
Genome-Wide Identification, Evolution, and Expression Pattern Analysis of the GATA Gene Family in Tartary Buckwheat (Fagopyrum tataricum)
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Identification of FtGATA Genes
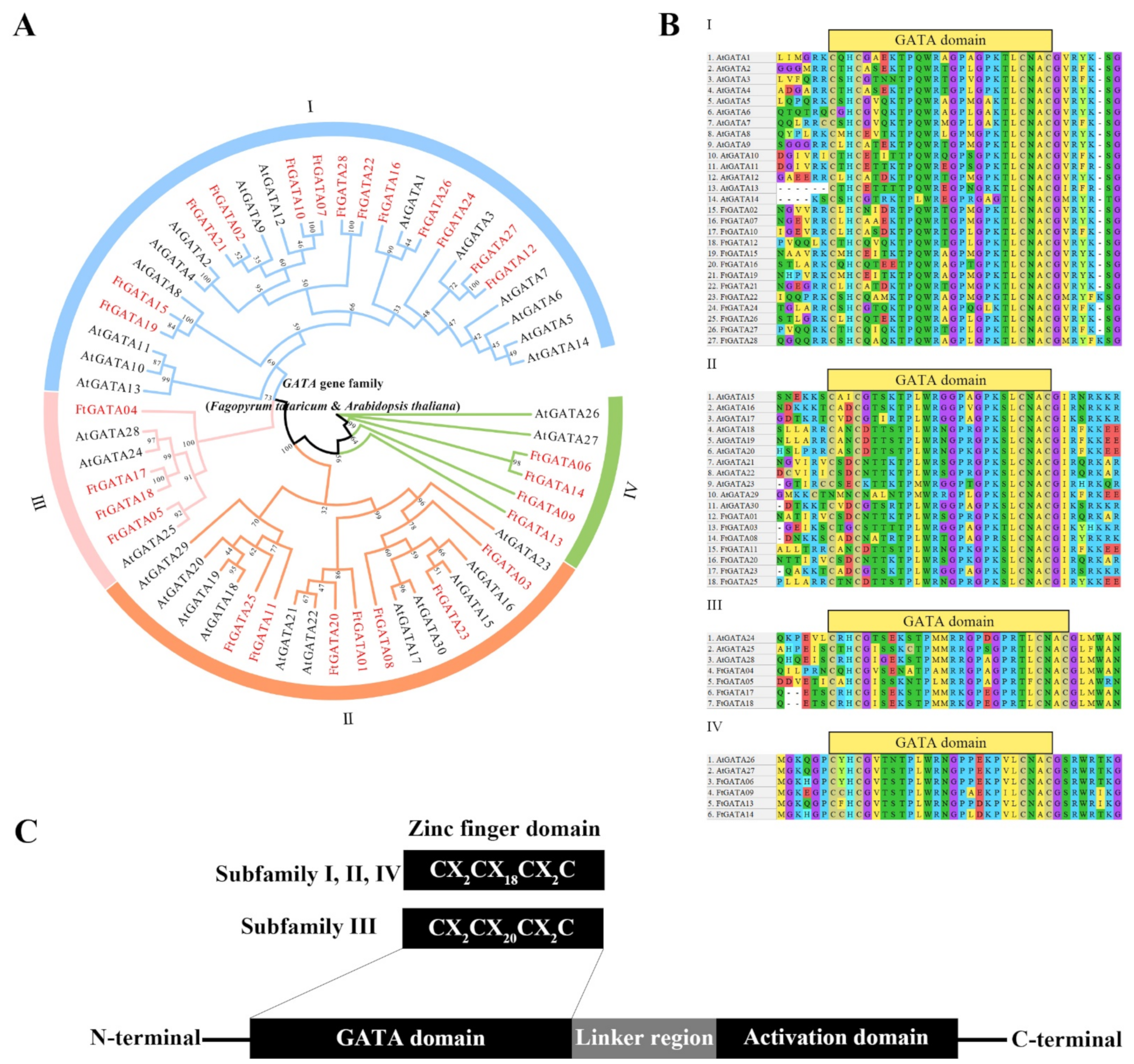
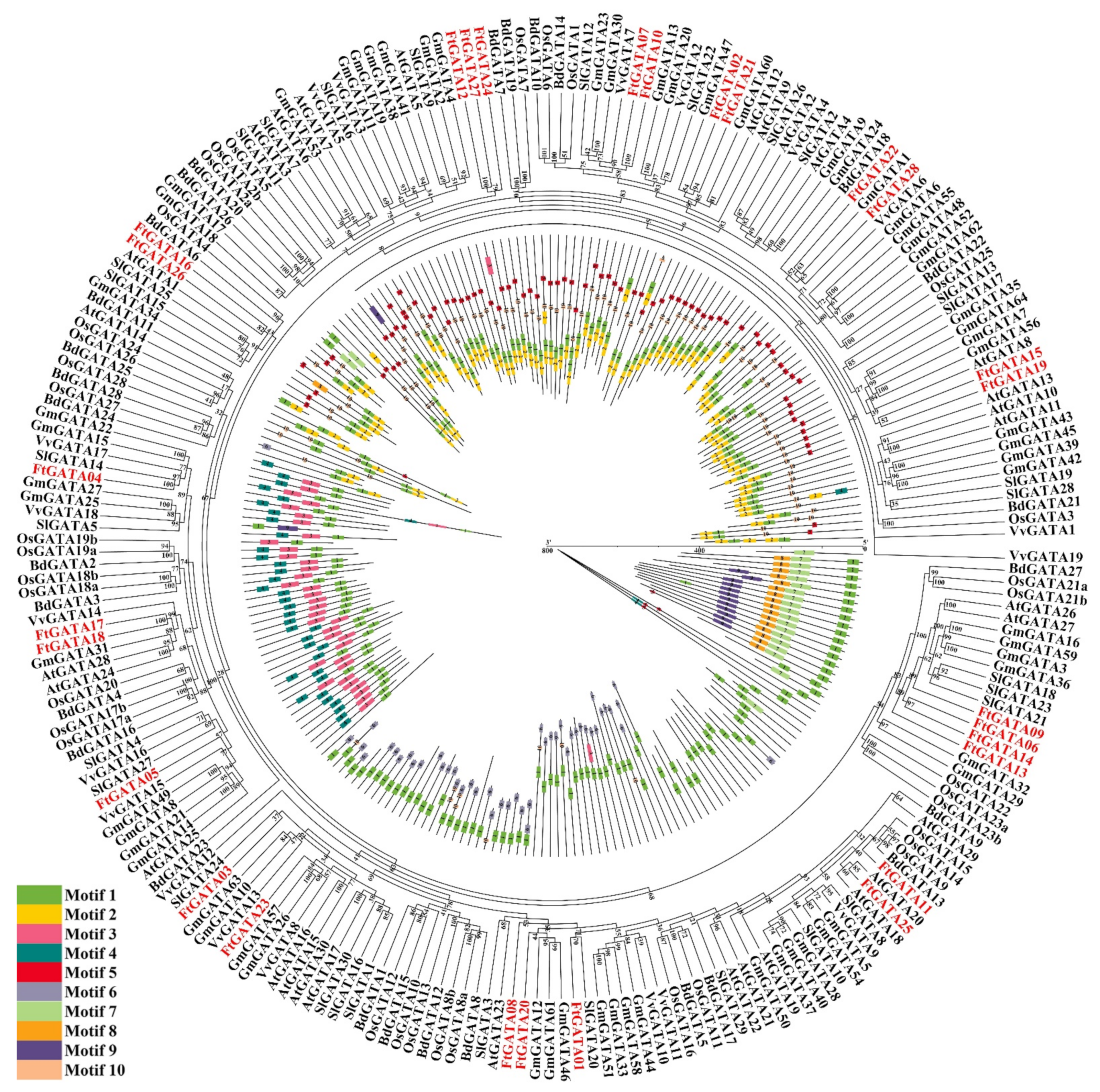
2.2. Phylogenetic Analysis, Classification, and Multiple Sequence Alignment of Ftgata Proteins
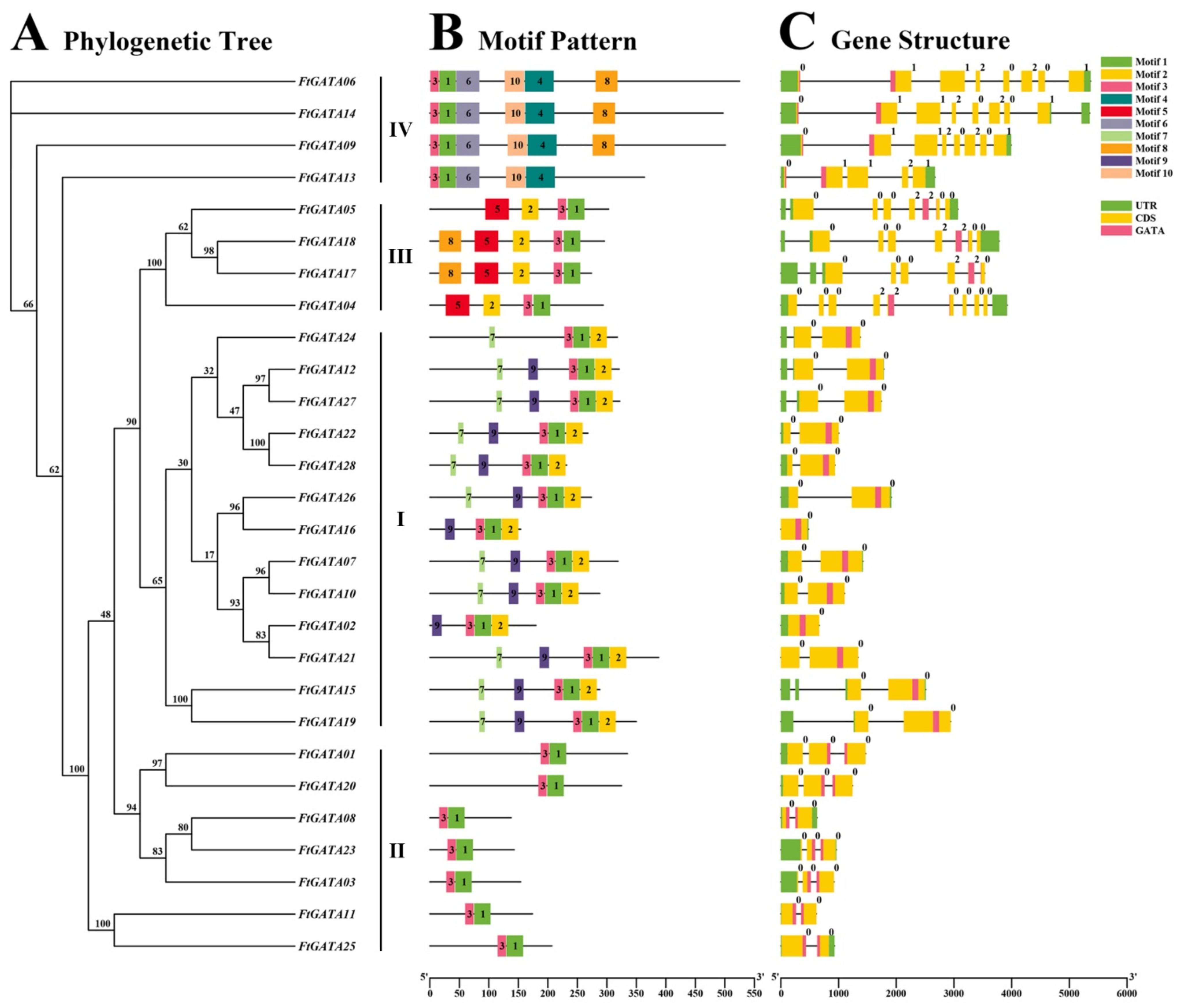
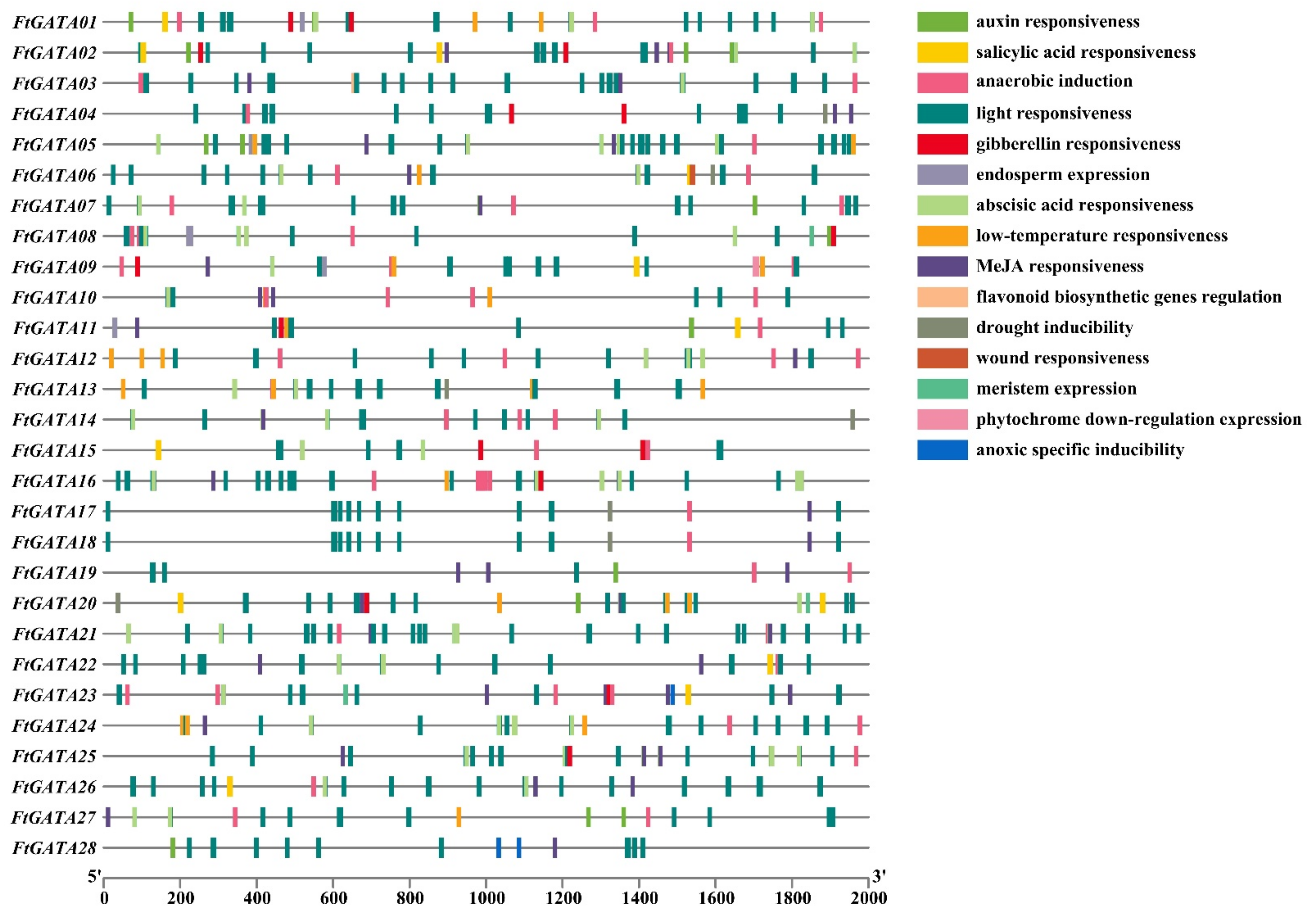
2.3. Gene Structures, Conserved Motifs, and Cis-Acting Element Analysis of the Ftgata Gene Family
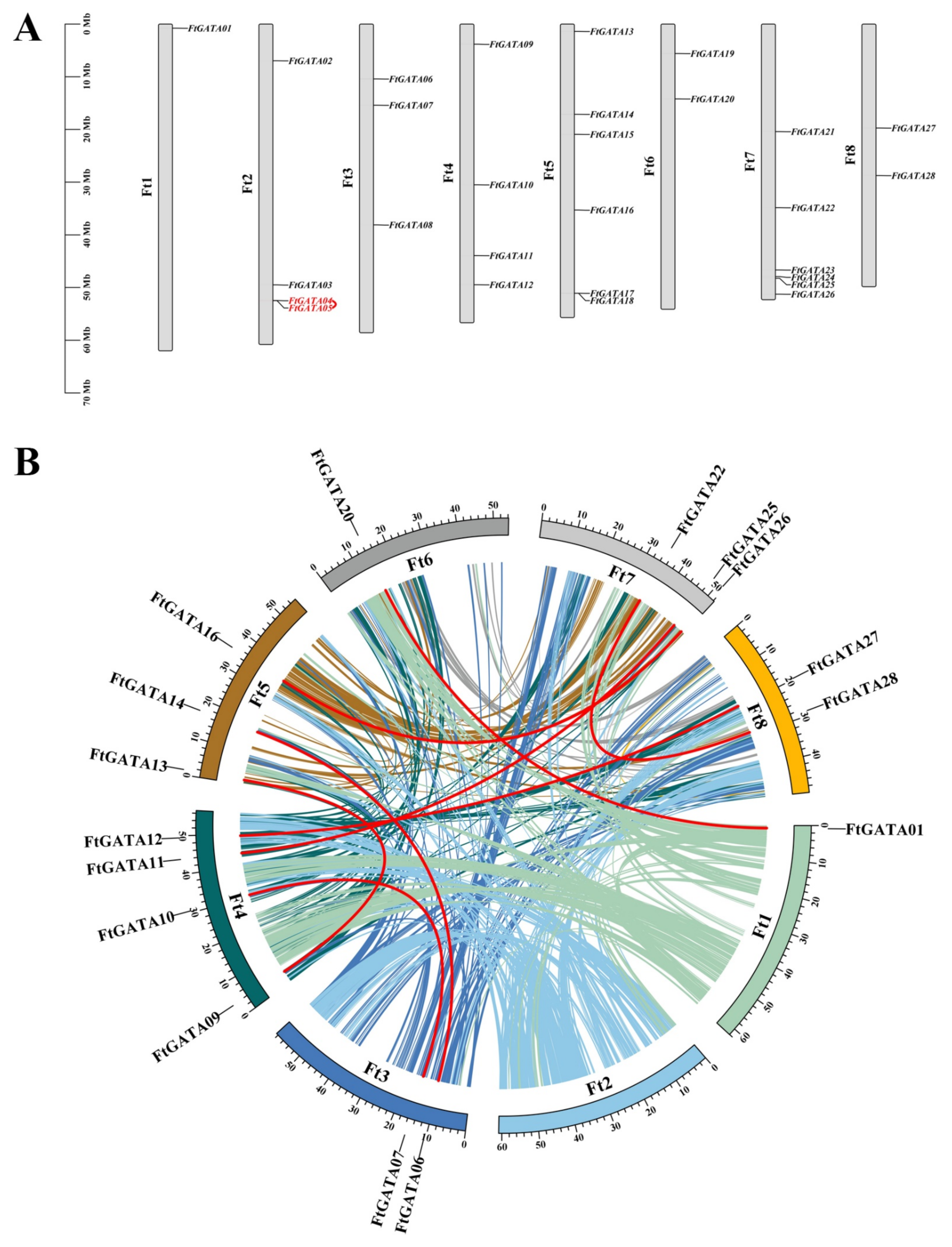
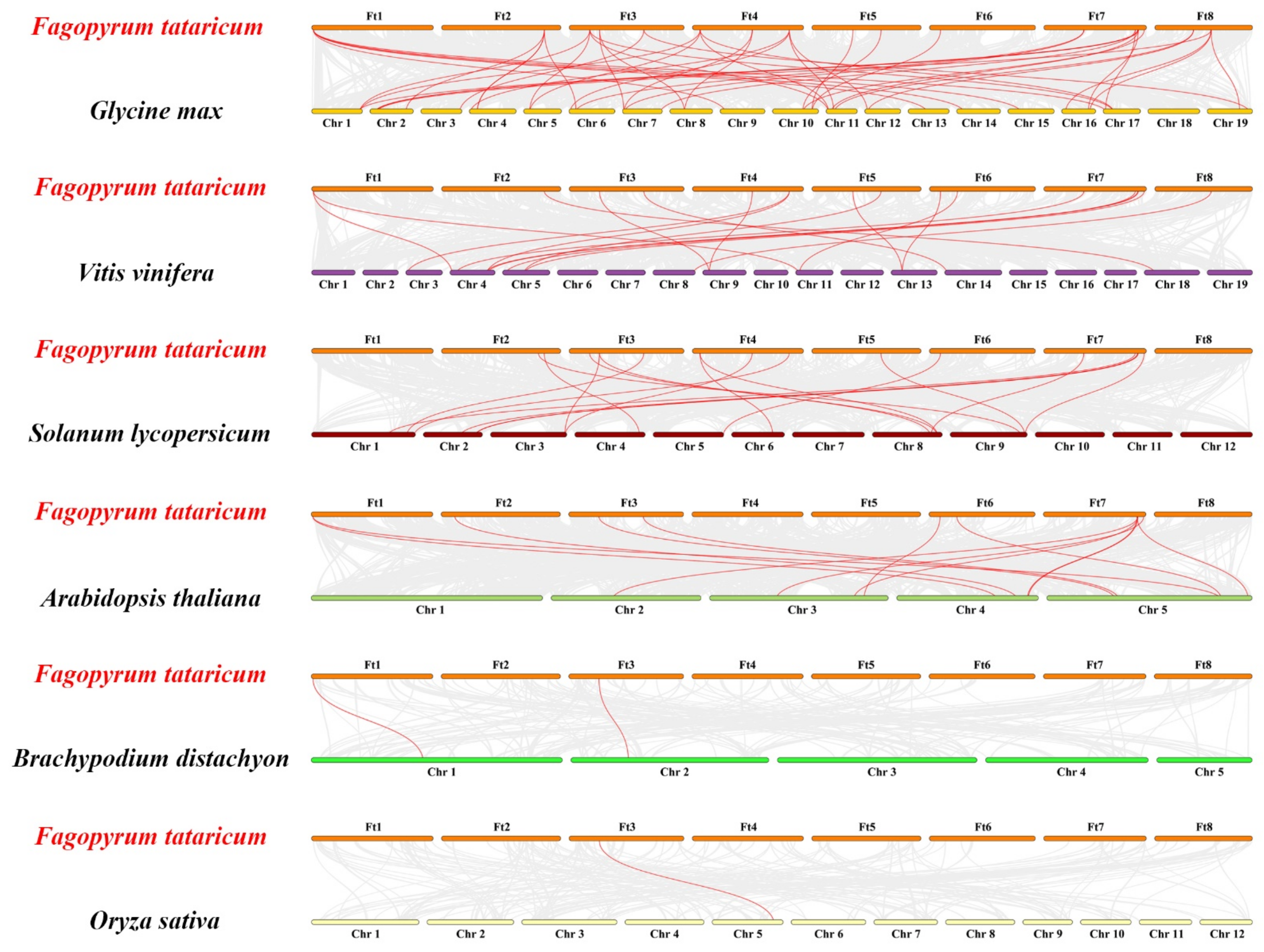
2.4. Chromosomal Distribution-, Gene Duplication-, and Collinear Analysis of Ftgata Genes
2.5. Evolutionary Analysis of FtGATA Proteins and GATA Proteins of Different Plants
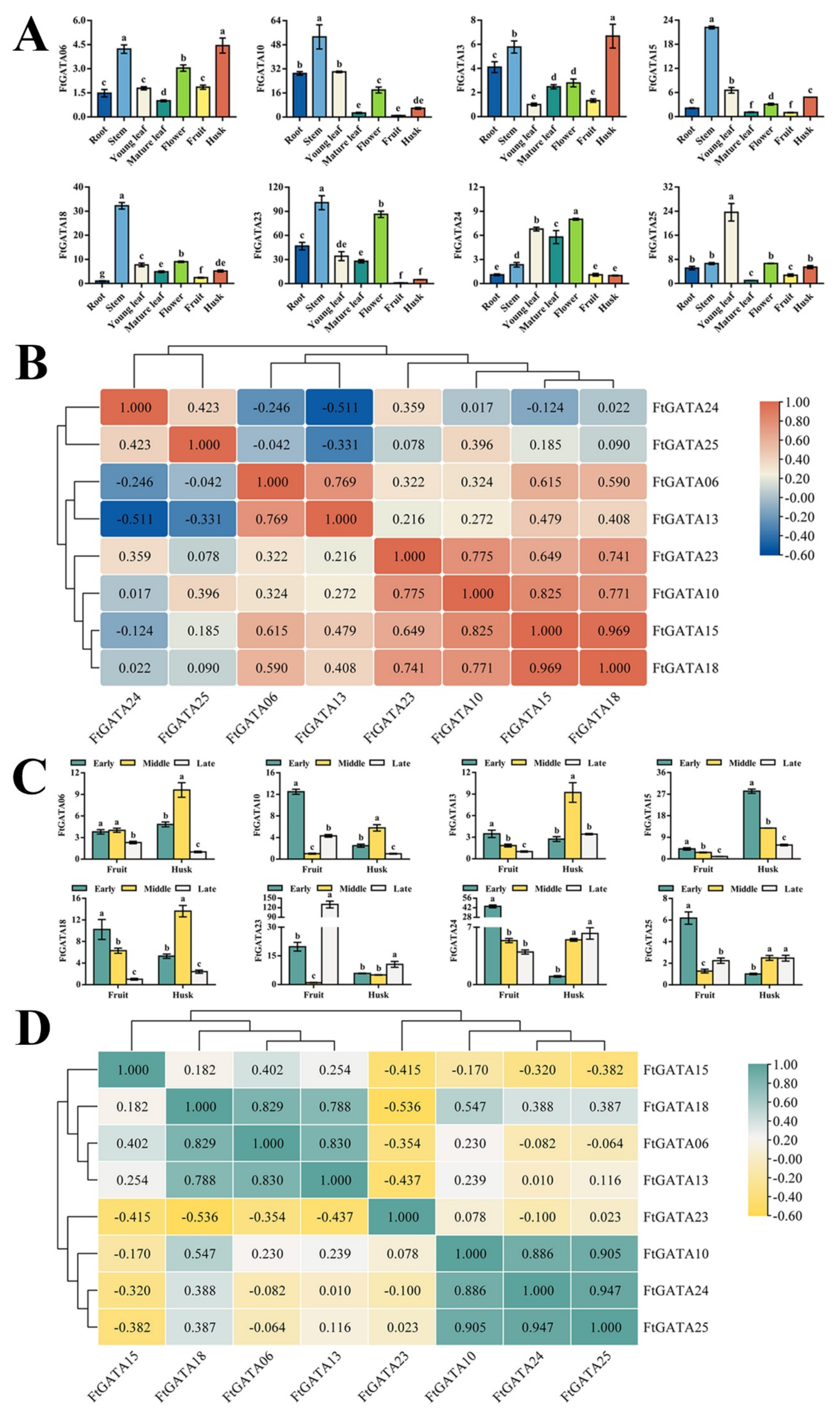
2.6. Expression Patterns of FtGATAs in Different Plant Tissues and Fruit Development
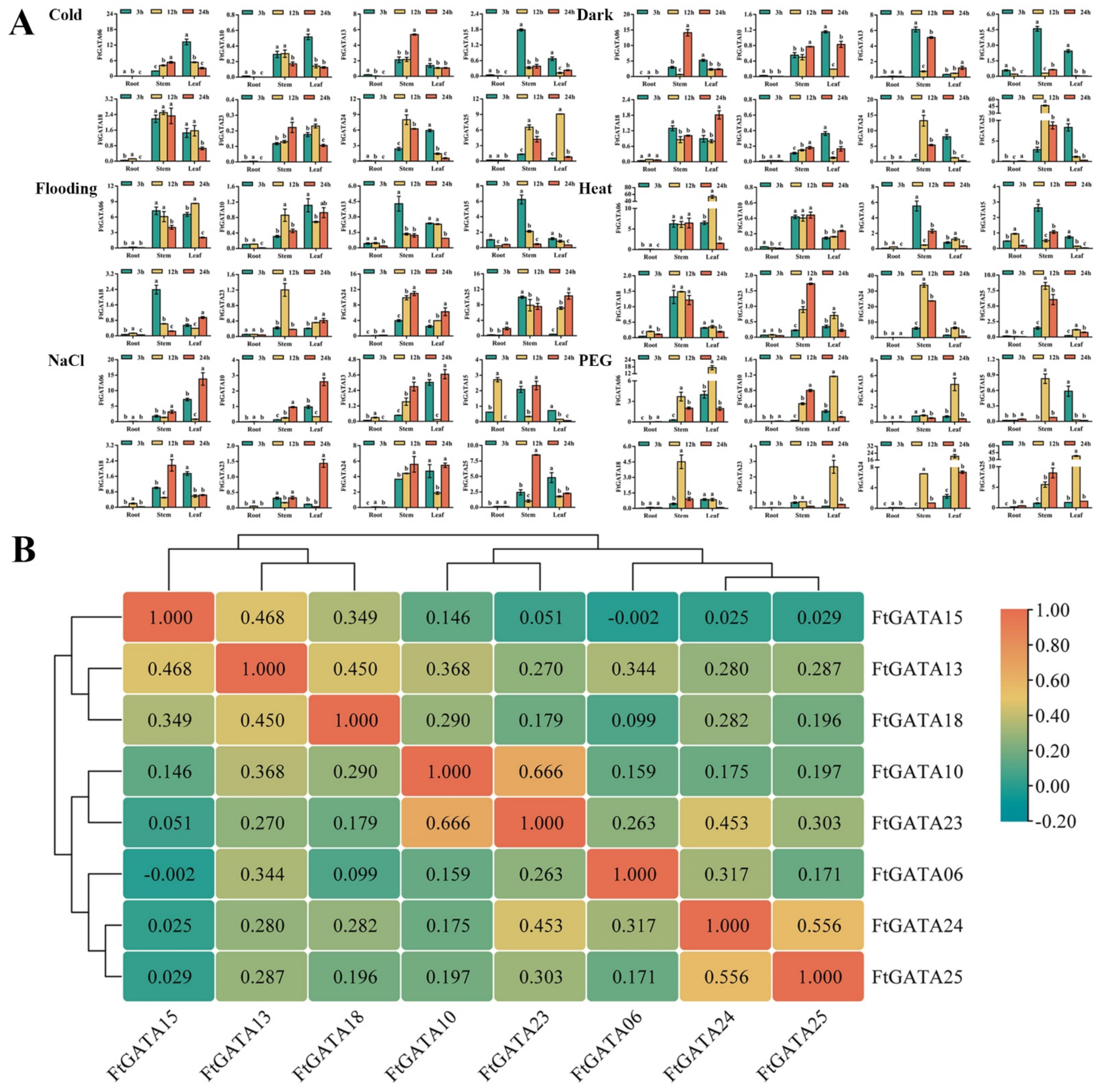
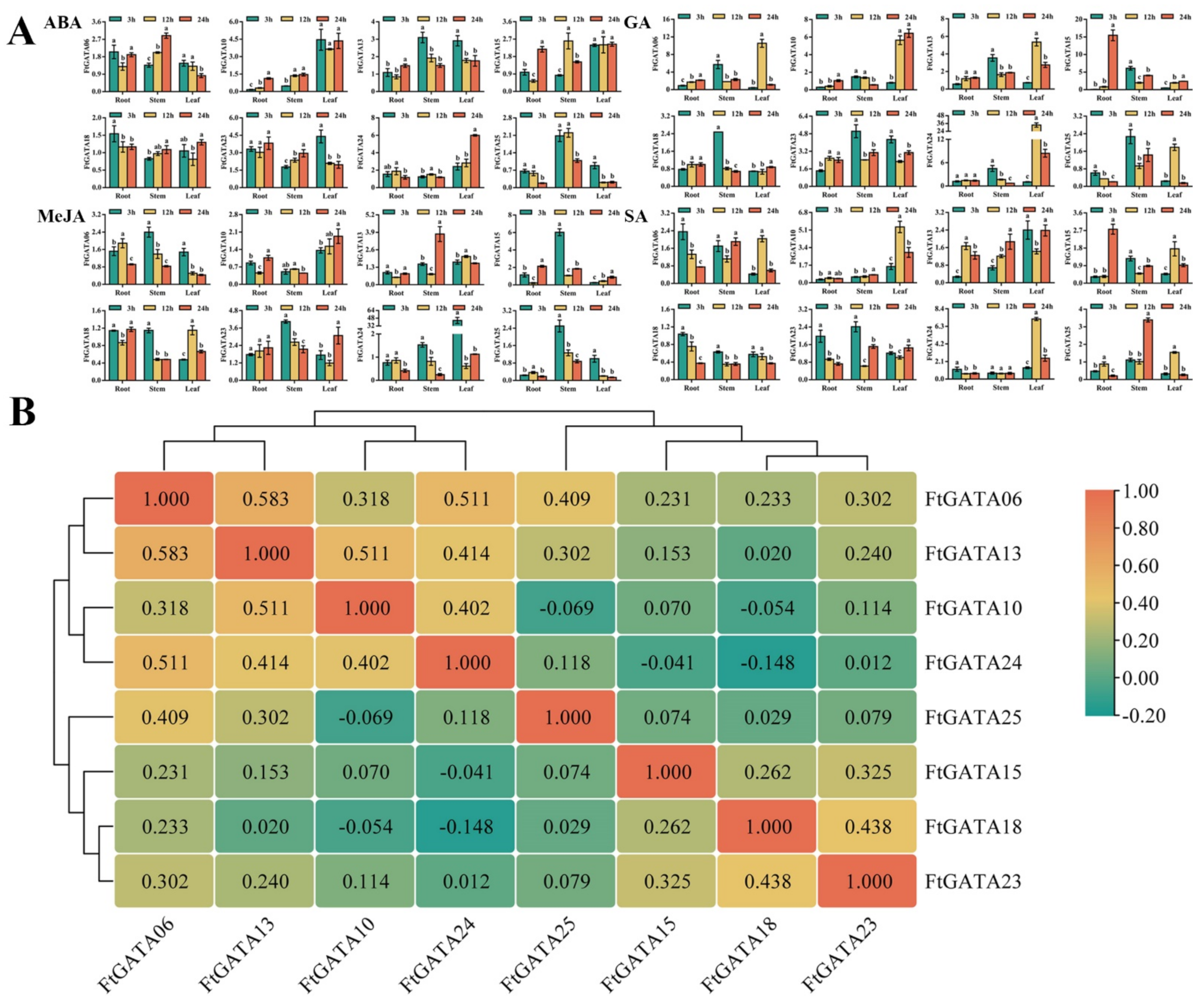
2.7. Expression Patterns of FtGATA Genes in Response to Abiotic Treatments
2.8. Expression Patterns of FtGATA Genes in Response to Hormone Treatments
3. Discussion
3.1. Characteristics of FtGATA Genes
3.2. Evolution of FtGATA Genes
3.3. Spatiotemporal Expression Patterns of FtGATA Genes
4. Materials and Methods
4.1. Materials, Experiment Design, and Treatments
4.2. Total RNA Extraction, Reverse Transcription PCR (RT-PCR), and Quantitative Real-Time PCR (qRT-PCR)
4.3. Genome-Wide Identification of FtGATA Genes in Fagopyrum tataricum
4.4. GATA Gene Structure, Conserved Motifs, Cis-Acting Elements, and Protein Interactions
4.5. Chromosomal Distribution of FtGATA Genes
4.6. Phylogenetic Evolution and Classification of the FtGATA Gene Family
4.7. Statistics and Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Riechmann, J.L.; Heard, J.; Martin, G.; Reuber, L.; Jiang, C.; Keddie, J.; Adam, L.; Pineda, O.; Ratcliffe, O.J.; Samaha, R.R.; et al. Arabidopsis transcription factors: Genome-wide comparative analysis among eukaryotes. Science 2000, 290, 2105–2110. [Google Scholar] [CrossRef]
