
Physiological and Pathological Remodeling of Cerebral Microvessels
 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
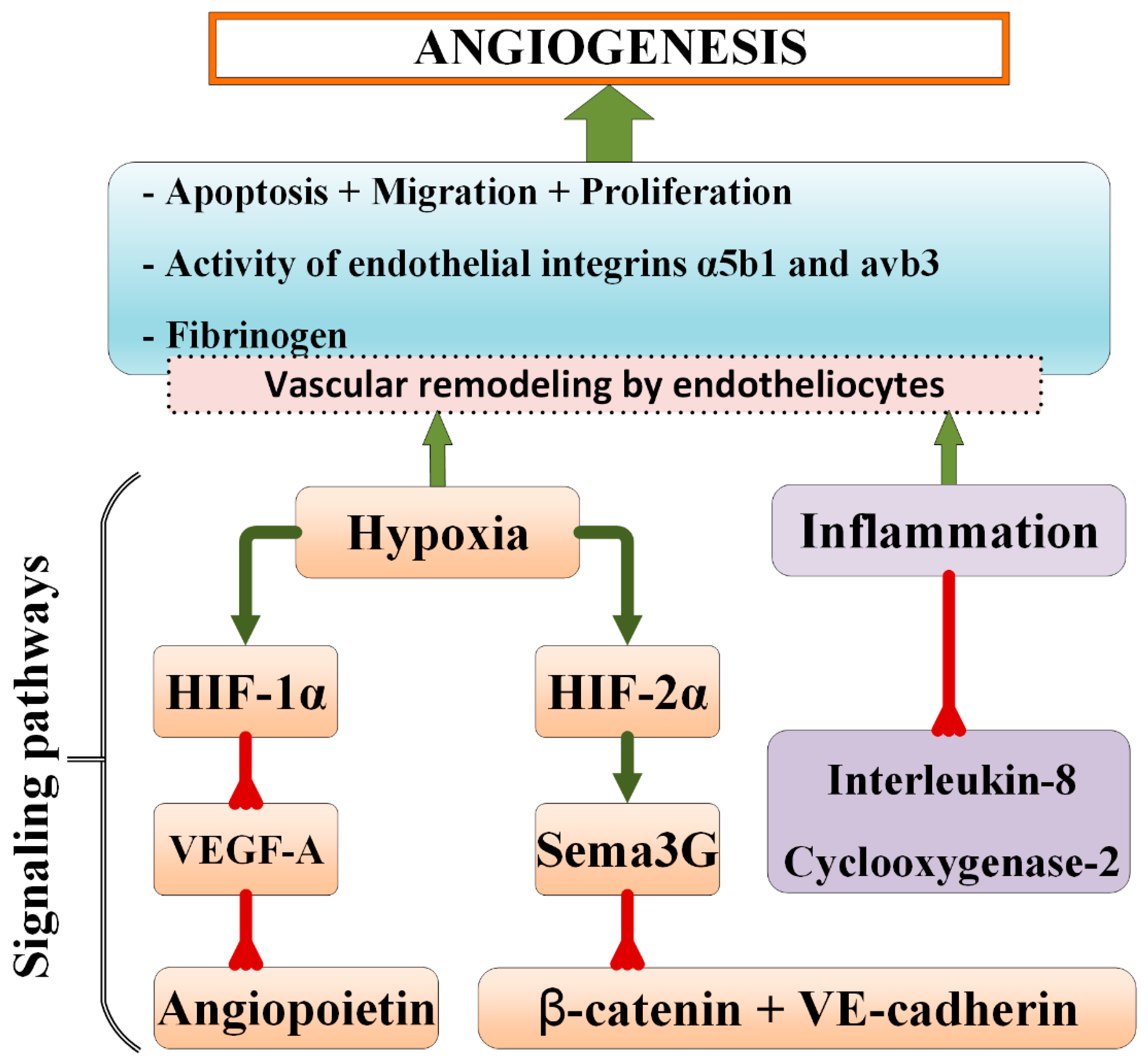
2. General Mechanisms of Microvasculature Remodeling
3. Functional Competence of Cerebral Endothelial Cells and Their Role in Brain Vessel Remodeling
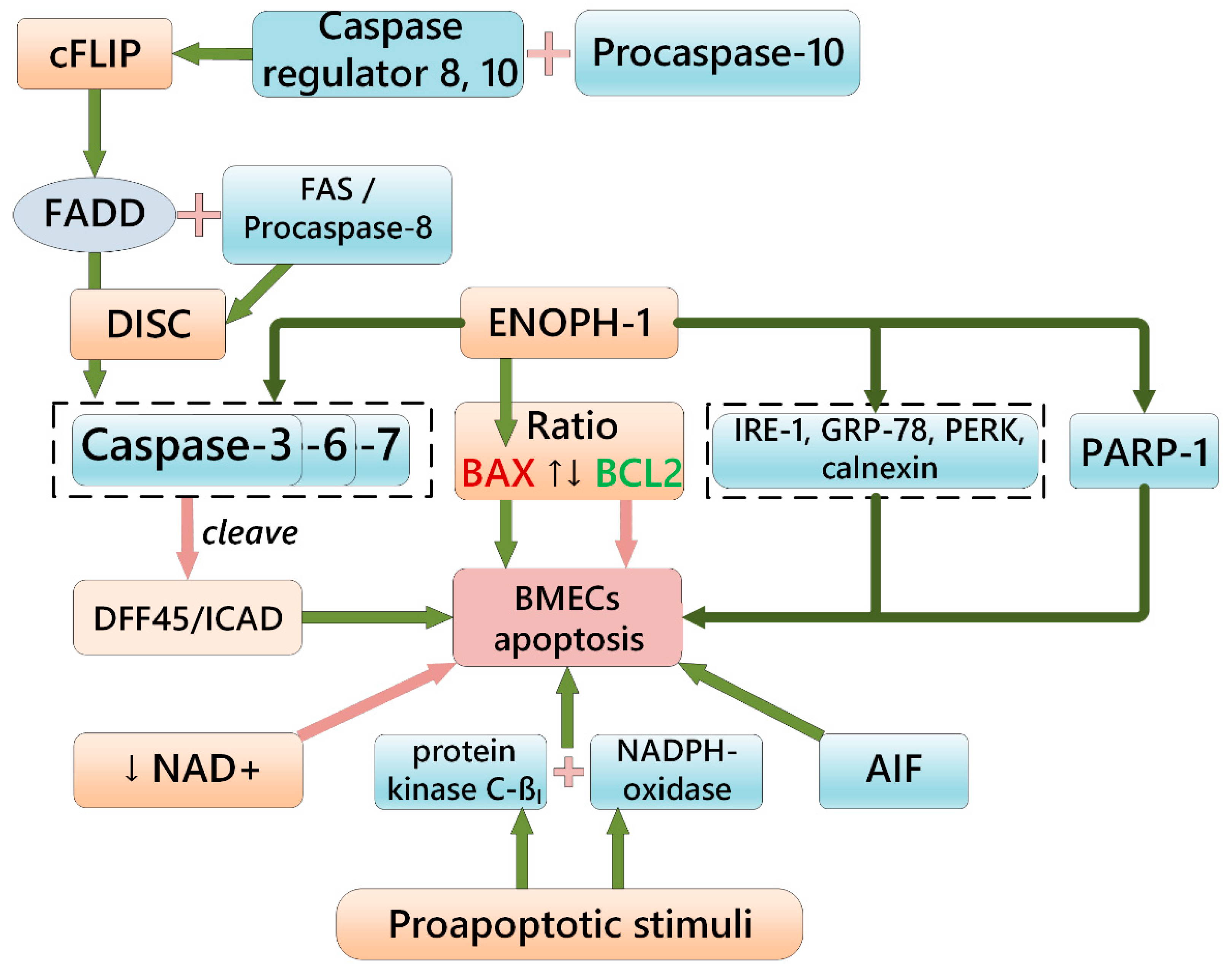
4. Brief Overview of BMEC Apoptosis and Microvessel Pruning
5. Regulation and Outcomes of Vascular Regression and Pruning in the Brain
6. Aberrant Microvessel Remodeling in Alzheimer’s-Type Neurodegeneration
7. Some Perspectives of Targeting Abnormal Microvascular Remodeling in Brain Diseases
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AIF | apoptosis inducing factor |
| Ang | angiopoietin |
| Ang2 | angiopoietin-2 |
| APAF1 | apoptotic protease activating factor 1 |
| APOE | apolipoprotein E |
| ATP | adenosinetrisphosphate |
| BAX | bcl2-associated x protein |
| BBB | blood-brain barrier |
| BCL-2 | b-cell lymphoma 2 |
| BMECs | brain microvessel endothelial cells |
| cFLIP | cellular FADD-like IL-1β-converting enzyme)-inhibitory protein |
| CNS | central nervous system |
| ColIV | collagen V |
| COX-2 | cyclooxygenase-2 |
| CXCR3 | activation of CXC chemokine receptor 3 |
| CytC | cytochrome c |
| DISC | death-inducing signaling complex |
| DLL4 | delta like canonical notch ligand 4 |
| ENOPH1 | enolase-phosphatase 1 |
| FADD | FAS-associated protein with death domain |
| FGF2 | fibroblast growth factor 2 |
| GLUT1 | glucose transporter 1 |
| HBMEC | human brain microvascular endothelial cells |
| HIF-1α | hypoxia-inducible factor 1-alpha |
| IL-8 iPSCs | interleukin-8 induced pluripotent stem cells |
| JAG1 | jagged1 |
| JNKs | c-jun n-terminal kinases |
| LRP1 | low density lipoprotein receptor-related protein 1 |
| MAP kinase | mitogen-activated protein kinase |
| MCP1 | monocyte chemotactic protein 1 |
| MEOX2 | mesenchyme homeobox 2 |
| NAD+ | nicotinamide adenine dinucleotide |
| Notch 3 | neurogenic locus notch homolog protein 3 |
| NRF2 | NF-e2-related factor 2 |
| NRP1 | neuropilin-1 |
| PARP-1 | poly [ADP-ribose] polymerase 1 |
| PDGF | platelet-derived growth factor |
| PGFD | prox1-gfp/flt1-dsred |
| RSPO3 | R-spondin 3 |
| S1PR1 | sphingosine-1-phosphate receptor 1 |
| Sema | semaphorin |
| TIMPs | tissue inhibitors of metalloproteinases |
| TRPA1 | transient receptor potential cation channel A1 |
| TWEAK | TNF-like weak inducer of apoptosis |
| VE-cadherin | vascular endothelial cadherin |
| VEGF | vascular endothelial growth factor |
| VEGF-A | vascular endothelial growth factor A |
| VEGFR | vascular endothelial growth factor receptor |
References
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uspenskaya, Y.A.; Morgun, A.V.; Osipova, E.D.; Pozhilenkova, E.A.; Salmina, A.B. Mechanisms of cerebral angiogenesis in health and brain pathology. Neurosci. Behav. Physiol. 2022, 52, 453–461. [Google Scholar] [CrossRef]
- Korn, C.; Augustin, H.G. Mechanisms of vessel pruning and regression. Dev. Cell 2015, 34, 5–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef] [Green Version]
- Erdener, Ş.E.; Dalkara, T. Small Vessels Are a Big Problem in Neurodegeneration and Neuroprotection. Front. Neurol. 2019, 10, 889. [Google Scholar] [CrossRef]
- Elabi, O.; Gaceb, A.; Carlsson, R.; Padel, T.; Soylu-Kucharz, R.; Cortijo, I.; Li, W.; Li, J.-Y.; Paul, G. Human alpha-synuclein overexpression in a mouse model of Parkinson’s disease leads to vascular pathology, blood brain barrier leakage and pericyte activation. Sci. Rep. 2021, 11, 1120. [Google Scholar] [CrossRef]
- Khromova, N.; Kopnin, P.; Rybko, V.; Kopnin, B.P. Downregulation of VEGF-C expression in lung and colon cancer cells decelerates tumor growth and inhibits metastasis via multiple mechanisms. Oncogene 2012, 31, 1389–1397. [Google Scholar] [CrossRef] [Green Version]
- Petrova, T.V.; Koh, G.Y. Biological functions of lymphatic vessels. Science 2020, 369, eaax4063. [Google Scholar] [CrossRef] [PubMed]
- Masood, F.; Bhattaram, R.; Rosenblatt, M.I.; Kazlauskas, A.; Chang, J.H.; Azar, D.T. Lymphatic Vessel Regression and Its Therapeutic Applications: Learning From Principles of Blood Vessel Regression. Front. Physiol. 2022, 13, 846936. [Google Scholar] [CrossRef]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef]
- Goligorsky, M.S. Microvascular rarefaction: The decline and fall of blood vessels. Organogenesis 2010, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Melincovici, C.S.; Boşca, A.B.; Şuşman, S.; Mărginean, M.; Mihu, C.; Istrate, M.; Moldovan, I.M.; Roman, A.L.; Mihu, C.M. Vascular endothelial growth factor (VEGF)—Key factor in normal and pathological angiogenesis. Rom. J. Morphol. Embryol. 2018, 59, 455–467. [Google Scholar] [PubMed]
- Lobov, I.B.; Cheung, E.; Wudali, R.; Cao, J.; Halasz, G.; Wei, Y.; Economides, A.; Lin, H.C.; Papadopoulos, N.; Yancopoulos, G.D.; et al. The Dll4/Notch pathway controls postangiogenic blood vessel remodeling and regression by modulating vasoconstriction and blood flow. Blood 2011, 117, 6728–6737. [Google Scholar] [CrossRef] [PubMed]
- Korn, C.; Scholz, B.; Hu, J.; Srivastava, K.; Wojtarowicz, J.; Arnsperger, T.; Adams, R.H.; Boutros, M.; Augustin, H.; Augustin, I. Endothelial cell-derived non-canonical Wnt ligands control vascular pruning in angiogenesis. Development 2014, 141, 1757–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholz, B.; Korn, C.; Wojtarowicz, J.; Mogler, C.; Augustin, I.; Boutros, M.; Niehrs, C.; Augustin, H.G. Endothelial RSPO3 Controls Vascular Stability and Pruning through Non-canonical WNT/Ca(2+)/NFAT Signaling. Dev. Cell 2016, 36, 79–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peghaire, C.; Bats, M.L.; Sewduth, R.; Jeanningros, S.; Jaspard, B.; Couffinhal, T.; Duplàa, C.; Dufourcq, P. Fzd7 (Frizzled-7) Expressed by Endothelial Cells Controls Blood Vessel Formation Through Wnt/β-Catenin Canonical Signaling. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2369–2380. [Google Scholar] [CrossRef] [Green Version]
