
Novel Class of Proteasome Inhibitors: In Silico and In Vitro Evaluation of Diverse Chloro(trifluoromethyl)aziridines
 , , , ,
, , , ,  , ,
, ,  , and
, and
Abstract
:
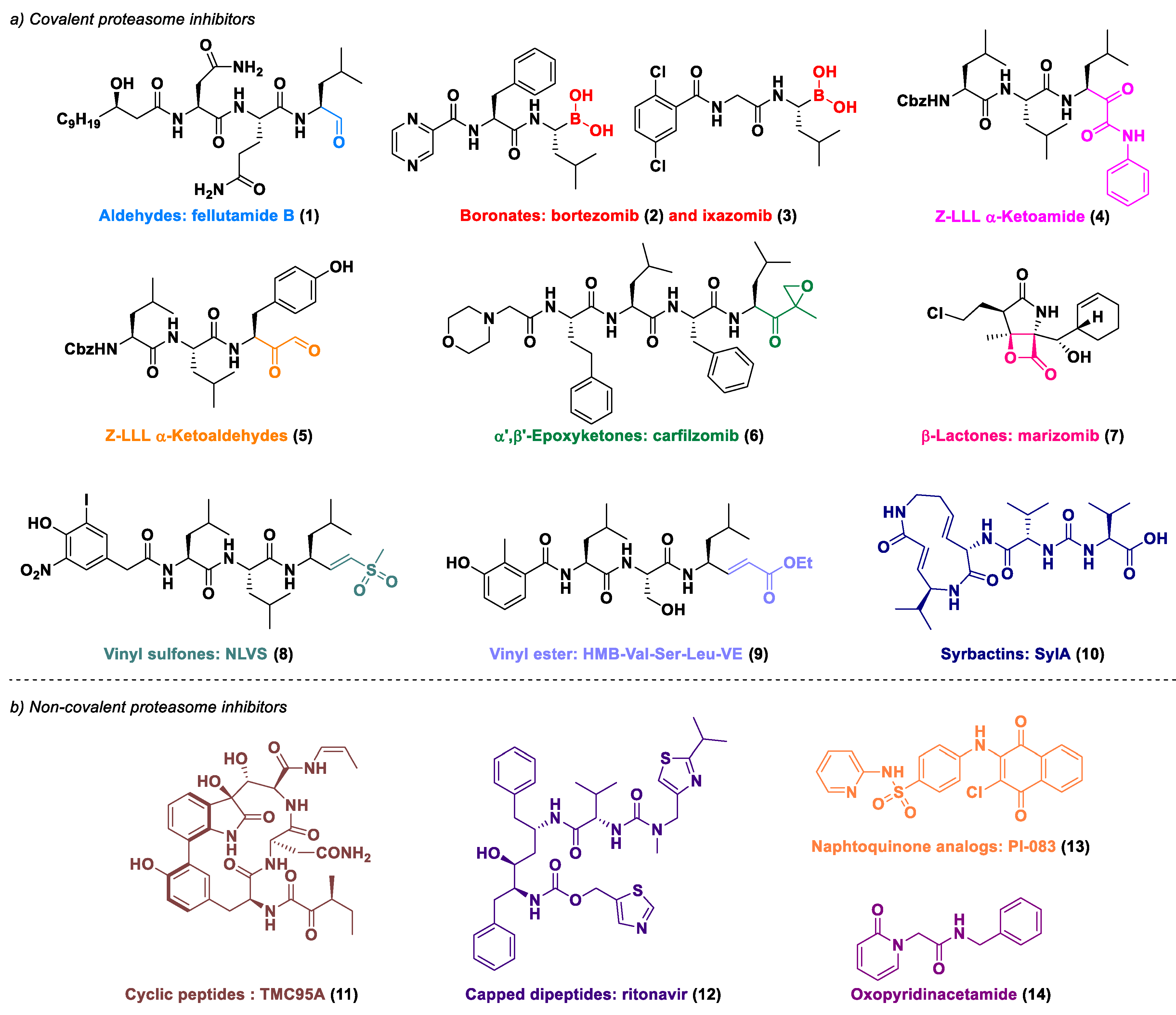
1. Introduction
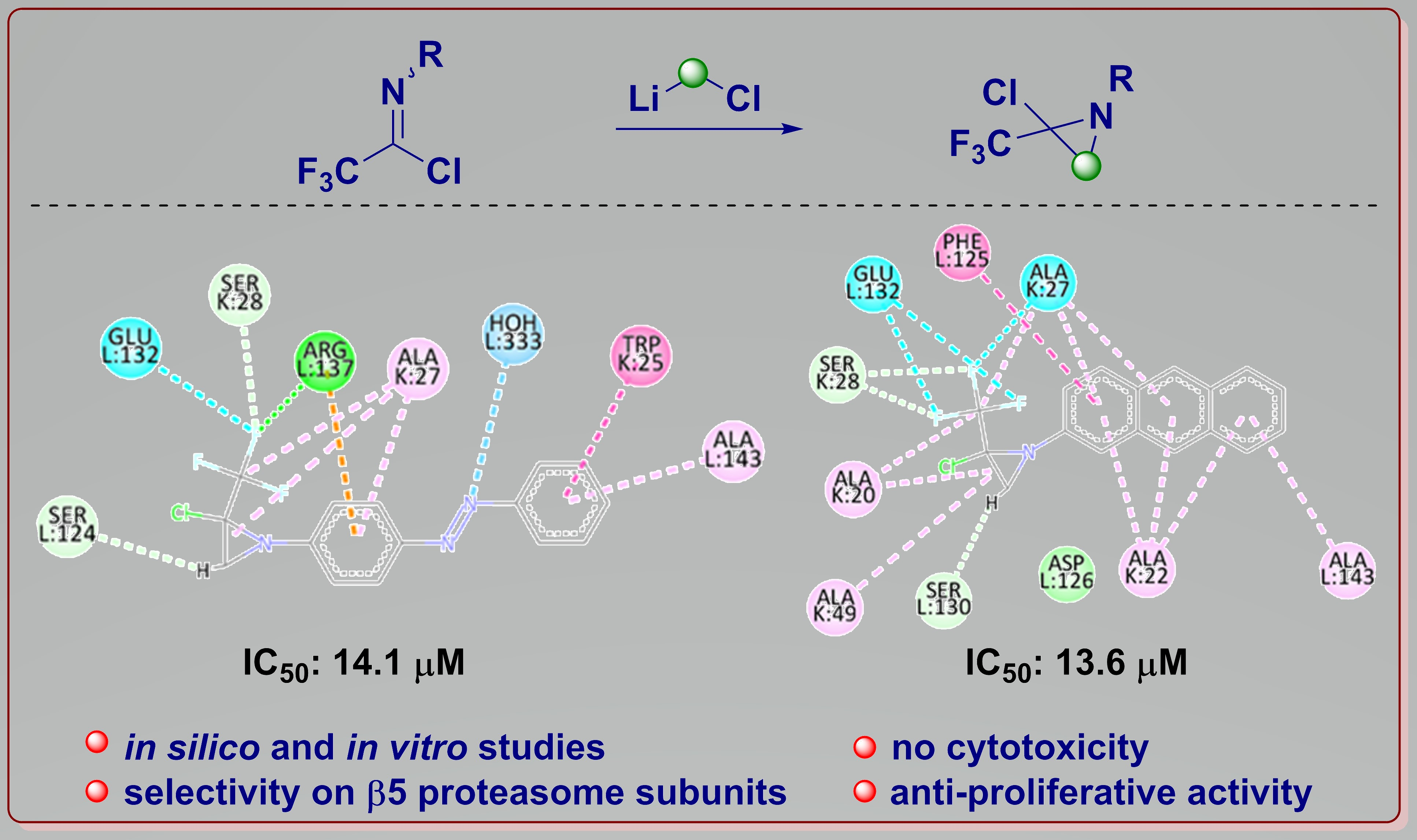
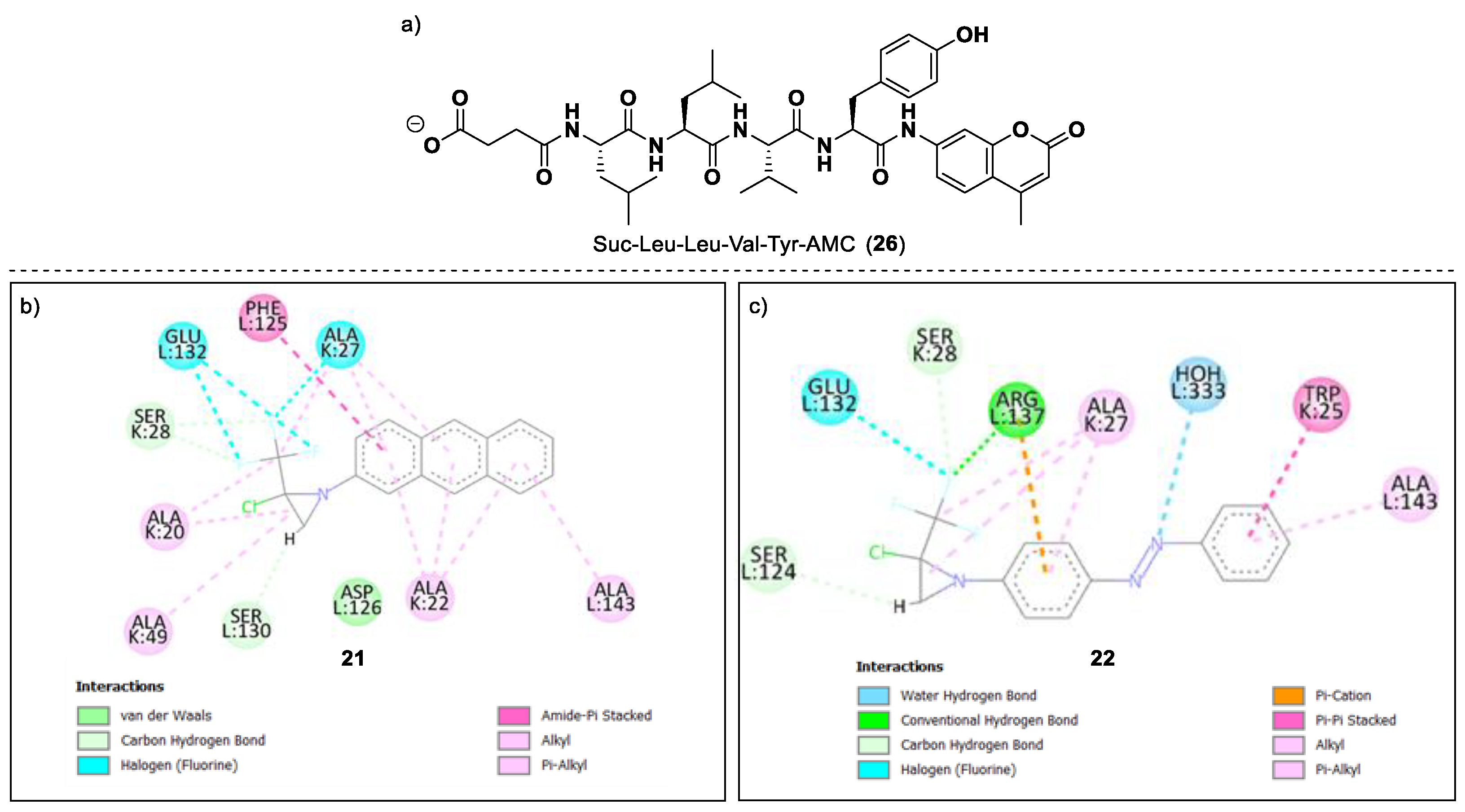
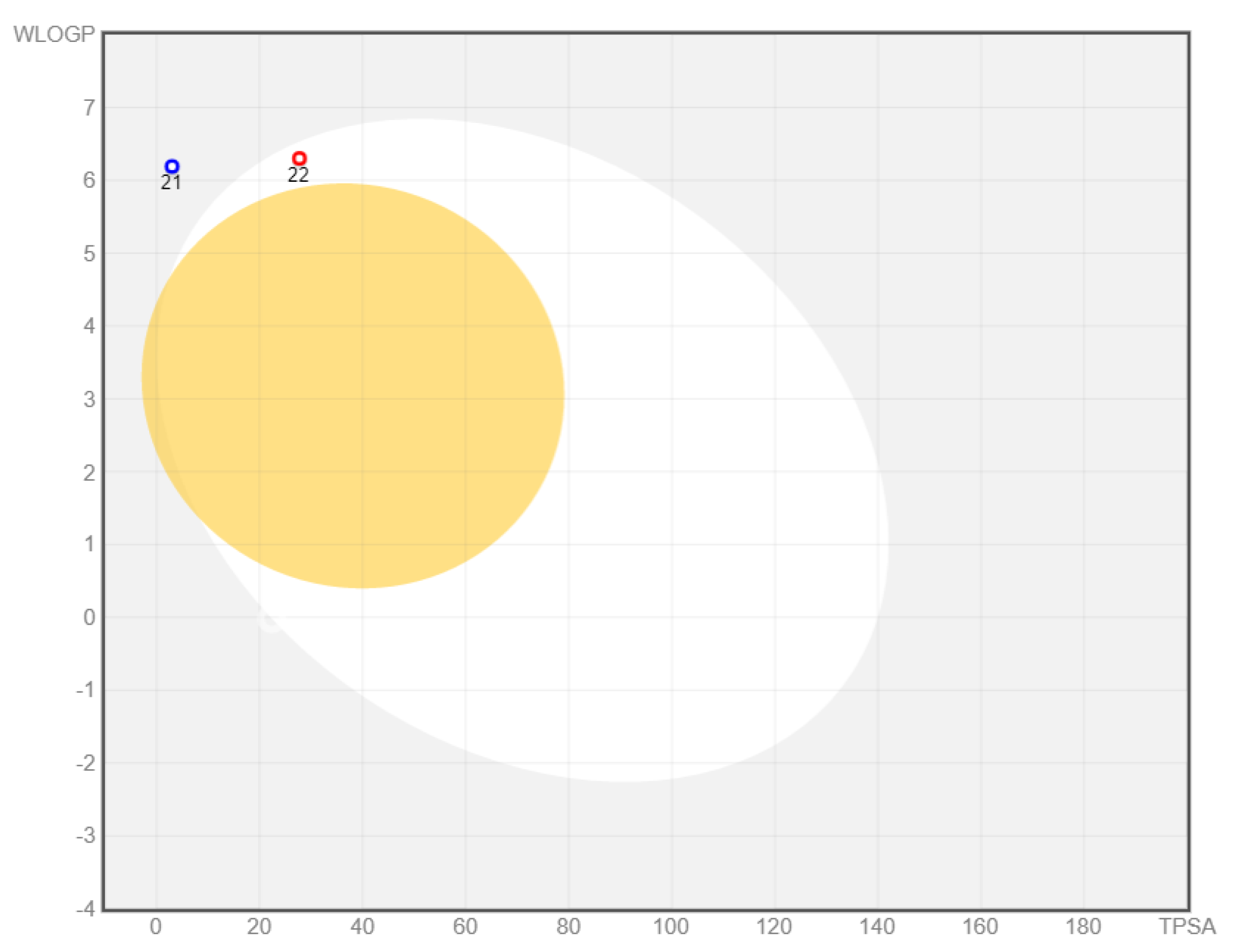
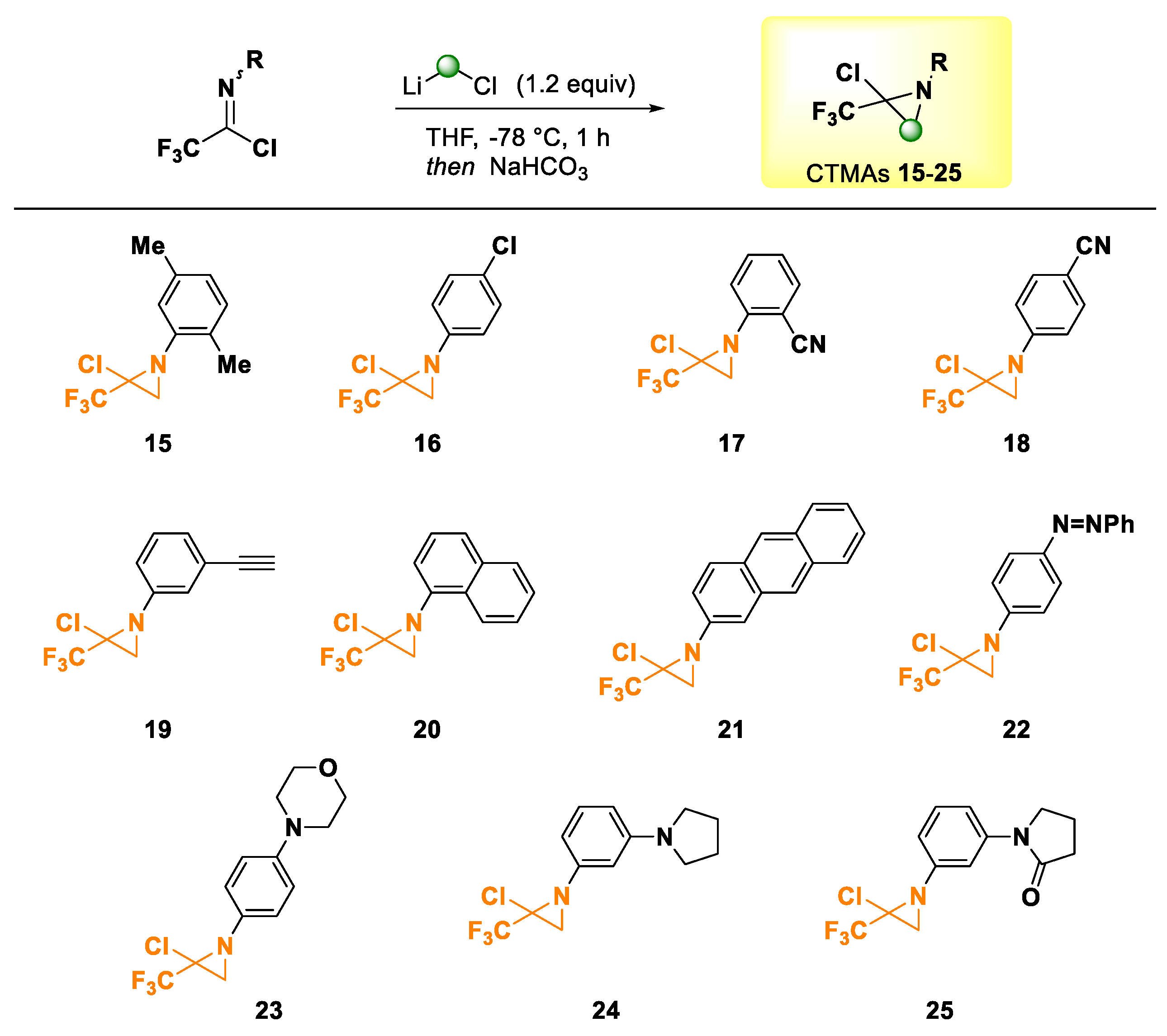
2. Results and Discussion
3. Materials and Methods
3.1. General Methods
3.2. General Procedures
3.2.1. General Procedure for Preparing Trifluoroacetimidoyl Chlorides [79]
3.2.2. General Procedure for Preparing Chloro-Trifluoromethylaziridines (CTMAs) [56]
3.3. In Silico Studies
3.4. Biological Assays
3.4.1. Inhibition Assay for the Chymotrypsin-like Activity of the 20S Proteasome
