
Comprehensive Comparative Analysis and Development of Molecular Markers for Dianthus Species Based on Complete Chloroplast Genome Sequences

 ,
,
Abstract
:1. Introduction
2. Results
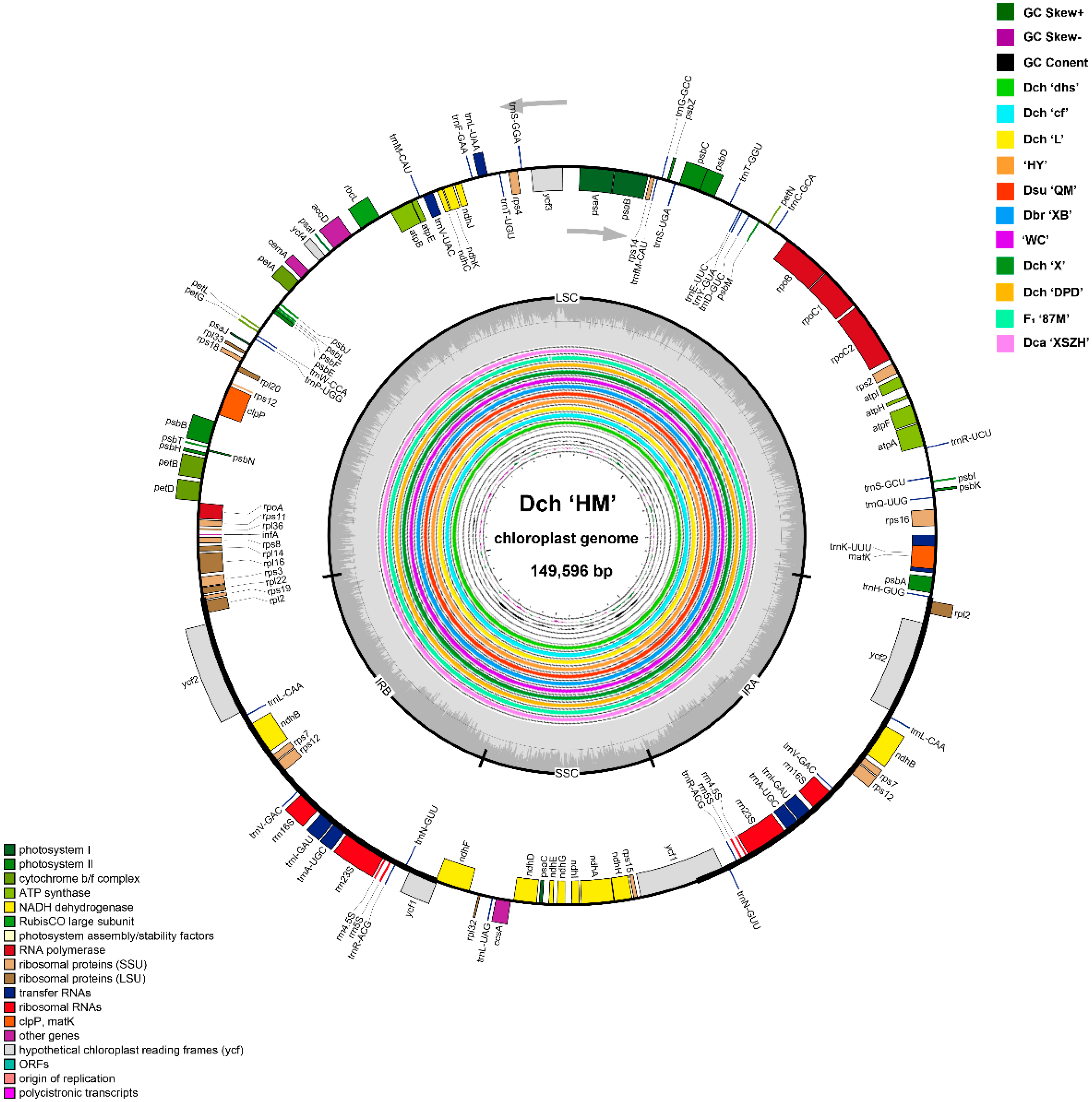
2.1. Features and Characteristics of the Dianthus Complete Chloroplast Genomes
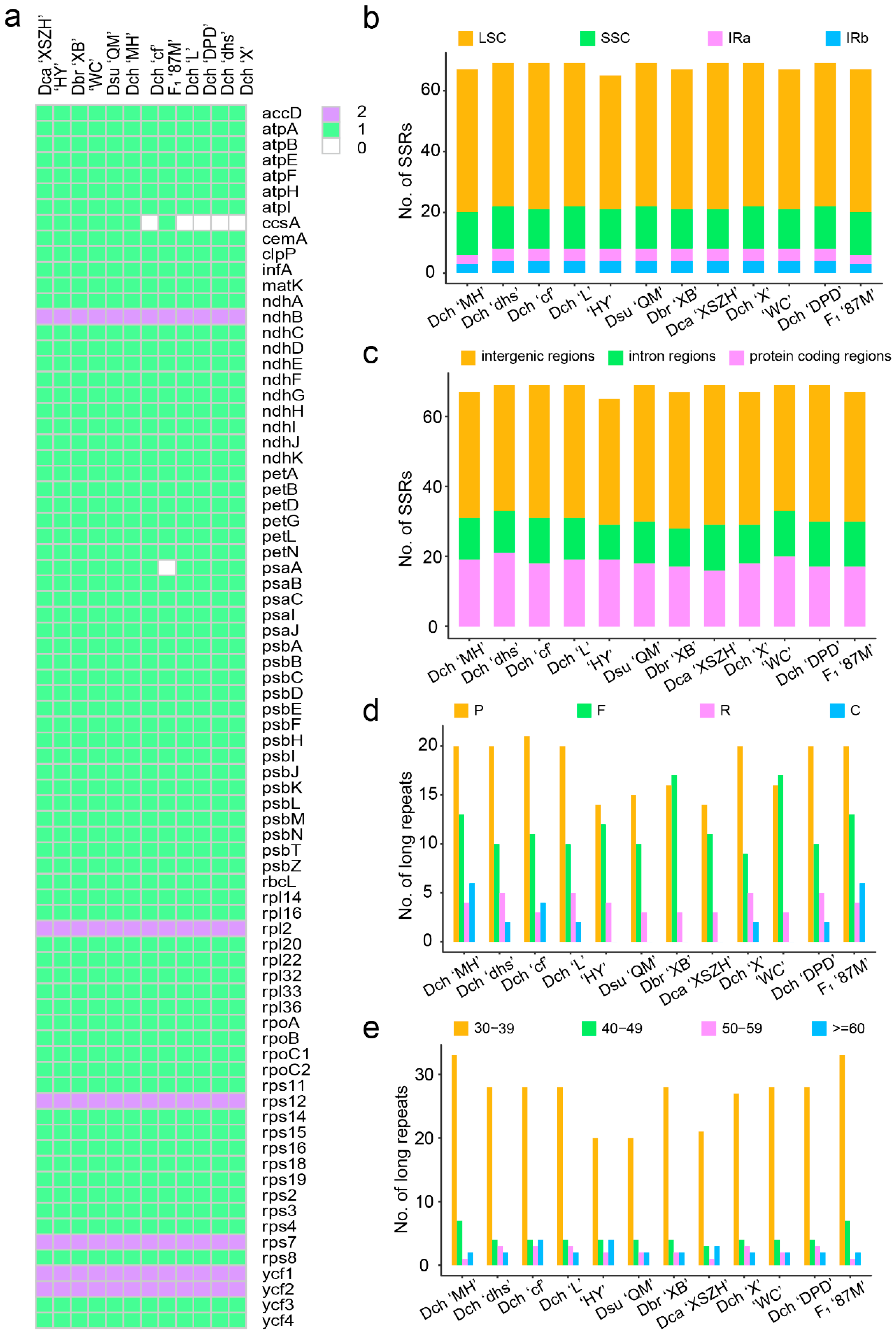
2.2. Sequence Repetition in the Dianthus Complete Chloroplast Genomes
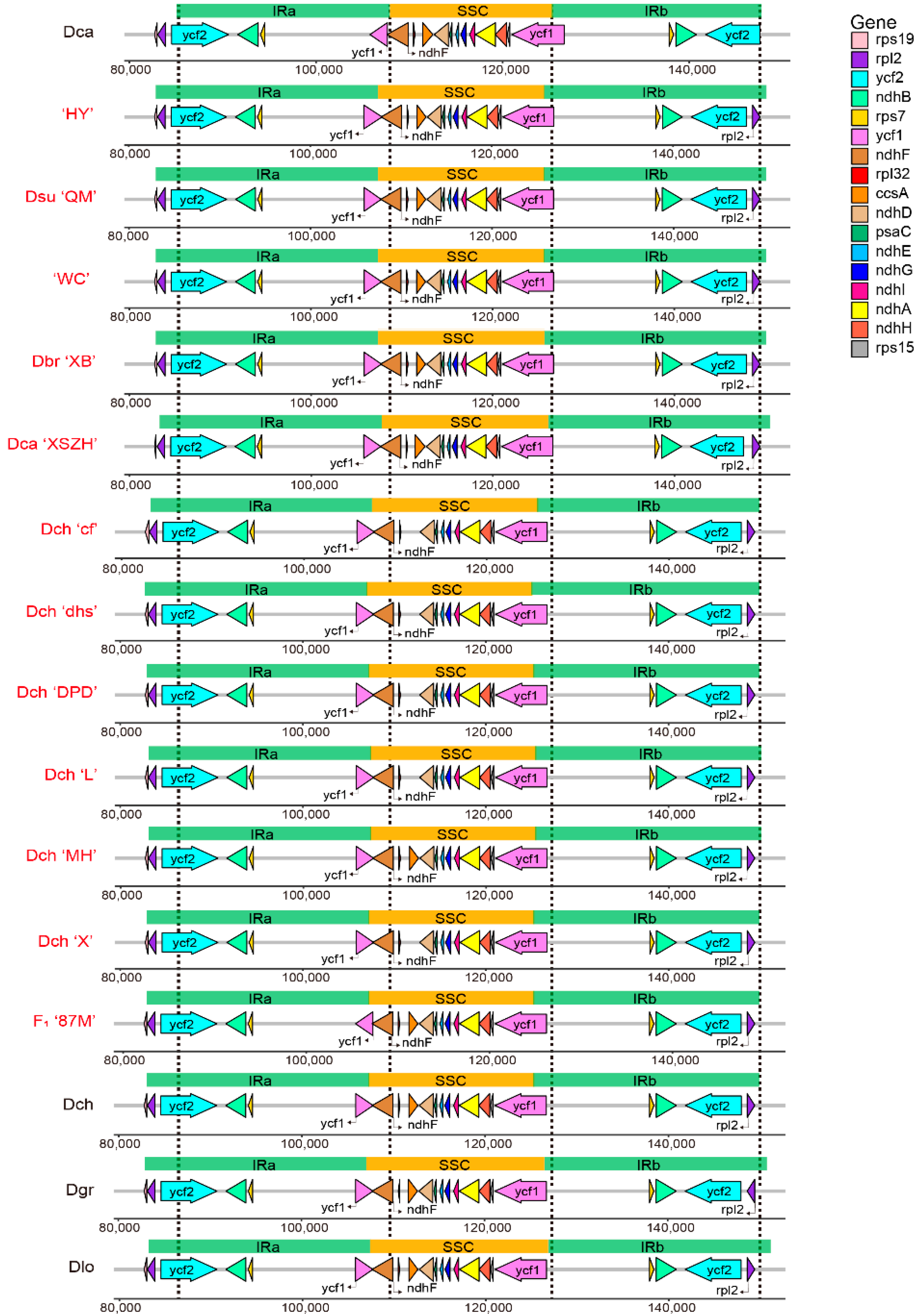
2.3. Contraction and Expansion of IR
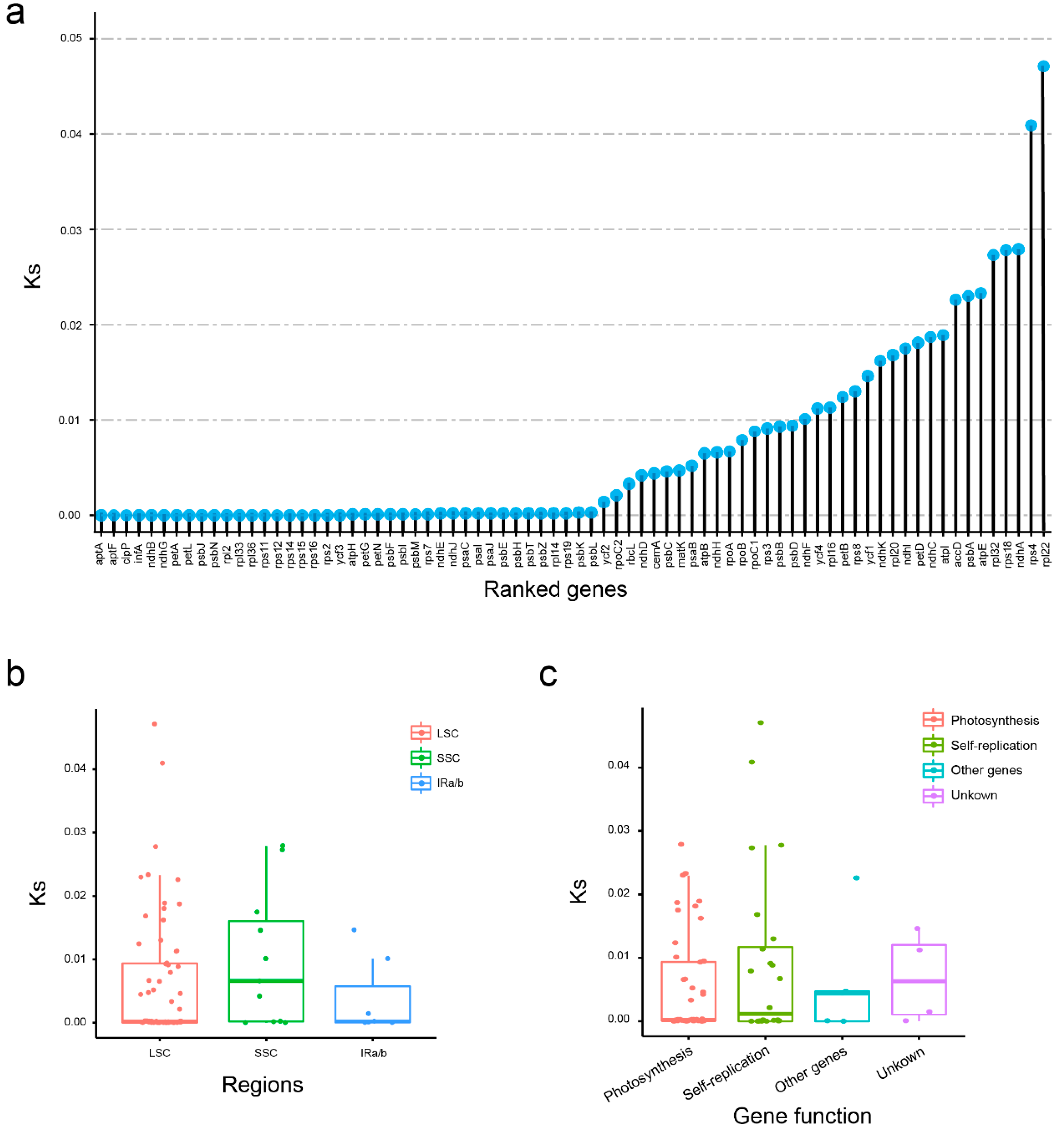
2.4. Evolutionary Rates Analyses
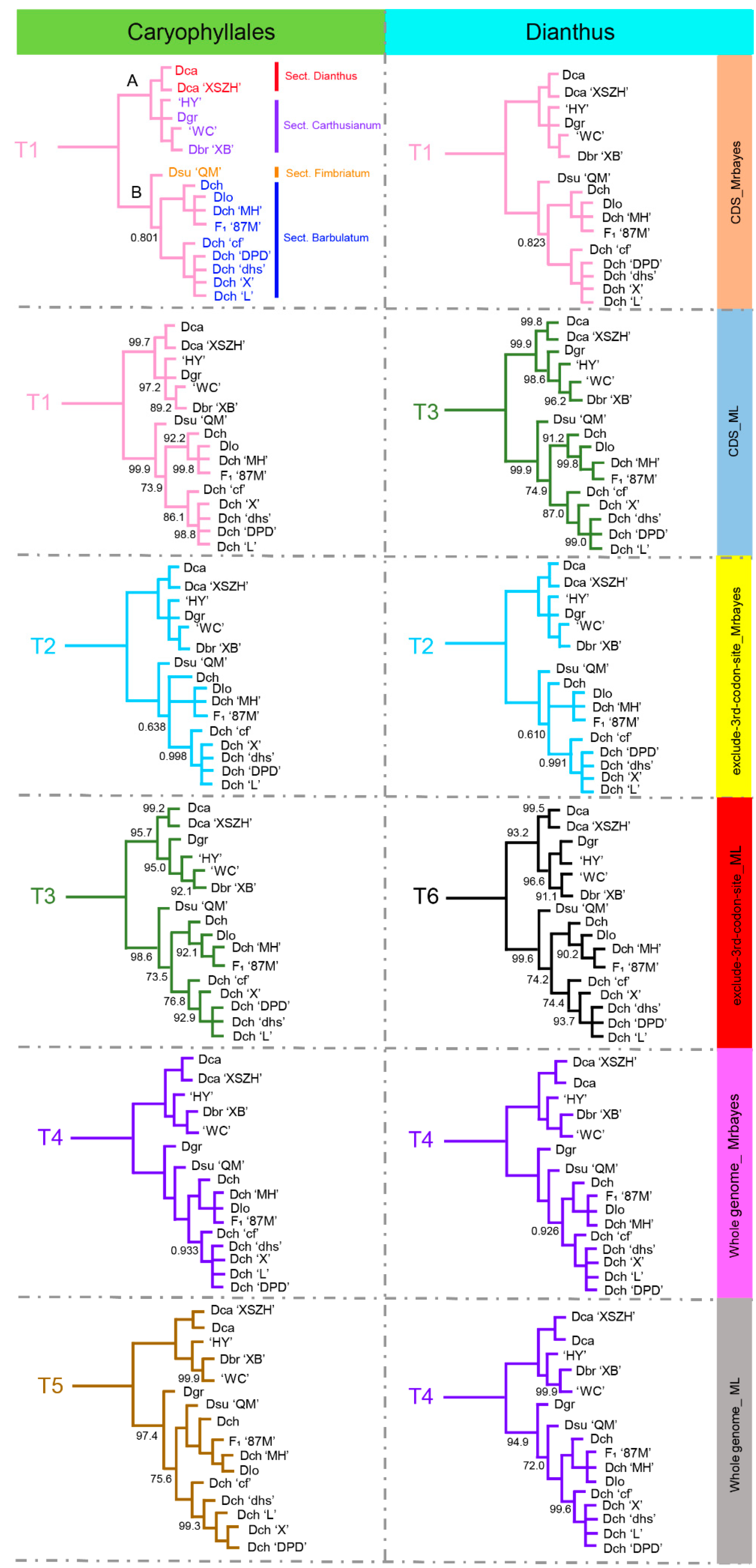
2.5. Phylogenetic Analyses

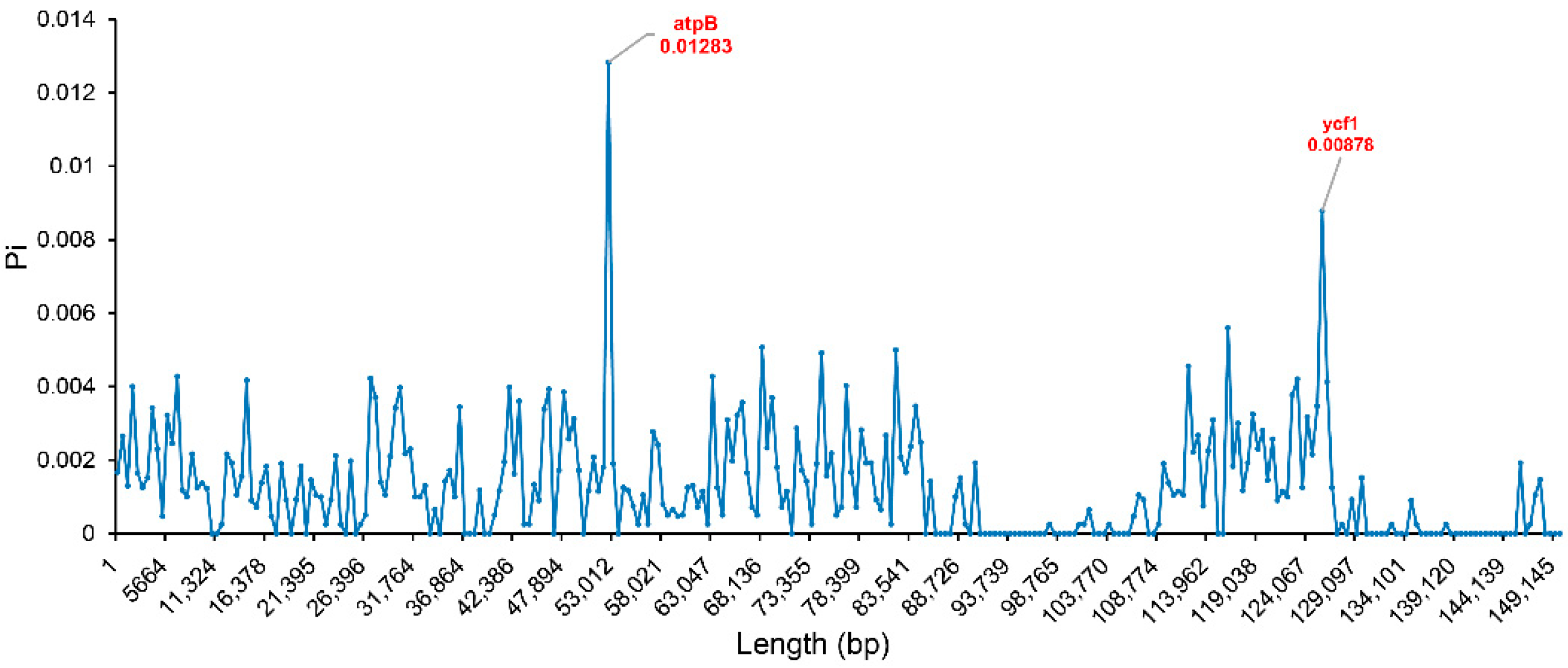
2.6. Hypervariable Regions
2.7. Molecular Marker Development Based on Dianthus Plastomes
3. Discussion
4. Materials and Methods
4.1. Plant Materials, DNA Extraction, and Sequencing
4.2. De Novo Genome Assembly and Annotation
4.3. Repeated Sequence Analysis and Comparison of Genome Structures
4.4. Evolutionary Rates Analysis
4.5. Phylogenomic Analysis
4.6. Identification of the Hypervariable Regions
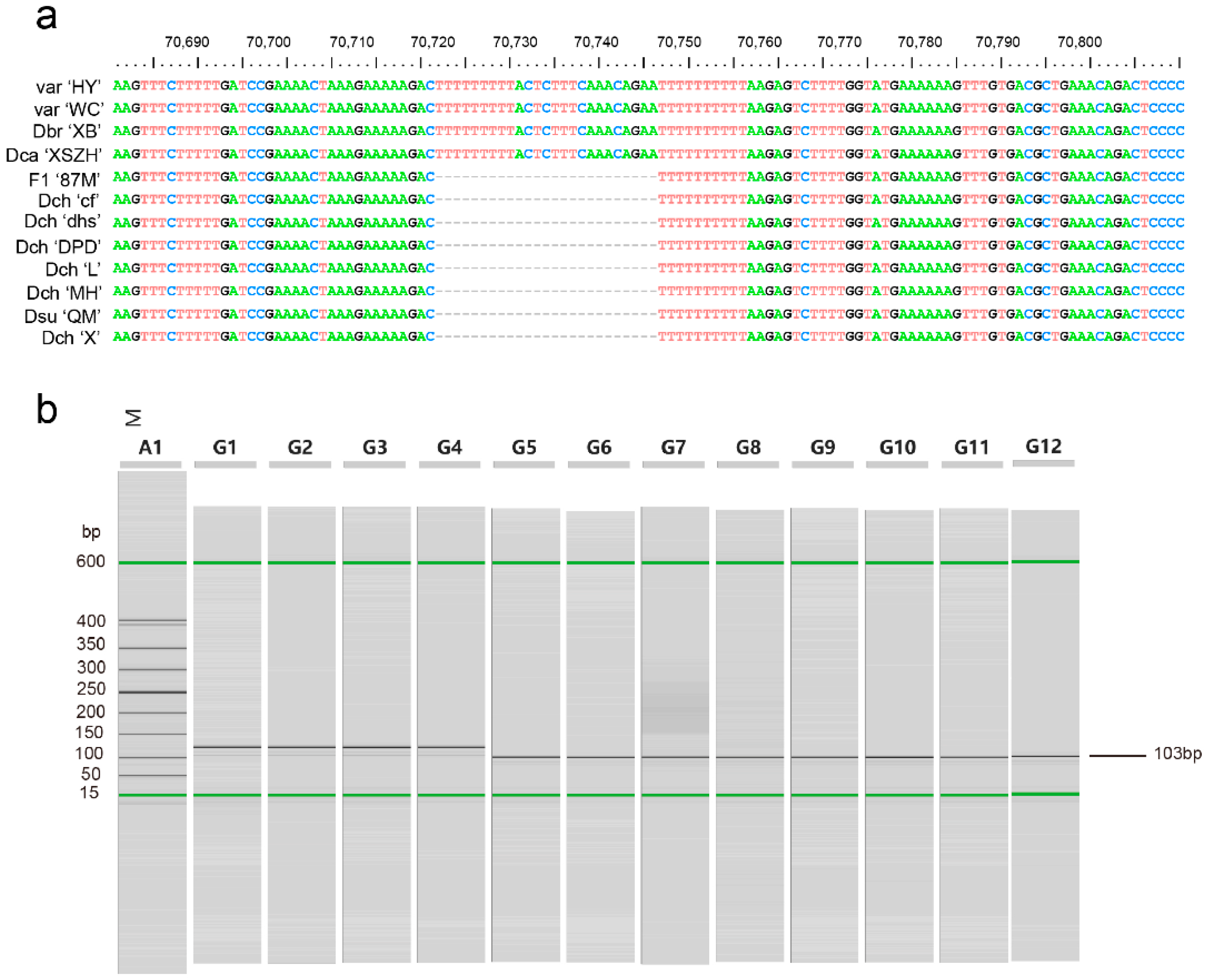
4.7. Identification and Validation of Molecular Markers for Species Discrimination
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, Z.; Wang, X.; Wang, D.; Xue, P.; Miao, N. The Complete Chloroplast Genome Sequence of Medicinal Plant: Dianthus chinensis (Caryophyllaceae). Mitochondrial DNA Part B 2021, 6, 327–328. [Google Scholar] [CrossRef] [PubMed]
- Casanova, E.; Valdés, A.E.; Zuker, A.; Fernández, B.; Vainstein, A.; Trillas, M.I.; Moysset, L. RolC-Transgenic Carnation Plants: Adventitious Organogenesis and Levels of Endogenous Auxin and Cytokinins. Plant Sci. 2004, 167, 551–560. [Google Scholar] [CrossRef]
- Flora of China Editorial Committee. Flora of China; Missouri Botanical Garden and Harvard University Herbaria: St. Louis, MO, USA, 2018. [Google Scholar]
- Yagi, M. Recent Progress in Genomic Analysis of Ornamental Plants, with a Focus on Carnation. Hortic. J. 2015, 84, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.; Park, J.; Kim, H.; Jin, H.-G.; Kim, H.; Ahn, Y.; Kim, Y.; Lee, H.; Lee, Y.; Kang, D. Dianthus Superbus Improves Glomerular Fibrosis and Renal Dysfunction in Diabetic Nephropathy Model. Nutrients 2019, 11, 553. [Google Scholar] [CrossRef] [Green Version]
- Nile, S.H.; Kim, D.H.; Nile, A.; Park, G.S.; Gansukh, E.; Kai, G. Probing the Effect of Quercetin 3-Glucoside from Dianthus Superbus L against Influenza Virus Infection- In Vitro and in Silico Biochemical and Toxicological Screening. Food Chem. Toxicol. 2020, 135, 110985. [Google Scholar] [CrossRef]