- Kazanskaya, O.; Ohkawara, B.; Heroult, M.; Wu, W.; Maltry, N.; Augustin, H.; Niehrs, C. The Wnt signaling regulator R-spondin 3 promotes angioblast and vascular development. Development 2008, 35, 3655–3664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, J.R.; Fortunato, I.C.; Fonseca, C.G.; Pezzarossa, A.; Barbacena, P.; Dominguez-Cejudo, M.A.; Vasconcelos, F.F.; Santos, N.C.; Carvalho, F.A.; Franco, C.A. Non-canonical Wnt signaling regulates junctional mechanocoupling during angiogenic collective cell migration. Elife 2019, 8, e45853. [Google Scholar] [CrossRef]
- Franco, C.A.; Jones, M.L.; Bernabeu, O.M.; Vion, A.-C.; Barbacena, P.; Fan, J.; Mathivet, T.; Fonseca, C.; Ragab, A.; Yamaguchi, T.P.; et al. Non-canonical Wntsignalling modulates the endothelial shear stress flow sensor in vascular remodeling. Elife 2016, 5, e07727. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Baccouche, B.; Olayinka, O.; Serikbaeva, A.; Kazlauskas, A. The Role of the Wnt Pathway in VEGF/Anti-VEGF-Dependent Control of the Endothelial Cell Barrier. Investig. Ophthalmol. Vis. Sci. 2021, 62, 17. [Google Scholar] [CrossRef] [PubMed]
- Foulquier, S.; Daskalopoulos, E.P.; Lluri, G.; Hermans, K.C.M.; Deb, A.; Blankesteijn, W.M. WNT Signaling in Cardiac and Vascular Disease. Pharmacol. Rev. 2018, 70, 68–141. [Google Scholar] [CrossRef] [Green Version]
- Swaminathan, B.; Youn, S.-W.; Naiche, L.A.; Du, J.; Villa, S.R.; Metz, J.B.; Feng, H.; Zhang, C.; Kopan, R.; Sims, P.A.; et al. Endothelial Notch signaling directly regulates the small GTPase RND1 to facilitate Notch suppression of endothelial migration. Sci. Rep. 2022, 12, 1655. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Wong, F.; Niessen, K.; Karsan, A. Notch activation promotes endothelial survival through a PI3K-Slug axis. Microvasc. Res. 2013, 89, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Hellström, M.; Phng, L.K.; Gerhardt, H. VEGF and Notch signaling: The yin and yang of angiogenic sprouting. Cell Adh. Migr. 2007, 1, 133–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, W.; Sweeney, C.; Walsh, C.A.; Rooney, P.; McCormick, J.; Veale, D.; Fearon, U. Notch signalling pathways mediate synovial angiogenesis in response to vascular endothelial growth factor and angiopoietin 2. Ann. Rheum. Dis. 2013, 72, 1080–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallah, J.; Rini, B.I. HIF inhibitors: Status of current clinical development. Curr. Oncol. Rep. 2019, 21, 6. [Google Scholar] [CrossRef]
- Burnier, L.; Boroujerdi, A.; Fernández, J.A.; Welser-Alves, J.V.; Griffin, J.H.; Milner, R. Physiological cerebrovascular remodeling in response to chronic mild hypoxia: A role for activated protein C. Exp. Neurol. 2016, 283 Pt A, 396–403. [Google Scholar] [CrossRef] [Green Version]
- Cheng, T.; Liu, D.; Griffin, J.; Fernandez, J.; Castellino, F.; Rosen, E.D.; Fukudome, K.; Zlokovic, B.V. Activated protein C blocks p53-mediated apoptosis in ischemic human brain endothelium and is neuroprotective. Nat. Med. 2003, 9, 338–342. [Google Scholar] [CrossRef]
- Claxton, S.; Fruttiger, M. Role of arteries in oxygen induced vaso-obliteration. Exp. Eye Res. 2003, 77, 305–311. [Google Scholar] [CrossRef]
- Zhong, W.; Gao, X.; Wang, S.; Han, K.; Ema, M.; Adams, S.; Adams, R.H.; Rosenblatt, M.I.; Chang, J.H.; Azar, D.T. Prox1-GFP/Flt1-DsRed transgenic mice: An animal model for simultaneous live imaging of angiogenesis and lymphangiogenesis. Angiogenesis 2017, 20, 581–598. [Google Scholar] [CrossRef]
- Sajib, S.; Zahra, F.T.; Lionakis, M.S.; German, N.A.; Mikelis, C.M. Mechanisms of angiogenesis in microbe-regulated inflammatory and neoplastic conditions. Angiogenesis 2018, 21, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Logie, J.J.; Ali, S.; Marshall, K.M.; Heck, M.M.; Walker, B.R.; Hadoke, P.W. Glucocorticoid-mediated inhibition of angiogenic changes in human endothelial cells is not caused by reductions in cell proliferation or migration. PLoS ONE 2010, 5, e14476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, G.E.; Saunders, W.B. Molecular balance of capillary tube formation versus regression in wound repair: Role of matrix metalloproteinases and their inhibitors. J. Investig. Dermatol. Symp. Proc. 2006, 11, 44–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lingen, M.W.; Polverini, P.J.; Bouck, N.P. Retinoic acid induces cells cultured from oral squamous cell carcinomas to become anti-angiogenic. Am. J. Pathol. 1996, 149, 247–258. [Google Scholar]
- Gosain, A.; Matthies, A.M.; Dovi, J.V.; Barbul, A.; Gamelli, R.L.; DiPietro, L.A. Exogenous pro-angiogenic stimuli cannot prevent physiologic vessel regression. J. Surg. Res. 2006, 135, 218–225. [Google Scholar] [CrossRef]
- Watanabe, K.; Hasegawa, Y.; Yamashita, H.; Shimizu, K.; Ding, Y.; Abe, M.; Ohta, H.; Imagawa, K.; Hojo, K.; Maki, H.; et al. Vasohibin as an endothelium-derived negative feedback regulator of angiogenesis. J. Clin. Investig. 2004, 114, 898–907. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Sonoda, H. The vasohibin family: A negative regulatory system of angiogenesis genetically programmed in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 37–41. [Google Scholar] [CrossRef] [Green Version]
- Wietecha, M.S.; Chen, L.; Ranzer, M.J.; Anderson, K.; Ying, C.; Patel, T.B.; DiPietro, L.A. Sprouty2 downregulates angiogenesis during mouse skin wound healing. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H459–H467. [Google Scholar] [CrossRef] [Green Version]