- Fan, Y.; Yan, J.; Lai, D.; Yang, H.; Xue, G.; He, A.; Guo, T.; Chen, L.; Cheng, X.B.; Xiang, D.B.; et al. Genome-wide identification, expression analysis, and functional study of the GRAS transcription factor family and its response to abiotic stress in sorghum [Sorghum bicolor (L.) Moench]. BMC Genom. 2021, 22, 509. [Google Scholar] [CrossRef] [PubMed]
- Stevanovic, M.; Stanisavljevic Ninkovic, D.; Mojsin, M.; Drakulic, D.; Schwirtlich, M. Interplay of SOX transcription factors and microRNAs in the brain under physiological and pathological conditions. Neural Regen. Res. 2022, 17, 2325–2334. [Google Scholar] [CrossRef] [PubMed]
- Rueda-López, M.; Cañas, R.A.; Canales, J.; Cánovas, F.M.; Ávila, C. The overexpression of the pine transcription factor PpDof5 in Arabidopsis leads to increased lignin content and affects carbon and nitrogen metabolism. Physiol. Plant. 2015, 155, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yuan, L.; Guo, W.; Wu, W. Transcription factor TERF1 promotes seed germination under osmotic conditions by activating gibberellin acid signaling. Plant Sci. 2022, 322, 111350. [Google Scholar] [CrossRef] [PubMed]
- Zha, Q.; Xi, X.; He, Y.; Yin, X.; Jiang, A. Interaction of VvbZIP60s and VvHSP83 in response to high-temperature stress in grapes. Gene 2022, 810, 146053. [Google Scholar] [CrossRef] [PubMed]
- Darigh, F.; Iranbakhsh, A.; Oraghi Ardebili, Z.; Ebadi, M.; Hassanpour, H. Simulated microgravity contributed to modification of callogenesis performance and secondary metabolite production in Cannabis indica. Plant Physiol. Biochem. 2022, 186, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Schwechheimer, C.; Zourelidou, M.; Bevan, M.W. Plant Transcription Factor Studies. Annu. Rev. Plant Phys. Plant Mol. Biol. 1998, 49, 127–150. [Google Scholar] [CrossRef] [Green Version]
- Lowry, J.A.; Atchley, W.R. Molecular evolution of the GATA family of transcription factors: Conservation within the DNA-binding domain. J. Mol. Evol. 2000, 50, 103–115. [Google Scholar] [CrossRef]