- Nimura, M. Breeding of Interspecific Hybridization Among Carnation (Dianthus caryophyllus L.), Dianthus japonicus Thunb., and Dianthus × Isensis Hirahata et Kitam. In The Carnation Genome; Onozaki, T., Yagi, M., Eds.; Compendium of Plant Genomes; Springer: Singapore, 2020; pp. 159–176. ISBN 9789811582615. [Google Scholar]
- Gao, C.; Wu, C.; Zhang, Q.; Zhao, X.; Wu, M.; Chen, R.; Zhao, Y.; Li, Z. Characterization of Chloroplast Genomes From Two Salvia Medicinal Plants and Gene Transfer Among Their Mitochondrial and Chloroplast Genomes. Front. Genet. 2020, 11, 574962. [Google Scholar] [CrossRef] [PubMed]
- Santos, V.; Almeida, C. The Complete Chloroplast Genome Sequences of Three Spondias Species Reveal Close Relationship among the Species. Genet. Mol. Biol. 2019, 42, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Pang, X.; Xu, Y.; Jiang, W.; Liao, B.; Yu, J.; Xu, J.; Song, J.; Chen, S. Plastid Genome Data Provide New Insights into the Phylogeny and Evolution of the Genus Epimedium. J. Adv. Res. 2022, 36, 175–185. [Google Scholar] [CrossRef]
- Niu, E.; Jiang, C.; Wang, W.; Zhang, Y.; Zhu, S. Chloroplast Genome Variation and Evolutionary Analysis of Olea europaea L. Genes 2020, 11, 879. [Google Scholar] [CrossRef]
- Henriquez, C.L.; Abdullah; Ahmed, I.; Carlsen, M.M.; Zuluaga, A.; Croat, T.B.; McKain, M.R. Evolutionary Dynamics of Chloroplast Genomes in Subfamily Aroideae (Araceae). Genomics 2020, 112, 2349–2360. [Google Scholar] [CrossRef]
- Park, I.; Song, J.; Yang, S.; Choi, G.; Moon, B. A Comprehensive Study of the Genus Sanguisorba (Rosaceae) Based on the Floral Micromorphology, Palynology, and Plastome Analysis. Genes 2021, 12, 1764. [Google Scholar] [CrossRef]
- Chen, S.; Xu, Z.; Zhao, Y.; Zhong, X.; Li, C.; Yang, G. Structural Characteristic and Phylogenetic Analysis of the Complete Chloroplast Genome of Dianthus caryophyllus. Mitochondrial DNA Part B 2018, 3, 1131–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stothard, P.; Wishart, D.S. Circular Genome Visualization and Exploration Using CGView. Bioinformatics 2005, 21, 537–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, A.; Guo, W.; Gupta, S.; Fan, W.; Mower, J.P. Evolutionary Dynamics of the Plastid Inverted Repeat: The Effects of Expansion, Contraction, and Loss on Substitution Rates. New Phytol. 2016, 209, 1747–1756. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Guisinger, M.; Kim, H.