- Dimmeler, S.; Zeiher, A.M. Endothelial cell apoptosis in angiogenesis and vessel regression. Circ. Res. 2000, 87, 434–439. [Google Scholar] [CrossRef] [Green Version]
- Watson, E.C.; Grant, Z.L.; Coultas, L. Endothelial cell apoptosis in angiogenesis and vessel regression. Cell Mol. Life Sci. 2017, 74, 4387–4403. [Google Scholar] [CrossRef]
- Franco, C.A.; Jones, M.L.; Bernabeu, M.O.; Geudens, I.; Mathivet, T.; Rosa, A.; Lopes, F.M.; Lima, A.P.; Ragab, A.; Collins, R.T.; et al. Dynamic Endothelial Cell Rearrangements Drive Developmental Vessel Regression. PLoS Biol. 2015, 13, e1002125. [Google Scholar] [CrossRef]
- Ito, M.; Yoshioka, M. Regression of the hyaloid vessels and pupillary membrane of the mouse. Anat. Embryol. 1999, 200, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Madigan, M.C.; van Driel, D.; Maslimb, J.; Billson, F.A.; Mprovisab, J.; Penfold, P.L. The Human Hyaloid System: Cell Death and Vascular Regression. Exp. Eye Res. 2000, 70, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, J.H.; Yu, Y.S.; Mun, J.Y.; Kim, K.-W. Autophagy-induced regression of hyaloid vessels in early ocular development. Autophagy 2010, 6, 922–928. [Google Scholar] [CrossRef]
- Schafer, C.M.; Gurley, J.M.; Kurylowicz, K.; Lin, P.K.; Chen, W.; Elliott, M.H.; Davis, G.E.; Bhatti, F.; Griffin, C.T. An inhibitor of endothelial ETS transcription factors promotes physiologic and therapeutic vessel regression. Proc. Natl. Acad. Sci. USA 2020, 117, 26494–26502. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, R.J.; Yates, C.C.; Rodgers, M.E.; Du, X.; Wells, A. IP-10 induces dissociation of newly formed blood vessels. J. Cell Sci. 2009, 122 Pt 12, 2064–2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodnar, R.J.; Rodgers, M.E.; Chen, W.C.; Wells, A. Pericyte regulation of vascular remodeling through the CXC receptor 3. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2818–2829. [Google Scholar] [CrossRef] [Green Version]
- Yates-Binder, C.C.; Rodgers, M.; Jaynes, J.; Wells, A.; Bodnar, R.J.; Turner, T. An IP-10 (CXCL10)-derived peptide inhibits angiogenesis. PLoS ONE 2012, 7, e40812. [Google Scholar] [CrossRef] [Green Version]
- Meredith, J., Jr.; Mu, Z.; Saido, T.; Du, X. Cleavage of the cytoplasmic domain of the integrin beta3 subunit during endothelial cell apoptosis. J. Biol. Chem. 1998, 273, 19525–19531. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.; Zhu, L. Semaphorins and Their Receptors: From Axonal Guidance to Atherosclerosis. Front. Physiol. 2018, 9, 1236. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Gong, J.; Xu, Z.; Thimmulappa, R.K.; Mitchell, K.L.; Welsbie, D.S.; Biswal, S.; Duh, E.J. Nrf2 in ischemic neurons promotes retinal vascular regeneration through regulation of semaphorin 6A. Proc. Natl. Acad. Sci. USA 2015, 112, E6927–E6936. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.J.; Hu, J.; Uemura, A.; Tetzlaff, F.; Augustin, H.G.; Fischer, A. Semaphorin-3C signals through Neuropilin-1 and PlexinD1 receptors to inhibit pathological angiogenesis. EMBO Mol. Med. 2015, 7, 1267–1284. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Uemura, A.; Fukushima, Y.; Yoshida, Y.; Hirashima, M. Semaphorin 3G Provides a Repulsive Guidance Cue to Lymphatic Endothelial Cells via Neuropilin-2/PlexinD1. Cell Rep. 2016, 17, 2299–2311. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.Y.; Sun, N.H.; Chen, X.; Gong, J.J.; Yuan, S.T.; Hu, Z.Z.; Lu, N.N.; Körbelin, J.; Fukunaga, K.; Liu, Q.H.; et al. Endothelium-derived semaphorin 3G attenuates ischemic retinopathy by coordinating β-catenin-dependent vascular remodeling. J. Clin. Investig. 2021, 131, e135296. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Pan, H.; Wang, H.; Li, X.; Bu, X.; Wang, Q.; Gao, Y.; Wen, G.; Zhou, Y.; Cong, Z.; et al. Interplay between VEGF and Nrf2 regulates angiogenesis due to intracranial venous hypertension. Sci. Rep. 2016, 6, 37338. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Moore, A.N.; Redell, J.B.; Dash, P.K. Enhancing expression of Nrf2-driven genes protects the blood brain barrier after brain injury. J. Neurosci. 2007, 27, 10240–10248. [Google Scholar] [CrossRef] [Green Version]
- Zlokovic, B.V. Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci. 2005, 28, 202–208. [Google Scholar] [CrossRef]
- Pozhilenkova, E.A.; Lopatina, O.L.; Komleva, Y.K.; Salmin, V.V.; Salmina, A.B. Blood-brain barrier-supported neurogenesis in healthy and diseased brain. Rev. Neurosci. 2017, 28, 397–415. [Google Scholar] [CrossRef]
- DeStefano, J.G.; Xu, Z.S.; Williams, A.J.; Yimam, N.; Searson, P.C. Effect of shear stress on iPSC-derived human brain microvascular endothelial cells (dhBMECs). Fluids Barriers CNS 2017, 14, 20. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Rarick, K.R.; Ramchandran, R. Established, New and Emerging Concepts in Brain Vascular Development. Front. Physiol. 2021, 12, 636736. [Google Scholar] [CrossRef]
- Boriushkin, E.; Fancher, I.S.; Levitan, I. Shear-Stress Sensitive Inwardly-Rectifying K+ Channels Regulate Developmental Retinal Angiogenesis by Vessel Regression. Cell Physiol. Biochem. 2019, 52, 1569–1583. [Google Scholar] [CrossRef] [PubMed]
- Vion, A.-C.; Perovic, T.; Petit, C.; Hollfinger, I.; Bartels-Klein, E.; Frampton, E.; Gordon, E.; Claesson-Welsh, L.; Gerhardt, H. Endothelial Cell Orientation and Polarity Are Controlled by Shear Stress and VEGF Through Distinct Signaling Pathways. Front. Physiol. 2021, 11, 623769. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Cha, Y.R.; Pham, V.N.; Sakurai, A.; Roman, B.L.; Gutkind, J.S.; Weinstein, B.M. Assembly and patterning of the vascular network of the vertebrate hindbrain. Development 2011, 138, 1705–1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulrich, F.; Ma, L.H.; Baker, R.G.; Torres-Vázquez, J. Neurovascular development in the embryonic zebrafish hindbrain. Dev. Biol. 2011, 357, 134–151. [Google Scholar] [CrossRef] [Green Version]