- Teakle, G.R.; Manfield, I.W.; Graham, J.F.; Gilmartin, P.M. Arabidopsis thaliana GATA factors: Organisation, expression and DNA-binding characteristics. Plant Mol. Biol. 2002, 50, 43–57. [Google Scholar] [CrossRef]
- Behringer, C.; Schwechheimer, C. B-GATA transcription factors-insights into their structure, regulation, and role in plant development. Front. Plant Sci. 2015, 6, 90. [Google Scholar] [CrossRef] [Green Version]
- Ravindran, P.; Verma, V.; Stamm, P.; Kumar, P.P. A Novel RGL2-DOF6 Complex Contributes to Primary Seed Dormancy in Arabidopsis thaliana by Regulating a GATA Transcription Factor. Mol. Plant. 2017, 10, 1307–1320. [Google Scholar] [CrossRef] [Green Version]
- Hudson, D.; Guevara, D.R.; Hand, A.J.; Xu, Z.; Hao, L.; Chen, X.; Zhu, T.; Bi, Y.M.; Rothstein, S.J. Rice cytokinin GATA transcription Factor1 regulates chloroplast development and plant architecture. Plant Physiol. 2013, 162, 132–144. [Google Scholar] [CrossRef] [Green Version]
- An, Y.; Zhou, Y.; Han, X.; Shen, C.; Wang, S.; Liu, C.; Yin, W.; Xia, X. The GATA transcription factor GNC plays an important role in photosynthesis and growth in poplar. J. Exp. Bot. 2020, 71, 1969–1984. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Yu, Q.; Zeng, J.; He, X.; Liu, W. Genome-wide identification and characterization of GATA family genes in wheat. BMC Plant Biol. 2022, 22, 372. [Google Scholar] [CrossRef] [PubMed]
- Naito, T.; Kiba, T.; Koizumi, N.; Yamashino, T.; Mizuno, T. Characterization of a unique GATA family gene that responds to both light and cytokinin in Arabidopsis thaliana. Biosci. Biotechnol. Biochem. 2007, 71, 1557–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, R.; Behringer, C.; Zourelidou, M.; Schwechheimer, C. Convergence of auxin and gibberellin signaling on the regulation of the GATA transcription factors GNC and GNL in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2013, 110, 13192–13197. [Google Scholar] [CrossRef] [Green Version]