-G.; Ruck, E.; Blazier, J.C.; McMurtry, V.; Kuehl, J.V.; Boore, J.; Jansen, R.K. Extensive Reorganization of the Plastid Genome of Trifolium subterraneum (Fabaceae) Is Associated with Numerous Repeated Sequences and Novel DNA Insertions. J. Mol. Evol. 2008, 67, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Li, D.-M.; Li, J.; Wang, D.-R.; Xu, Y.-C.; Zhu, G.-F. Molecular Evolution of Chloroplast Genomes in Subfamily Zingiberoideae (Zingiberaceae). BMC Plant Biol. 2021, 21, 558. [Google Scholar] [CrossRef]
- Wu, H.-Y.; Wong, K.-H.; Kong, B.L.-H.; Siu, T.-Y.; But, G.W.-C.; Tsang, S.S.-K.; Lau, D.T.-W.; Shaw, P.-C. Comparative Analysis of Chloroplast Genomes of Dalbergia Species for Identification and Phylogenetic Analysis. Plants 2022, 11, 1109. [Google Scholar] [CrossRef]
- Shi, H.; Yang, M.; Mo, C.; Xie, W.; Liu, C.; Wu, B.; Ma, X. Complete Chloroplast Genomes of Two Siraitia Merrill Species: Comparative Analysis, Positive Selection and Novel Molecular Marker Development. PLoS ONE 2019, 14, e0226865. [Google Scholar] [CrossRef]
- de Souza, U.J.B.; Nunes, R.; Targueta, C.P.; Diniz-Filho, J.A.F.; Telles, M.P.D.C. The Complete Chloroplast Genome of Stryphnodendron adstringens (Leguminosae—Caesalpinioideae): Comparative Analysis with Related Mimosoid Species. Sci. Rep. 2019, 9, 14206. [Google Scholar] [CrossRef] [Green Version]
- Abdullah; Mehmood, F.; Shahzadi, I.; Waseem, S.; Mirza, B.; Ahmed, I.; Waheed, M.T. Chloroplast Genome of Hibiscus Rosa-Sinensis (Malvaceae): Comparative Analyses and Identification of Mutational Hotspots. Genomics 2020, 112, 581–591. [Google Scholar] [CrossRef]
- Ndunge, J.; Dong, X.; Yang, J.-X.; Mbandi, M.; Wanga, V.; Oulo, M.; Saina, J.; Musili, P.; Hu, G.-W. Complete Chloroplast Genomes of Chlorophytum comosum and Chlorophytum gallabatense: Genome Structures, Comparative and Phylogenetic Analysis. Plants 2020, 9, 296. [Google Scholar] [CrossRef] [Green Version]
- Zheng, G.; Wei, L.; Li, M.; Wu, Z.; Cuihua, G.; Chen, K. Comparative Analyses of Chloroplast Genomes from 13 Lagerstroemia (Lythraceae) Species: Identification of Highly Divergent Regions and Inference of Phylogenetic Relationships. Plant Mol. Biol. 2020, 102, 659–676. [Google Scholar] [CrossRef]
- Weng, M.-L.; Blazier, J.C.; Govindu, M.; Jansen, R.K. Reconstruction of the Ancestral Plastid Genome in Geraniaceae Reveals a Correlation between Genome Rearrangements, Repeats, and Nucleotide Substitution Rates. Mol. Biol. Evol. 2013, 31, 645–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wambugu, P.W.; Brozynska, M.; Furtado, A.; Waters, D.L.; Henry, R.J. Relationships of Wild and Domesticated Rices (Oryza AA Genome Species) Based upon Whole Chloroplast