- Boström, K.I.; Yao, J.; Wu, X.; Yao, Y. Endothelial Cells May Have Tissue-Specific Origins. J. Cell Biol. Histol. 2018, 11, 104. [Google Scholar]
- Zhao, X.; Xu, Z.; Xiao, L.; Shi, T.; Xiao, H.; Wang, Y.; Li, Y.; Xue, F.; Zeng, W. Review on the Vascularization of Organoids and Organoids-on-a-Chip. Front. Bioeng. Biotechnol. 2021, 9, 637048. [Google Scholar] [CrossRef]
- Autiero, M.; De Smet, F.; Claes, F.; Carmeliet, P. Role of neural guidance signals in blood vessel navigation. Cardiovasc. Res. 2005, 65, 629–638. [Google Scholar] [CrossRef]
- Liu, J.; Li, J. Astrocytes Protect Human Brain Microvascular Endothelial Cells from Hypoxia Injury by Regulating VEGF Expression. J. Healthc. Eng. 2022, 18, 1884959. [Google Scholar] [CrossRef]
- Thakore, P.; Alvarado, M.G.; Ali, S.; Mughal, A.; Pires, P.W.; Yamasaki, E.; Pritchard, H.A.; Isakson, B.E.; Tran, C.H.T.; Earley, S. Brain endothelial cell TRPA1 channels initiate neurovascular coupling. Elife 2021, 10, e63040. [Google Scholar] [CrossRef]
- Bernardini, M.; Brossa, A.; Chinigo, G.; Grolez, G.P.; Trimaglio, G.; Allart, L.; Hulot, A.; Marot, G.; Genova, T.; Joshi, A.; et al. Transient Receptor Potential Channel Expression Signatures in Tumor-Derived Endothelial Cells: Functional Roles in Prostate Cancer Angiogenesis. Cancers 2019, 11, 956. [Google Scholar] [CrossRef] [Green Version]
- Malinovskaya, N.A.; Komleva, Y.K.; Salmin, V.V.; Morgun, A.V.; Shuvaev, A.N.; Panina, Y.A.; Boitsova, E.B.; Salmina, A.B. Endothelial Progenitor Cells Physiology and Metabolic Plasticity in Brain Angiogenesis and Blood-Brain Barrier Modeling. Front. Physiol. 2016, 7, 599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yetkin-Arik, B.; Vogels, I.M.C.; Nowak-Sliwinska, P.; Weiss, A.; Houtkooper, R.H.; Van Noorden, C.J.F.; Klaassen, I.; Schlingemann, R.O. The role of glycolysis and mitochondrial respiration in the formation and functioning of endothelial tip cells during angiogenesis. Sci. Rep. 2019, 9, 12608. [Google Scholar] [CrossRef] [PubMed]
- Yetkin-Arik, B.; Vogels, I.M.C.; Neyazi, N.; Van Duinen, V.; Houtkooper, R.H.; Van Noorden, C.J.F.; Klaassen, I.; Schlingemann, R.O. Endothelial tip cells in vitro are less glycolytic and have a more flexible response to metabolic stress than non-tip cells. Sci. Rep. 2019, 9, 10414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munji, R.N.; Soung, A.L.; Weiner, G.A.; Sohet, F.; Semple, B.D.; Trivedi, A.; Gimlin, K.; Kotoda, M.; Korai, M.; Aydin, S.; et al. Profiling the mouse brain endothelial transcriptome in health and disease models reveals a core blood-brain barrier dysfunction module. Nat. Neurosci. 2019, 22, 1892–1902. [Google Scholar] [CrossRef]
- Al Mamun, A.; Hayashi, H.; Yamamura, A.; Nayeem, J.; Sato, M. Hypoxia induces the translocation of glucose transporter 1 to the plasma membrane in vascular endothelial cells. J. Physiol. Sci. 2020, 70, 44. [Google Scholar] [CrossRef]
- Chen, M.B.; Yang, A.C.; Yousef, H.; Lee, D.; Chen, W.; Schaum, N.; Lehallier, B.; Quake, S.R.; Wyss-Coray, T. Brain Endothelial Cells Are Exquisite Sensors of Age-Related Circulatory Cues. Cell Rep. 2020, 30, 4418–4432.e4. [Google Scholar] [CrossRef]
- Eelen, G.; de Zeeuw, P.; Treps, L.; Harjes, U.; Wong, B.W.; Carmeliet, P. Endothelial Cell Metabolism. Physiol. Rev. 2018, 98, 3–58. [Google Scholar] [CrossRef] [Green Version]
- Leung, S.W.S.; Shi, Y. The glycolytic process in endothelial cells and its implications. Acta Pharmacol. Sin. 2022, 43, 251–259. [Google Scholar] [CrossRef]
- McDonald, C.J.; Blankenheim, Z.J.; Drewes, L.R. Brain Endothelial Cells: Metabolic Flux and Energy Metabolism. In Handbook of Experimental Pharmacology; Springer: Cham, Switzerland, 2021. [Google Scholar] [CrossRef]
- Stamatovic, S.M.; Keep, R.F.; Andjelkovic, A.V. Brain endothelial cell-cell junctions: How to “open” the blood brain barrier. Curr. Neuropharmacol. 2008, 6, 179–192. [Google Scholar] [CrossRef] [Green Version]
- Stamatovic, S.M.; Dimitrijevic, O.B.; Keep, R.F.; Andjelkovic, A.V. Protein kinase Calpha-RhoAcross-talk in CCL2-induced alterations in brain endothelial permeability. J. Biol. Chem. 2006, 281, 8379–8388. [Google Scholar] [CrossRef] [Green Version]
- Demeule, M.; Régina, A.; Jodoin, J.; Laplante, A.; Dagenais, C.; Berthelet, F.; Moghrabi, A.; Béliveau, R. Drug transport to the brain: Key roles for the efflux pump P-glycoprotein in the blood-brain barrier. Vascul. Pharmacol. 2002, 38, 339–348. [Google Scholar] [CrossRef]
- Singh, A.K.; Jiang, Y.; Gupta, S.; Benlhabib, E. Effects of chronic ethanol drinking on the blood brain barrier and ensuing neuronal toxicity in alcohol-preferring rats subjected to intraperitoneal LPS injection. Alcohol Alcohol. 2007, 42, 385–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruzaeva, V.A.; Morgun, A.; Khilazheva, E.D.; Kuvacheva, N.V.; Pozhilenkova, E.; Boitsova, E.B.; Martynova, G.P.; Taranushenko, T.E.; Salmina, A.B. Features of blood-brain barrier formation affected by the modulation of HIF activity in astroglial and neuronal cells in vitro. Biochem. (Moscow) Suppl. Ser. B Biomed. Chem. 2017, 11, 81–86. [Google Scholar] [CrossRef]
- Biswas, S.; Cottarelli, A.; Agalliu, D. Neuronal and glial regulation of CNS angiogenesis and barriergenesis. Development 2020, 147, dev182279. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Jiang, L.; Li, C.; Hu, D.; Bu, J.-W.; Cai, D.; Du, J.-L. Haemodynamics-Driven Developmental Pruning of Brain Vasculature in Zebrafish. PLoS Biol. 2012, 10, e1001374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biron, K.E.; Dickstein, D.L.; Gopaul, R.; Jefferies, W.A. Amyloid triggers extensive cerebral angiogenesis causing blood brain barrier permeability and hypervascularity in Alzheimer’s disease. PLoS ONE 2011, 6, e23789. [Google Scholar] [CrossRef] [Green Version]