3.4.2. Inhibition Assay for the Post-Glutamyl Peptide Hydrolyzing (PGPH or Caspase-like) Activity of the 20S Proteasome
3.4.3. Inhibition Assay for Trypsin-like Activity of the 20S Proteasome
3.4.4. Cell Culture
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Leonardo-Sousa, C.; Carvalho, A.N.; Guedes, R.A.; Fernandes, P.M.P.; Aniceto, N.; Salvador, J.A.R.; Gama, M.J.; Guedes, R.C. Revisiting Proteasome Inhibitors: Molecular Underpinnings of Their Development, Mechanisms of Resistance and Strategies to Overcome Anti-Cancer Drug Resistance. Molecules 2022, 27, 2201. [Google Scholar] [CrossRef]
- Kisselev, A.F.; Goldberg, A.L. Proteasome inhibitors: From research tools to drug candidates. Chem. Biol. 2001, 8, 739–758. [Google Scholar] [CrossRef] [Green Version]
- Thompson, S.J.; Loftus, L.T.; Ashley, M.D.; Meller, R. Ubiquitin-proteasome system as a modulator of cell fate. Curr. Opin. Pharmacol. 2008, 8, 90–95. [Google Scholar] [CrossRef] [Green Version]
- Bhat, K.P.; Greer, S.F. Proteolytic and non-proteolytic roles of ubiquitin and the ubiquitin proteasome system in transcriptional regulation. Biochim. Biophys. Acta 2011, 1809, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L.; Gramm, C.; Rothstein, L.; Clark, K.; Stein, R.; Dick, L.; Hwang, D.; Goldberg, A.L. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 1994, 78, 761–771. [Google Scholar] [CrossRef]
- Craiu, A.; Gaczynska, M.; Akopian, T.; Gramm, C.F.; Fenteany, G.; Goldberg, A.L.; Rock, K.L. Lactacystin and clasto-lactacystin beta-lactone modify multiple proteasome beta-subunits and inhibit intracellular protein degradation