Genome Sequences. Sci. Rep. 2015, 5, 13957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, L.; Xie, D.; Xiao, Q.; Peng, C.; Yu, Y.; He, X. Sequencing and Analyses on Chloroplast Genomes of Tetrataenium candicans and Two Allies Give New Insights on Structural Variants, DNA Barcoding and Phylogeny in Apiaceae Subfamily Apioideae. PeerJ 2019, 7, e8063. [Google Scholar] [CrossRef] [Green Version]
- Cauz-Santos, L.A.; da Costa, Z.P.; Callot, C.; Cauet, S.; Zucchi, M.I.; Bergès, H.; van den Berg, C.; Vieira, M.L.C. A Repertory of Rearrangements and the Loss of an Inverted Repeat Region in Passiflora Chloroplast Genomes. Genome Biol. Evol. 2020, 12, 1841–1857. [Google Scholar] [CrossRef]
- Song, Y.; Zhang, Y.; Xu, J.; Li, W.; Li, M. Characterization of the Complete Chloroplast Genome Sequence of Dalbergia Species and Its Phylogenetic Implications. Sci. Rep. 2019, 9, 20401. [Google Scholar] [CrossRef] [Green Version]
- Smulders, M.; Rus-Kortekaas, W.; Vosman, B. Microsatellite Markers Useful throughout the Genus Dianthus. Genome 2000, 43, 208–210. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Yae, B.W.; Kim, K.S. Segregation Patterns of Several Morphological Characters and RAPD Markers in Interspecific Hybrids between Dianthus giganteus and D. carthusianorum. Sci. Hortic. 2005, 105, 53–64. [Google Scholar] [CrossRef]
- Fu, X.; Ning, G.; Gao, L.; Bao, M. Genetic Diversity of Dianthus Accessions as Assessed Using Two Molecular Marker Systems (SRAPs and ISSRs) and Morphological Traits. Sci. Hortic. 2008, 117, 263–270. [Google Scholar] [CrossRef]
- He, D.; Gichira, A.W.; Li, Z.; Nzei, J.M.; Guo, Y.; Wang, Q.; Chen, J. Intergeneric Relationships within the Early-Diverging Angiosperm Family Nymphaeaceae Based on Chloroplast Phylogenomics. Int. J. Mol. Sci. 2018, 19, 3780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zuo, Y.; Zhu, X.; Liao, S.; Ma, J. Complete Chloroplast Genomes and Comparative Analysis of Sequences Evolution among Seven Aristolochia (Aristolochiaceae) Medicinal Species. Int. J. Mol. Sci. 2019, 20, 1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Duan, W.; Zhao, J.; Jing, Y.; Feng, M.; Kuang, B.; Wei, N.; Chen, B.; Yang, X. Comparative Analysis of Chloroplast Genome in Saccharum Spp. and Related Members of ‘Saccharum Complex.’. Int. J. Mol. Sci. 2022, 23, 7661. [Google Scholar] [CrossRef]
- Lloyd Evans, D.; Joshi, S.V.; Wang, J. Whole Chloroplast Genome and Gene Locus Phylogenies Reveal the Taxonomic Placement and Relationship of Tripidium (Panicoideae: Andropogoneae) to Sugarcane. BMC Evol. Biol. 2019, 19, 33. [Google Scholar] [CrossRef] [Green Version]