- Klermund, C.; Ranftl, Q.L.; Diener, J.; Bastakis, E.; Richter, R.; Schwechheimer, C. LLM-Domain B-GATA Transcription Factors Promote Stomatal Development Downstream of Light Signaling Pathways in Arabidopsis thaliana Hypocotyls. Plant Cell. 2016, 28, 646–660. [Google Scholar] [CrossRef] [Green Version]
- Du, K.; Xia, Y.; Zhan, D.; Xu, T.; Lu, T.; Yang, J.; Kang, X. Genome-Wide Identification of the Eucalyptus urophylla GATA Gene Family and Its Diverse Roles in Chlorophyll Biosynthesis. Int. J. Mol. Sci. 2022, 23, 5251. [Google Scholar] [CrossRef]
- Nutan, K.K.; Singla-Pareek, S.L.; Pareek, A. The Saltol QTL-localized transcription factor OsGATA8 plays an important role in stress tolerance and seed development in Arabidopsis and rice. J. Exp. Bot. 2020, 71, 684–698. [Google Scholar] [CrossRef]
- Shaikhali, J.; de Dios Barajas-Lopéz, J.; Ötvös, K.; Kremnev, D.; Garcia, A.S.; Srivastava, V.; Wingsle, G.; Bako, L.; Strand, Å. The CRYPTOCHROME1-Dependent Response to Excess Light is Mediated through the Transcriptional Activators Zinc Finger Protein Expressed in Inflorescence Meristem LIKE1 and ZML2 in Arabidopsis. Plant Cell. 2012, 24, 3009–3025. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.M.; Lin, W.H.; Zhu, S.; Zhu, J.Y.; Sun, Y.; Fan, X.Y.; Cheng, M.; Hao, Y.; Oh, E.; Tian, M.; et al. Integration of light- and brassinosteroid-signaling pathways by a GATA transcription factor in Arabidopsis. Dev Cell. 2010, 19, 872–883. [Google Scholar] [CrossRef] [Green Version]
- Yao, X.; Zhou, M.; Ruan, J.; Peng, Y.; Yang, H.; Tang, Y.; Gao, A.; Cheng, J. Pretreatment with H2O2 Alleviates the Negative Impacts of NaCl Stress on Seed Germination of Tartary Buckwheat (Fagopyrum tataricum). Plants 2021, 10, 1784. [Google Scholar] [CrossRef]
- Sun, Z.; Wang, X.; Liu, R.; Du, W.; Ma, M.; Han, Y.; Li, H.; Liu, L.; Hou, S. Comparative transcriptomic analysis reveals the regulatory mechanism of the gibberellic acid pathway of Tartary buckwheat (Fagopyrum tataricum (L.) Gaertn.) dwarf mutants. BMC Plant Biol. 2021, 21, 206. [Google Scholar] [CrossRef]