- Ridder, D.A.; Wenzel, J.; Müller, K.; Töllner, K.; Tong, X.-K.; Assmann, J.C.; Stroobants, S.; Weber, T.; Niturad, C.; Fischer, L.; et al. Brain endothelial TAK1 and NEMO safeguard the neurovascular unit. J. Exp. Med. 2015, 212, 1529–1549. [Google Scholar] [CrossRef] [Green Version]
- Zille, M.; Ikhsan, M.; Jiang, Y.; Lampe, J.; Wenzel, J.; Schwaninger, M. The impact of endothelial cell death in the brain and its role after stroke: A systematic review. Cell Stress 2019, 3, 330–347. [Google Scholar] [CrossRef] [Green Version]
- Errede, M.; Mangieri, D.; Longo, G.; Girolamo, F.; de Trizio, I.; Vimercati, A.; Serio, G.; Frei, K.; Perris, R.; Virgintino, D. Tunneling nanotubes evoke pericyte/endothelial communication during normal and tumoral angiogenesis. Fluids Barriers CNS 2018, 15, 28. [Google Scholar] [CrossRef] [Green Version]
- Brown, W.R. A review of string vessels or collapsed, empty basement membrane tubes. J. Alzheimer’s. Dis. 2010, 21, 725–739. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xu, B.; Chen, Q.; Yan, Y.; Du, J.; Du, X. Apoptosis of Endothelial Cells Contributes to Brain Vessel Pruning of Zebrafish During Development. Front. Mol. Neurosci. 2018, 11, 222. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-Q.; Chen, P.; Haimovitz-Friedman, A.; Reilly, R.M.; Wong, C.S. Endothelial Apoptosis Initiates Acute Blood–Brain Barrier Disruption after Ionizing Radiation1. Cancer Res. 2003, 63, 5950–5956. [Google Scholar] [PubMed]
- Basuroy, S.; Leffler, C.W.; Parfenova, H. CORM-A1 prevents blood-brain barrier dysfunction caused by ionotropic glutamate receptor-mediated endothelial oxidative stress and apoptosis. Am. J. Physiol. Cell Physiol. 2013, 304, C1105–C1115. [Google Scholar] [CrossRef]
- Mahajan, S.D.; Tutino, V.M.; Redae, Y.; Meng, H.; Siddiqui, A.; Woodruff, T.M.; Jarvis, J.N.; Hennon, T.; Schwartz, S.; Quigg, R.J.; et al. C5a induces caspase-dependent apoptosis in brain vascular endothelial cells in experimental lupus. Immunology 2016, 148, 407–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Choi, S.-M.; Whitcomb, D.J.; Kim, B.C. Adiponectin controls the apoptosis and the expression of tight junction proteins in brain endothelial cells through AdipoR1 under beta amyloid toxicity. Cell Death Dis. 2017, 8, e3102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Li, X.; Qiao, S.; Yang, D.; Li, Z.; Xu, J.; Li, W.; Su, L.; Liu, W. Occludin degradation makes brain microvascular endothelial cells more vulnerable to reperfusion injury in vitro. J. Neurochem. 2021, 156, 352–366. [Google Scholar] [CrossRef]
- Anasooya, S.C.; Robinson, B.D.; Yeager, A.; Beeram, M.R.; Davis, M.L.; Isbell, C.L.; Huang, J.H.; Tharakan, B. The Tri-phasic Role of Hydrogen Peroxide in Blood-Brain Barrier Endothelial cells. Sci. Rep. 2019, 9, 133. [Google Scholar] [CrossRef] [Green Version]
- Shao, B.; Bayraktutan, U. Hyperglycaemia promotes human brain microvascular endothelial cell apoptosis via induction of protein kinase C-ßI and prooxidant enzyme NADPH oxidase. Redox. Biol. 2014, 2, 694–701. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, T.; Yang, K.; Xu, J.; Ren, L.; Li, W.; Liu, W. Cerebral Microvascular Endothelial Cell Apoptosis after Ischemia: Role of Enolase-Phosphatase 1 Activation and Aci-Reductone Dioxygenase 1 Translocation. Front. Mol. Neurosci. 2016, 9, 79. [Google Scholar] [CrossRef] [Green Version]
- Rakkar, K.; Bayraktutan, K. Increases in intracellular calcium perturb blood–brain barrier via protein kinase C-alpha and apoptosis. Biochim. Et Biophys. Acta (BBA) Mol. Basis Dis. 2016, 1862, 56–71. [Google Scholar] [CrossRef]
- Huang, P.; Rani, M.R.; Ahluwalia, M.S.; Bae, E.; Prayson, R.A.; Weil, R.J.; Nowacki, A.S.; Hedayat, H.; Sloan, A.E.; Lathia, J.D.; et al. Endothelial expression of TNF receptor-1 generates a proapoptotic signal inhibited by integrin α6β1 in glioblastoma. Cancer Res. 2012, 72, 1428–1437. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Ozaki, T.; Takahashi, A.; Miyauchi, M.; Ono, S.; Takada, N. DFF45/ICAD restores cisplatin-induced nuclear fragmentation but not DNA cleavage in DFF45-deficient neuroblastoma cells. Oncogene 2007, 26, 5669–5673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dittmar, M.S.; Petermichl, W.; Lindner, R.; Sinner, B.; Graf, B.M.; Schlachetzki, F.; Gruber, M. In Vitro Induction of Endothelial Apoptosis of the Post-Hypoxic Blood-Brain Barrier by Isoflurane but Not by Sevoflurane and Midazolam. PLoS ONE 2015, 10, e0130408. [Google Scholar] [CrossRef]
- Csiszar, A.; Tarantini, S.; Yabluchanskiy, A.; Balasubramanian, P.; Kiss, T.; Farkas, E.; Baur, J.A.; Ungvari, Z. Role of endothelial NAD+ deficiency in age-related vascular dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H1253–H1266. [Google Scholar] [CrossRef]
- Deng, X.; Liang, X.; Yang, H.; Huang, Z.; Huang, X.; Liang, C.; Kuang, Y.; Qin, Y.; Lin, F.; Luo, Z. Nicotinamide mononucleotide (NMN) protects bEnd.3 cells against H2 O2 -induced damage via NAMPT and the NF-κB p65 signalling pathway. FEBS Open Bio 2021, 11, 866–879. [Google Scholar] [CrossRef]
- Lobov, I.B.; Rao, S.; Carroll, T.J.; Vallance, J.E.; Ito, M.; Ondr, J.K.; Kurup, S.; Glass, D.A.; Patel, M.S.; Shu, W.; et al. WNT7b mediates macrophage-induced programmed cell death in patterning of the vasculature. Nature 2005, 437, 417–421. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.-Y.; Lai, Y.-L.; Liu, K.-H.; Lin, S.; Chen, H.-Y.; Liang, C.-H.; Wu, H.-M.; Hsu, K.-S. TNFα-mediated necroptosis in brain endothelial cells as a potential mechanism of increased seizure susceptibility in mice following systemic inflammation. J. Neuroinflam. 2022, 19, 29. [Google Scholar] [CrossRef]