- Park, I.; Song, J.-H.; Yang, S.; Kim, W.J.; Choi, G.; Moon, B.C. Cuscuta Species Identification Based on the Morphology of Reproductive Organs and Complete Chloroplast Genome Sequences. Int. J. Mol. Sci. 2019, 20, 2726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, W.; Xu, C.; Liu, Y.; Shi, J.; Li, W.; Suo, Z. Chloroplast Phylogenomics and Divergence Times of Lagerstroemia (Lythraceae). BMC Genom. 2021, 22, 434. [Google Scholar] [CrossRef]
- Greenberg, A.K.; Donoghue, M.J. Molecular Systematics and Character Evolution in Caryophyllaceae. Taxon 2011, 60, 1637–1652. [Google Scholar] [CrossRef]
- Valente, L.M.; Savolainen, V.; Vargas, P. Unparalleled Rates of Species Diversification in Europe. Proc. R. Soc. B Biol. Sci. 2010, 277, 1489–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Deng, T.; Moore, M.J.; Ji, Y.; Lin, N.; Zhang, H.; Meng, A.; Wang, H.; Sun, Y.; Sun, H. Plastome Phylogenomics of Saussurea (Asteraceae: Cardueae). BMC Plant Biol. 2019, 19, 290. [Google Scholar] [CrossRef]
- Ma, P.-F.; Zhang, Y.-X.; Zeng, C.-X.; Guo, Z.-H.; Li, D.-Z. Chloroplast Phylogenomic Analyses Resolve Deep-Level Relationships of an Intractable Bamboo Tribe Arundinarieae (Poaceae). Syst. Biol. 2014, 63, 933–950. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Gao, H.; Jiang, W.; Zhang, H.; Yu, X.; Liu, E.; Tian, X. The Complete Chloroplast Genome of Gleditsia sinensis and Gleditsia japonica: Genome Organization, Comparative Analysis, and Development of Taxon Specific DNA Mini-Barcodes. Sci. Rep. 2020, 10, 16309. [Google Scholar] [CrossRef]
- Dong, W.-L.; Wang, R.-N.; Zhang, N.-Y.; Fan, W.-B.; Fang, M.-F.; Li, Z.-H. Molecular Evolution of Chloroplast Genomes of Orchid Species: Insights into Phylogenetic Relationship and Adaptive Evolution. Int. J. Mol. Sci. 2018, 19, 716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, C.; Ma, L.; Wu, Z.; Chen, K.; Wang, Y. Comparative Analyses of Chloroplast Genomes from 22 Lythraceae Species: Inferences for Phylogenetic Relationships and Genome Evolution within Myrtales. BMC Plant Biol. 2019, 19, 281. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, L.; Chen, H.; Jiang, M.; Wu, W.; Liu, S.; Wang, J.; Liu, C. Phylogenetic Analysis and Development of Molecular Markers for Five Medicinal Alpinia Species Based on Complete Plastome Sequences. BMC Plant Biol. 2021, 21, 431. [Google Scholar] [CrossRef]