- Wen, W.; Li, Z.; Shao, J.; Tang, Y.; Zhao, Z.; Yang, J.; Ding, M.; Zhu, X.; Zhou, M. The Distribution and Sustainable Utilization of Buckwheat Resources under Climate Change in China. Plants 2021, 10, 2081. [Google Scholar] [CrossRef]
- Holasova, M.; Fiedlerova, V.; Smrcinova, H.; Orsak, M.; Lachman, J.; Vavreinova, S. Buckwheat—The source of antioxidant activity in functional foods. Food Res. Int. 2002, 35, 207–211. [Google Scholar] [CrossRef]
- Chua, L.S. A review on plant-based rutin extraction methods and its pharmacological activities. J. Ethnopharmacol. 2013, 150, 805–817. [Google Scholar] [CrossRef]
- Ma, Z.; Liu, M.; Sun, W.; Huang, L.; Wu, Q.; Bu, T.; Li, C.; Chen, H. Genome-wide identification and expression analysis of the trihelix transcription factor family in Tartary buckwheat (Fagopyrum tataricum). BMC Plant Biol. 2019, 19, 344. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Ma, Z.; Zheng, T.; Wang, J.; Huang, L.; Sun, W.; Zhang, Y.; Jin, W.; Zhan, J.; Cai, Y.; et al. The Potential Role of Auxin and Abscisic Acid Balance and FtARF2 in the Final Size Determination of Tartary Buckwheat Fruit. Int. J. Mol. Sci. 2018, 19, 2755. [Google Scholar] [CrossRef] [Green Version]
- Zielińska, D.; Turemko, M.; Kwiatkowski, J.; Zieliński, H. Evaluation of flavonoid contents and antioxidant capacity of the aerial parts of common and Tartary buckwheat plants. Molecules 2012, 17, 9668–9682. [Google Scholar] [CrossRef]
- Reyes, J.C.; Muro-Pastor, M.I.; Florencio, F.J. The GATA family of transcription factors in Arabidopsis and rice. Plant Physiol. 2004, 134, 1718–1732. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.; Nutan, K.K.; Singla-Pareek, S.L.; Pareek, A. Abiotic Stresses Cause Differential Regulation of Alternative Splice Forms of GATA Transcription Factor in Rice. Front. Plant. Sci. 2017, 8, 1944. [Google Scholar] [CrossRef] [Green Version]
- Peng, W.; Li, W.; Song, N.; Tang, Z.; Liu, J.; Wang, Y.; Pan, S.; Dai, L.; Wang, B. Genome-Wide Characterization, Evolution, and Expression Profile Analysis of GATA Transcription Factors in Brachypodium distachyon. Int. J. Mol. Sci. 2021, 22, 2026. [Google Scholar] [CrossRef]