- Bisht, K.; Okojie, K.A.; Sharma, K.; Lentferink, D.H.; Sun, Y.-Y.; Chen, H.-R.; Uweru, J.O.; Amancherla, S.; Calcuttawala, Z.; Campos-Salazar, A.B.; et al. Capillary-associated microglia regulate vascular structure and function through PANX1-P2RY12 coupling in mice. Nat. Commun. 2021, 12, 5289. [Google Scholar] [CrossRef]
- Gomez-Arboledas, A.; Acharya, M.M.; Tenner, A.J. The Role of Complement in Synaptic Pruning and Neurodegeneration. Immunotargets Ther. 2021, 10, 373–386. [Google Scholar] [CrossRef]
- Gnanaguru, G.; Tabor, S.J.; Yuda, K.; Mukai, R.; Köhl, J.; Connor, K.M. Complement facilitates developmental microglial pruning of astrocyte and vascular networks. bioRxiv 2022. [Google Scholar] [CrossRef]
- Cheng, H.; Di, G.; Gao, C.C.; He, G.; Wang, X.; Han, Y.L.; Sun, L.A.; Zhou, M.L.; Jiang, X. FTY720 Reduces Endothelial Cell Apoptosis and Remodels Neurovascular Unit after Experimental Traumatic Brain Injury. Int. J. Med. Sci. 2021, 18, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Londoño, D.; Carvajal, J.; Strle, K.; Kim, K.S.; Cadavid, D. IL-10 Prevents apoptosis of brain endothelium during bacteremia. J. Immunol. 2011, 186, 7176–7186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammes, H.P.; Lin, J.; Wagner, P.; Feng, Y.; Vom Hagen, F.; Krzizok, T.; Renner, O.; Breier, G.; Brownlee, M.; Deutsch, U. Angiopoietin-2 causes pericyte dropout in the normal retina: Evidence for involvement in diabetic retinopathy. Diabetes 2004, 53, 1104–1110. [Google Scholar] [CrossRef] [Green Version]
- Benn, A.; Alonso, F.; Mangelschots, J.; Génot, E.; Lox, M.; Zwijsen, A. BMP-SMAD1/5 Signaling Regulates Retinal Vascular Development. Biomolecules 2020, 10, 488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favier, J.; Corvol, P. Angiogénèsephysiologique [Physiological angiogenesis]. Therapie 2001, 56, 455–463. [Google Scholar]
- Baffert, F.; Le, T.; Sennino, B.; Thurston, G.; Kuo, C.J.; Hu-Lowe, D.; McDonald, D.M. Cellular changes in normal blood capillaries undergoing regression after inhibition of VEGF signaling. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H547–H559. [Google Scholar] [CrossRef] [Green Version]
- Quaegebeur, A.; Lange, C.; Carmeliet, P. The neurovascular link in health and disease: Molecular mechanisms and therapeutic implications. Neuron 2011, 71, 406–424. [Google Scholar] [CrossRef] [Green Version]
- Chow, B.W.; Nunez, V.; Kaplan, L.; Granger, A.J.; Bistrong, K.; Zucker, H.L.; Kumar, P.; Sabatini, B.L.; Gu, C. Caveolae in CNS arterioles mediate neurovascular coupling. Nature 2020, 579, 106–110. [Google Scholar] [CrossRef]
- Coelho-Santos, V.; Shih, A.Y. Postnatal development of cerebrovascular structure and the neurogliovascular unit. Wiley Interdiscip. Rev. Dev. Biol. 2020, 9, e363. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, L.; Chow, B.W.; Gu, C. Neuronal regulation of the blood-brain barrier and neurovascular coupling. Nat. Rev. Neurosci. 2020, 21, 416–432. [Google Scholar] [CrossRef]
- Reeson, P.; Choi, K.; Brown, C.E. VEGF signaling regulates the fate of obstructed capillaries in mouse cortex. Elife 2018, 7, e33670. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.; Chang-Ling, T. Roles of endothelial cell migration and apoptosis in vascular remodeling during development of the central nervous system. Microcirculation 2000, 7, 317–333. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Agalliu, D.; Zhou, L.; Kuhnert, F.; Kuo, C.J.; Barres, B.A. Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 641–646. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Li, J.-L.; Chen, X.; Ci, B.; Chen, F.; Lu, N.; Shen, B.; Zheng, L.; Jia, J.; Yi, Y.; et al. Reduction of neuronal activity mediated by blood-vessel regression in the brain. bioRxiv 2020, 15, 262782. [Google Scholar] [CrossRef]
- Ma, S.; Kwon, H.J.; Huang, Z. A functional requirement for astroglia in promoting blood vessel development in the early postnatal brain. PLoS ONE 2012, 7, e48001. [Google Scholar] [CrossRef] [Green Version]
- Benderro, G.F.; LaManna, J.C. HIF-1α/COX-2 expression and mouse brain capillary remodeling during prolonged moderate hypoxia and subsequent re-oxygenation. Brain Res. 2014, 1569, 41–47. [Google Scholar] [CrossRef] [Green Version]
- Lynch, C.N.; Wang, Y.C.; Lund, J.K.; Chen, Y.-W.; Leal, J.A.; Wiley, S.R. TWEAK Induces Angiogenesis and Proliferation of Endothelial Cells. J. Biol. Chem. 1999, 274, 8455–8459. [Google Scholar] [CrossRef] [Green Version]
- Serini, G.; Tamagnone, L. Bad vessels beware! Semaphorins will sort you out! EMBO Mol. Med. 2015, 7, 1251–1253. [Google Scholar] [CrossRef]
- Zhang, C.L.; Hong, C.D.; Wang, H.L.; Chen, A.Q.; Zhou, Y.F.; Wan, Y.; Li, Y.N.; Hu, B. The role of semaphorins in small vessels of the eye and brain. Pharmacol. Res. 2020, 160, 105044. [Google Scholar] [CrossRef]
- Pichiule, P.; LaManna, J.C. Angiopoietin-2 and rat brain capillary remodeling during adaptation and deadaptation to prolonged mild hypoxia. J. Appl. Physiol. 2002, 93, 1131–1139. [Google Scholar] [CrossRef] [Green Version]
- Dore-Duffy, P.; LaManna, J.C. Physiologic angiodynamics in the brain. Antioxid. Redox Signal 2007, 9, 1363–1371. [Google Scholar] [CrossRef] [PubMed]
- Morland, C.; Andersson, K.A.; Haugen, Ø.P.; Hadzic, A.; Kleppa, L.; Gille, A.; Rinholm, J.E.; Palibrk, V.; Diget, E.H.; Kennedy, L.H.; et al. Exercise induces cerebral VEGF and angiogenesis via the lactate receptor HCAR1. Nat. Commun. 2017, 8, 15557. [Google Scholar] [CrossRef] [PubMed]
- Kerr, A.L.; Steuer, E.L.; Pochtarev, V.; Swain, R.A. Angiogenesis but not neurogenesis is critical for normal learning and memory acquisition. Neuroscience 2010, 171, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Salmina, A.B.; Gorina, Y.V.; Komleva, Y.K.; Panina, Y.A.; Malinovskaya, N.A.; Lopatina, O.L. Early Life Stress and Metabolic Plasticity of Brain Cells: Impact on Neurogenesis and Angiogenesis. Biomedicines 2021, 9, 1092. [Google Scholar] [CrossRef]