- Fu, C.-N.; Wu, C.-S.; Ye, L.-J.; Mo, Z.-Q.; Liu, J.; Chang, Y.-W.; Li, D.-Z.; Chaw, S.-M.; Gao, L.-M. Prevalence of Isomeric Plastomes and Effectiveness of Plastome Super-Barcodes in Yews (Taxus) Worldwide. Sci. Rep. 2019, 9, 2773. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.-H.; Lu, J.-M.; Wen, J.; Ebihara, A.; Li, D.-Z. Applying DNA Barcodes to Identify Closely Related Species of Ferns: A Case Study of the Chinese Adiantum (Pteridaceae). PLoS ONE 2016, 11, e0160611. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Jin, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; dePamphilis, C.W.; Yi, T.-S.; Li, D.-Z. GetOrganelle: A Fast and Versatile Toolkit for Accurate de Novo Assembly of Organelle Genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.; Wu, X.; Huang, L.; Liu, C. CPGAVAS2, an Integrated Plastome Sequence Annotator and Analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef]
- Thiel, T.; Michalek, W.; Varshney, R.K.; Graner, A. Exploiting EST Databases for the Development and Characterization of Gene-Derived SSR-Markers in Barley (Hordeum vulgare L.). TAG Theor. Appl. Genet. Theor. Angew. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-Web: A Web Server for Microsatellite Prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [Green Version]
- Benson, G. Tandem Repeats Finder: A Program to Analyze DNA Sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The Manifold Applications of Repeat Analysis on a Genomic Scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [Green Version]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An Online Program to Visualize the Junction Sites of Chloroplast Genomes. Bioinforma. Oxf. Engl. 2018, 34, 3030–3031. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML: A Program Package for Phylogenetic Analysis by Maximum Likelihood. Comput. Appl. Biosci. CABIOS 1997, 13, 555–556. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Talavera, G.; Castresana, J. Improvement of Phylogenies after Removing Divergent and Ambiguously Aligned Blocks from Protein Sequence Alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An Integrated and Scalable Desktop Platform for Streamlined Molecular Sequence Data Management and Evolutionary Phylogenetics Studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (ITOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Mayor, C.; Brudno, M.; Schwartz, J.R.; Poliakov, A.; Rubin, E.M.; Frazer, K.A.; Pachter, L.S.; Dubchak, I. VISTA: Visualizing Global DNA Sequence Alignments of Arbitrary Length. Bioinformatics 2000, 16, 1046–1047. [Google Scholar] [CrossRef] [Green Version]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational Tools for Comparative Genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [Green Version]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Koressaar, T.; Remm, M. Enhancements and Modifications of Primer Design Program Primer3. Bioinformatics 2007, 23, 1289–1291. [Google Scholar] [CrossRef] [Green Version]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New Capabilities and Interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Genome Size /bp | GC Content /% | LSC Length /bp | SSC Length /bp | IR Length /bp | Gene (Protein, rRNA, tRNA) |
|---|---|---|---|---|---|---|
| Dch ‘MH’ | 149,596 | 36.33 | 82,840 | 17,150 | 24,803 | 126 (84, 8, 34) |
| Dch ‘dhs’ | 149,641 | 36.31 | 82,886 | 17,139 | 24,808 | 125 (83, 8, 34) |
| Dch ‘cf’ | 149,602 | 36.32 | 82,837 | 17,149 | 24,808 | 125 (83, 8, 34) |
| Dch ‘L’ | 149,641 | 36.31 | 82,886 | 17,139 | 24,808 | 125 (83, 8, 34) |
| Dch ‘X’ | 149,641 | 36.31 | 82,883 | 17,139 | 24,808 | 125 (83, 8, 34) |
| Dch ‘DPD’ | 149,641 | 36.31 | 82,888 | 17,139 | 24,807 | 125 (83, 8, 34) |
| Dsu ‘QM’ | 149,726 | 36.30 | 82,954 | 17,135 | 24,818 | 126 (84, 8, 34) |
| Dbr ‘XB’ | 149,642 | 36.31 | 82,893 | 17,123 | 24,813 | 124 (84, 8, 33) |
| Dca ‘XSZH’ | 149,596 | 36.32 | 82,935 | 17,096 | 24,781 | 124 (84, 6, 34) |
| ‘WC’ | 149,660 | 36.31 | 82,909 | 17,123 | 24,814 | 126 (84, 8, 34) |
| ‘HY’ | 149,800 | 36.30 | 82,963 | 17,227 | 24,805 | 126 (84, 8, 34) |
| F1 ‘87M’ | 149,192 | 36.30 | 82,436 | 17,150 | 24,803 | 125 (83, 8, 34) |
| Category of Genes | Group of Genes | Name of Genes |
|---|---|---|
| Photosynthesis | Subunits of ATP synthase | atpA, atpB, atpE, atpF *, atpI, atpH |
| Subunits of photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbI, psbH, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | |
| Subunits of NADH-dehydrogenase | ndhA *, ndhB (×2) *, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | |
| Subunits of cytochrome b/f complex | petA, petB *, petD *, petG, petL, petN | |
| Subunits of photosystem I | psaA ②, psaB, psaC, psaI, psaJ, ycf3 | |
| Subunits of rubisco | rbcL | |
| Self-replication | Large subunit of ribosome | rpl2 (×2), rpl14, rpl16 *, rpl20, rpl22, rpl32, rpl33, rpl36 |
| DNA dependent RNA polymerase | rpoA, rpoB, rpoC1 *, rpoC2 | |
| Small subunit of ribosome | rps2, rps3, rps4, rps7 (×2), rps8, rps11, rps12 (×2) **, rps14, rps15, rps16 *, rps18, rps19 ④ | |
| Ribosomal RNAs | rrn4.5 (×2), rrn5 (×2), rrn16 (×2), rrn23 (×2) | |