- Zhang, C.; Hou, Y.; Hao, Q.; Chen, H.; Chen, L.; Yuan, S.; Shan, Z.; Zhang, X.; Yang, Z.; Qiu, D.; et al. Genome-wide survey of the soybean GATA transcription factor gene family and expression analysis under low nitrogen stress. PLoS ONE 2015, 10, e0125174. [Google Scholar] [CrossRef] [Green Version]
- Lai, D.; Yao, X.; Yan, J.; Gao, A.; Yang, H.; Xiang, D.; Ruan, J.; Fan, Y.; Cheng, J. Genome-wide identification, phylogenetic and expression pattern analysis of GATA family genes in foxtail millet (Setaria italica). BMC Genom. 2022, 23, 549. [Google Scholar] [CrossRef]
- Holub, E.B. The arms race is ancient history in Arabidopsis, the wildflower. Nat. Rev. Genet. 2001, 2, 516–527. [Google Scholar] [CrossRef]
- Cheng, X.; Tian, B.; Gao, C.; Gao, W.; Yan, S.; Yao, H.; Wang, X.; Jiang, Y.; Hu, L.; Pan, X.; et al. Identification and expression analysis of candidate genes related to seed dormancy and germination in the wheat GATA family. Plant Physiol. Biochem. 2021, 169, 343–359. [Google Scholar] [CrossRef]
- Manzoor, M.A.; Sabir, I.A.; Shah, I.H.; Wang, H.; Yu, Z.; Rasool, F.; Mazhar, M.Z.; Younas, S.; Abdullah, M.; Cai, Y. Comprehensive Comparative Analysis of the GATA Transcription Factors in Four Rosaceae Species and Phytohormonal Response in Chinese Pear (Pyrus bretschneideri) Fruit. Int. J. Mol. Sci. 2021, 22, 12492. [Google Scholar] [CrossRef]
- Hou, Y.; Lu, Q.; Su, J.; Jin, X.; Jia, C.; An, L.; Tian, Y.; Song, Y. Genome-Wide Analysis of the HDAC Gene Family and Its Functional Characterization at Low Temperatures in Tartary Buckwheat (Fagopyrum tataricum). Int. J. Mol. Sci. 2022, 23, 7622. [Google Scholar] [CrossRef]
- Shi, M.; Huang, Q.; Wang, Y.; Wang, C.; Zhu, R.; Zhang, S.; Kai, G. Genome-wide survey of the GATA gene family in camptothecin-producing plant Ophiorrhiza pumila. BMC Genom. 2022, 23, 256. [Google Scholar] [CrossRef]