- Pulga, A. Dynamics of the Cerebral Microvasculature during the Course of Memory Consolidation in the Rat: Physiological and Altered Conditions Induced by Hypertension and Hypergravity. Ph.D. Thesis, Neurons and Cognition Université de Bordeaux, Bordeaux, France, December 2016. [Google Scholar]
- LaManna, J.C. Cerebral Angioplasticity: The Anatomical Contribution to Ensuring Appropriate Oxygen Transport to Brain. Adv. Exp. Med. Biol. 2018, 1072, 3–6. [Google Scholar] [CrossRef]
- Kleszka, K.; Leu, T.; Quinting, T.; Jastrow, H.; Pechlivanis, S.; Fandrey, J.; Schreiber, T. Hypoxia-inducible factor-2α is crucial for proper brain development. Sci. Rep. 2020, 10, 19146. [Google Scholar] [CrossRef]
- Skuli, N.; Liu, L.; Runge, A.; Wang, T.; Yuan, L.; Patel, S.; Iruela-Arispe, L.; Simon, M.C.; Keith, B. Endothelial deletion of hypoxia-inducible factor-2alpha (HIF-2alpha) alters vascular function and tumor angiogenesis. Blood 2009, 114, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Pereira, A.C.; Gray, J.D.; Kogan, J.F.; Davidson, R.L.; Rubin, T.G.; Okamoto, M.; Morrison, J.H.; McEwen, B.S. Age and Alzheimer’s disease gene expression profiles reversed by the glutamate modulator riluzole. Mol. Psychiatry 2017, 22, 296–305. [Google Scholar] [CrossRef]
- Yang, Y.; Torbey, M.T. Angiogenesis and Blood-Brain Barrier Permeability in Vascular Remodeling after Stroke. Curr. Neuropharmacol. 2020, 18, 1250–1265. [Google Scholar] [CrossRef]
- Carmeliet, P. Fibroblast growth factor-1 stimulates branching and survival of myocardial arteries: A goal for therapeutic angiogenesis? Circ. Res. 2000, 87, 176–178. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.W.; Friedman, B.; Cheng, Q.; Lyden, P.D. Stroke-evoked angiogenesis results in a transient population of microvessels. J. Cereb. Blood Flow Metab. 2007, 27, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.K.; Milner, R. The impact of chronic mild hypoxia on cerebrovascular remodelling; uncoupling of angiogenesis and vascular breakdown. Fluids Barriers CNS 2021, 18, 50. [Google Scholar] [CrossRef] [PubMed]
- Farkas, E.; Luiten, P.G. Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog. Neurobiol. 2001, 64, 575–611. [Google Scholar] [CrossRef] [Green Version]
- Sagare, A.P.; Bell, R.D.; Zlokovic, B.V. Neurovascular dysfunction and faulty amyloid β-peptide clearance in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a011452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iturria-Medina, Y.I.; Sotero, R.C.; Toussaint, P.J.; Mateos-Pérez, J.M.; Evans, A.C. Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat. Commun. 2016, 7, 11934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Mesquita, S.; Louveau, A.; Vaccari, A.; Smirnov, I.; Cornelison, R.C.; Kingsmore, K.M.; Contarino, C.; Onengut-Gumuscu, S.; Farber, E.; Raper, D.; et al. Functional aspects of meningeal lymphatics in ageing and Alzheimer’s disease. Nature 2018, 560, 185–191. [Google Scholar] [CrossRef]
- Solis, E., Jr.; Hascup, K.N.; Hascup, E.R. Alzheimer’s Disease: The Link Between Amyloid-β and Neurovascular Dysfunction. J. Alzheimers. Dis. 2020, 76, 1179–1198. [Google Scholar] [CrossRef]
- Salmina, A.; Kharitonova, E.; Gorina, Y.; Teplyashina, E.; Malinovskaya, N.; Khilazheva, E.; Mosyagina, A.; Morgun, A.; Shuvaev, A.; Salmin, V.; et al. Blood–Brain Barrier and Neurovascular Unit In Vitro Models for Studying Mitochondria-Driven Molecular Mechanisms of Neurodegeneration. Int. J. Mol. Sci. 2021, 22, 4661. [Google Scholar] [CrossRef]
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [Green Version]
- Bell, R.D.; Zlokovic, B.V. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Qin, W.; Jia, X.; Wang, F.; Zuo, X.; Wu, L.; Zhou, A.; Li, D.; Min, B.; Wei, C.; Tang, Y.; et al. Elevated plasma angiogenesis factors in Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 45, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Bell, M.A.; Ball, M.J. Morphometric comparison of hippocampal microvasculature in ageing and demented people: Diameters and densities. Acta Neuropathol. 1981, 53, 299–318. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.R.; Thore, C.R. Review: Cerebral microvascular pathology in ageing and neurodegeneration. Neuropathol. Appl. Neurobiol. 2011, 37, 56–74. [Google Scholar] [CrossRef] [Green Version]
- Riddle, D.R.; Sonntag, W.E.; Lichtenwalner, R.J. Microvascular plasticity in aging. Ageing Res. Rev. 2002, 2, 149–168. [Google Scholar] [CrossRef]
- Crystal, H.A.; Schneider, J.A.; Bennett, D.A.; Leurgans, S.; Levine, S.R. Associations of cerebrovascular and Alzheimer’s disease pathology with brain atrophy. Curr. Alzheimer. Res. 2014, 11, 309–316. [Google Scholar] [CrossRef]
- Mimenza-Alvarado, A.; Aguilar-Navarro, S.G.; Yeverino-Castro, S.; Mendoza-Franco, C.; Ávila-Funes, J.A.; Román, G.C. Neuroimaging Characteristics of Small-Vessel Disease in Older Adults with Normal Cognition, Mild Cognitive Impairment, and Alzheimer Disease. Dement. Geriatr. Cogn. Dis. Extra 2018, 8, 199–206. [Google Scholar] [CrossRef]
- Finsterwalder, S.; Vlegels, N.; Gesierich, B.; Caballero, M.A.; Weaver, N.A.; Franzmeier, N.; Georgakis, M.K.; Konieczny, M.J.; Koek, H.L.; Karch, C.M.; et al. Small vessel disease more than Alzheimer’s disease determines diffusion MRI alterations in memory clinic patients. Alzheimer’s Dement. 2020, 16, 1504–1514. [Google Scholar] [CrossRef]
- Kester, M.I.; Goos, J.D.; Teunissen, C.E.; Benedictus, M.R.; Bouwman, F.H.; Wattjes, M.P.; Barkhof, F.; Scheltens, P.; van der Flier, W.M. Associations between cerebral small-vessel disease and Alzheimer disease pathology as measured by cerebrospinal fluid biomarkers. JAMA Neurol. 2014, 71, 855–862. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Braidy, N.; Poljak, A.; Chan, D.K.Y.; Sachdev, P. Cerebral small vessel disease and the risk of Alzheimer’s disease: A systematic review. Ageing Res. Rev. 2018, 47, 41–48. [Google Scholar] [CrossRef]
