: 12704. https://doi.org/10.3390/ijms232012704