- Shabalina, S.A.; Ogurtsov, A.Y.; Spiridonov, A.N.; Novichkov, P.S.; Spiridonov, N.A.; Koonin, E.V. Distinct patterns of expression and evolution of intronless and intron-containing mammalian genes. Mol. Biol. Evol. 2010, 27, 1745–1749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, M.; Khurana, P.; Tyagi, A.K.; Khurana, J.P. Genome-wide analysis of intronless genes in rice and Arabidopsis. Funct. Integr. Genom. 2008, 8, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Sang, Y.; Liu, Q.; Lee, J.; Ma, W.; McVey, D.S.; Blecha, F. Expansion of amphibian intronless interferons revises the paradigm for interferon evolution and functional diversity. Sci. Rep. 2016, 6, 29072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Patra, B.; Pattanaik, S.; Wang, Y.; Yuan, L. GATA and Phytochrome Interacting Factor Transcription Factors Regulate Light-Induced Vindoline Biosynthesis in Catharanthus roseus. Plant Physiol. 2019, 180, 1336–1350. [Google Scholar] [CrossRef] [Green Version]
- Buzby, J.S.; Yamada, T.; Tobin, E.M. A light-regulated DNA-binding activity interacts with a conserved region of a Lemna gibba rbcS promoter. Plant Cell. 1990, 2, 805–814. [Google Scholar]
- Song, X.M.; Liu, T.K.; Duan, W.K.; Ma, Q.H.; Ren, J.; Wang, Z.; Li, Y.; Hou, X.L. Genome-wide analysis of the GRAS gene family in Chinese cabbage (Brassica rapa ssp. pekinensis). Genomics 2014, 103, 135–146. [Google Scholar] [CrossRef] [Green Version]
- Richter, R.; Bastakis, E.; Schwechheimer, C. Cross-repressive interactions between SOC1 and the GATAs GNC and GNL/CGA1 in the control of greening, cold tolerance, and flowering time in Arabidopsis. Plant Physiol. 2013, 162, 1992–2004. [Google Scholar] [CrossRef] [Green Version]
- Zubo, Y.O.; Blakley, I.C.; Franco-Zorrilla, J.M.; Yamburenko, M.V.; Solano, R.; Kieber, J.J.; Loraine, A.E.; Schaller, G.E. Coordination of Chloroplast Development through the Action of the GNC and GLK Transcription Factor Families. Plant Physiol. 2018, 178, 130–147. [Google Scholar] [CrossRef] [Green Version]