- Nadkarni, N.K.; Tudorascu, D.; Campbell, E.; Snitz, B.E.; Cohen, A.D.; Halligan, E.; Mathis, C.A.; Aizenstein, H.J.; Klunk, W.E. Association Between Amyloid-β, Small-vessel Disease, and Neurodegeneration Biomarker Positivity, and Progression to Mild Cognitive Impairment in Cognitively Normal Individuals. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 1753–1760. [Google Scholar] [CrossRef]
- Stefaniak, J.D.; Su, L.; Mak, E.; Sheikh-Bahaei, N.; Wells, K.; Ritchie, K.; Waldman, A.; Ritchie, C.W.; O’Brien, J.T. Cerebral small vessel disease in middle age and genetic predisposition to late-onset Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.M.; Mahoney, E.; Dumitrescu, L.; De Jager, P.L.; Koran, M.E.I.; Petyuk, V.A.; Robinson, R.A.; Ruderfer, D.M.; Cox, N.J.; Schneider, J.A.; et al. APOE ε4-specific associations of VEGF gene family expression with cognitive aging and Alzheimer’s disease. Neurobiol. Aging 2020, 87, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Guo, H.; Chow, N.; Sallstrom, J.; Bell, R.D.; Deane, R.; Brooks, A.; Kanagala, S.; Rubio, A.; Sagare, A.; et al. Role of MEOX2 homeobox gene in neurovascular dysfunction in Alzheimer disease. Nat. Med. 2005, 11, 959–965. [Google Scholar] [CrossRef]
- Deane, R.; Wu, Z.; Zlokovic, B.V. RAGE (yin) versus LRP (yang) balance regulates alzheimer amyloid beta-peptide clearance through transport across the blood-brain barrier. Stroke 2004, 35 (Suppl. 1), 2628–2631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paris, D.; Patel, N.; DelleDonne, A.; Quadros, A.; Smeed, R.; Mullan, M. Impaired angiogenesis in a transgenic mouse model of cerebral amyloidosis. Neurosci. Lett. 2004, 366, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Paris, D.; Townsend, K.; Quadros, A.; Humphrey, J.; Sun, J.; Brem, S.; Wotoczek-Obadia, M.; DelleDonne, A.; Patel, N.; Obregon, D.F.; et al. Inhibition of angiogenesis by Abeta peptides. Angiogenesis 2004, 7, 75–85. [Google Scholar] [CrossRef]
- Lemańska-Perek, A.; Leszek, J.; Krzyzanowska-Gołab, D.; Radzik, J.; Katnik-Prastowska, I. Molecular status of plasma fibronectin as an additional biomarker for assessment of Alzheimer’s dementia risk. Dement. Geriatr. Cogn. Disord. 2009, 28, 338–342. [Google Scholar] [CrossRef]
- Faraco, G.; Park, L.; Zhou, P.; Luo, W.; Paul, S.; Anrather, J.; Iadecola, C. Hypertension enhances Aβ-induced neurovascular dysfunction, promotes β-secretase activity, and leads to amyloidogenic processing of APP. J. Cereb. Blood Flow Metab. 2015, 36, 241–252. [Google Scholar] [CrossRef] [Green Version]
- Callahan, C.M.; Apostolova, L.G.; Gao, S.; Risacher, S.L.; Case, J.; Saykin, A.J.; Lane, K.A.; Swinford, C.G.; Yoder, M.C. Novel Markers of Angiogenesis in the Setting of Cognitive Impairment and Dementia. J. Alzheimer’s Dis. 2020, 75, 959–969. [Google Scholar] [CrossRef]
- Ali, M.; Bracko, O. VEGF Paradoxically Reduces Cerebral Blood Flow in Alzheimer’s Disease Mice. Neurosci. Insights 2022, 17, 26331055221109254. [Google Scholar] [CrossRef]
- Durrant, C.S.; Ruscher, K.; Sheppard, O.; Coleman, M.P.; Özen, I. Beta secretase 1-dependent amyloid precursor protein processing promotes excessive vascular sprouting through NOTCH3 signalling. Cell Death Dis. 2020, 11, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, C.S.; Choi, K.B.; Munro, L.; Wang, H.Y.; Pfeifer, C.G.; Jefferies, W.A. Reversing pathology in a preclinical model of Alzheimer’s disease by hacking cerebrovascular neoangiogenesis with advanced cancer therapeutics. EBioMedicine 2021, 71, 103503. [Google Scholar] [CrossRef] [PubMed]
- Poh, L.; Sim, W.L.; Jo, D.-G.; Dinh, Q.N.; Drummond, G.R.; Sobey, C.G.; Chen, C.L.-H.; Lai, M.K.P.; Fann, D.Y.; Arumugam, T.V. The role of inflammasomes in vascular cognitive impairment. Mol. Neurodegener. 2022, 17, 4. [Google Scholar] [CrossRef] [PubMed]
- Binet, F.; Cagnone, G.; Crespo-Garcia, S.; Hata, M.; Neault, M.; Dejda, A.; Wilson, A.M.; Buscarlet, M.; Mawambo, G.T.; Howard, J.P.; et al. Neutrophil extracellular traps target senescent vasculature for tissue remodeling in retinopathy. Science 2020, 369, eaay5356. [Google Scholar] [CrossRef]
- Klohs, J. An Integrated View on Vascular Dysfunction in Alzheimer’s Disease. Neurodegener. Dis. 2019, 19, 109–127. [Google Scholar] [CrossRef]
- Menet, R.; Lecordier, S.; ElAli, A. Wnt Pathway: An Emerging Player in Vascular and Traumatic Mediated Brain Injuries. Front. Physiol. 2020, 11, 565667. [Google Scholar] [CrossRef]
- Komleva, Y.K.; Potapenko, I.V.; Lopatina, O.L.; Gorina, Y.V.; Chernykh, A.; Khilazheva, E.D.; Salmina, A.B.; Shuvaev, A.N. NLRP3 Inflammasome Blocking as a Potential Treatment of Central Insulin Resistance in Early-Stage Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 11588. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tregub, P.P.; Averchuk, A.S.; Baranich, T.I.; Ryazanova, M.V.; Salmina, A.B. Physiological and Pathological Remodeling of Cerebral Microvessels. Int. J. Mol. Sci. 2022, 23, 12683. https://doi.org/10.3390/ijms232012683
Tregub PP, Averchuk AS, Baranich TI, Ryazanova MV, Salmina AB. Physiological and Pathological Remodeling of Cerebral Microvessels. International Journal of Molecular Sciences. 2022; 23(20):12683. https://doi.org/10.3390/ijms232012683
Chicago/Turabian StyleTregub, Pavel P., Anton S. Averchuk, Tatyana I. Baranich, Maria V. Ryazanova, and Alla B. Salmina. 2022. "Physiological and Pathological Remodeling of Cerebral Microvessels" International Journal of Molecular Sciences 23, no. 20: 12683. https://doi.org/10.3390/ijms232012683