| Transfer RNAs | trnA-UGC (×2) *, trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnfM-CAU, trnG-GCC, trnH-GUG ⑤, trnI-GAU (×2) *, trnK-UUU*, trnL-CAA (×2), trnL-UAA *, trnL-UAG, trnM-CAU, trnN-GUU (×2), trnP-UGG, trnQ-UUG, trnR-ACG (×2), trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC (×2), trnV-UAC *, trnW-CCA, trnY-GUA | |
| Other genes | Subunit of acetyl-CoA-carboxylase | accD |
| c-type cytocgrom synthesis gene | ccsA ① | |
| Envelop membrane protein | cemA | |
| Translational initiation factor | infA | |
| Protease | clpP ** | |
| Maturase | matK | |
| Unkown | Conserved open reading frames | ycf1 (×2) ③, ycf2 (×2), ycf3 **, ycf4 |
| Type | Repeat Unit | Numbers of Repeats | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dch ‘MH’ | Dch ‘dhs’ | Dch ‘cf’ | Dch ‘L’ | Dch ‘X’ | Dch ‘DPD’ | Dsu ‘QM’ | Dbr ‘XB’ | Dca ‘XSZH’ | ‘HY’ | ‘WC’ | F1 ‘87M’ | ||
| Mono- | A/T | 62 | 64 | 64 | 64 | 64 | 64 | 63 | 62 | 66 | 61 | 62 | 64 |
| C/G | 1 | 2 | 1 | 2 | 2 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | |
| Di- | AT/AT | 3 | 3 | 3 | 3 | 3 | 3 | 2 | 3 | 2 | 2 | 3 | 3 |
| Tri- | AAT/ATT | 9 | 7 | 6 | 7 | 7 | 7 | 8 | 8 | 8 | 7 | 8 | 9 |
| Tetra- | AAAT/ATTT | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 1 |
| Penta- | AACAC/GTGTT | 0 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 0 |
| AATAC/ATTGT | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |
| Hexa- | AAATAT/ATATTT | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| AATATG/ATATTC | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 0 | |
| AATATT/AATATT | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | |
| Total No. | 77 | 80 | 78 | 80 | 80 | 80 | 79 | 78 | 80 | 75 | 78 | 79 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, S.; Liu, J.; He, X.; Wang, J.; Wang, Z.; Zhang, X.; Bao, M.; Fu, X. Comprehensive Comparative Analysis and Development of Molecular Markers for Dianthus Species Based on Complete Chloroplast Genome Sequences. Int. J. Mol. Sci. 2022, 23, 12567. https://doi.org/10.3390/ijms232012567
Lin S, Liu J, He X, Wang J, Wang Z, Zhang X, Bao M, Fu X. Comprehensive Comparative Analysis and Development of Molecular Markers for Dianthus Species Based on Complete Chloroplast Genome Sequences. International Journal of Molecular Sciences. 2022; 23(20):12567. https://doi.org/10.3390/ijms232012567
Chicago/Turabian StyleLin, Shengnan, Jianyi Liu, Xingqun He, Jie Wang, Zehao Wang, Xiaoni Zhang, Manzhu Bao, and Xiaopeng Fu. 2022. "Comprehensive Comparative Analysis and Development of Molecular Markers for Dianthus Species Based on Complete Chloroplast Genome Sequences" International Journal of Molecular Sciences 23, no. 20: 12567. https://doi.org/10.3390/ijms232012567