- Richter, R.; Behringer, C.; Müller, I.K.; Schwechheimer, C. The GATA-Type Transcription Factors GNC and GNL/CGA1 Repress Gibberellin Signaling Downstream from DELLA Proteins and Phytochrome-Interacting Factors. Genes. Dev. 2010, 24, 2093–2104. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhou, Y.; Ding, L.; Wu, Z.; Liu, R.; Meyerowitz, E.M. Transcription repressor HANABA TARANU controls flower development by integrating the actions of multiple hormones, floral organ specification genes, and GATA3 family genes in Arabidopsis. Plant cell. 2013, 25, 83–101. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018, 46, D493–D496. [Google Scholar] [CrossRef]
- Yang, M.; Derbyshire, M.K.; Yamashita, R.A.; Marchler-Bauer, A. NCBI’s Conserved Domain Database and Tools for Protein Domain Analysis. Curr. Protoc. Bioinform. 2020, 69, e90. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.D.; Gibson, T.J.; Higgins, D.G. Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinform. 2003, 1, 2–3. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant. 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genom. Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, X.; Zhou, M.; Ruan, J.; He, A.; Ma, C.; Wu, W.; Lai, D.; Fan, Y.; Gao, A.; Weng, W.; et al. Genome-Wide Identification, Evolution, and Expression Pattern Analysis of the GATA Gene Family in Tartary Buckwheat (Fagopyrum tataricum). Int. J. Mol. Sci. 2022, 23, 12434. https://doi.org/10.3390/ijms232012434
Yao X, Zhou M, Ruan J, He A, Ma C, Wu W, Lai D, Fan Y, Gao A, Weng W, et al. Genome-Wide Identification, Evolution, and Expression Pattern Analysis of the GATA Gene Family in Tartary Buckwheat (Fagopyrum tataricum). International Journal of Molecular Sciences. 2022; 23(20):12434. https://doi.org/10.3390/ijms232012434
Chicago/Turabian StyleYao, Xin, Meiliang Zhou, Jingjun Ruan, Ailing He, Chao Ma, Weijiao Wu, Dili Lai, Yu Fan, Anjing Gao, Wenfeng Weng, and et al. 2022. "Genome-Wide Identification, Evolution, and Expression Pattern Analysis of the GATA Gene Family in Tartary Buckwheat (Fagopyrum tataricum)" International Journal of Molecular Sciences 23, no. 20: 12434. https://doi.org/10.3390/ijms232012434