and major histocompatibility complex class I antigen presentation. J. Biol. Chem. 1997, 272, 13437–13445. [Google Scholar] [CrossRef] [Green Version]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef]
- Ciechanover, A.; Brundin, P. The ubiquitin proteasome system in neurodegenerative diseases: Sometimes the chicken, sometimes the egg. Neuron 2003, 40, 427–446. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, A.L.; Ciechanover, A. Targeting proteins for destruction by the ubiquitin system: Implications for human pathobiology. Annu Rev. Pharmacol. Toxicol. 2009, 49, 73–96. [Google Scholar] [CrossRef]
- Unno, M.; Mizushima, T.; Morimoto, Y.; Tomisugi, Y.; Tanaka, K.; Yasuoka, N.; Tsukihara, T. The structure of the mammalian 20S proteasome at 2.75 A resolution. Structure 2002, 10, 609–618. [Google Scholar] [CrossRef] [Green Version]
- Demartino, G.N.; Gillette, T.G. Proteasomes: Machines for all reasons. Cell 2007, 129, 659–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kisselev, A.F.; Callard, A.; Goldberg, A.L. Importance of the different proteolytic sites of the proteasome and the efficacy of inhibitors varies with the protein substrate. J. Biol. Chem. 2006, 281, 8582–8590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacco, A.; Aujay, M.; Morgan, B.; Azab, A.K.; Maiso, P.; Liu, Y.; Zhang, Y.; Azab, F.; Ngo, H.T.; Issa, G.C.; et al. Carfilzomib-dependent selective inhibition of the chymotrypsin-like activity of the proteasome leads to antitumor activity in Waldenstrom’s Macroglobulinemia. Clin. Cancer Res. 2011, 17, 1753–1764. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, A.L. Development of proteasome inhibitors as research tools and cancer drugs. J. Cell. Biol. 2012, 199, 583–588. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.M.; Yu, Y.; Cheng, Y. Structure characterization of the 26S proteasome. Biochim. Biophys. Acta 2011, 1809, 67–79. [Google Scholar] [CrossRef] [Green Version]
- Kisselev, A.F.; van der Linden, W.A.; Overkleeft, H.S. Proteasome inhibitors: An expanding army attacking a unique target. Chem. Biol. 2012, 19, 99–115. [Google Scholar] [CrossRef] [Green Version]
- Groll, M.; Ditzel, L.; Lowe, J.; Stock, D.; Bochtler, M.; Bartunik, H.D.; Huber, R. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature 1997, 386, 463–471. [Google Scholar] [CrossRef]
- Weissman, A.M.; Shabek, N.; Ciechanover, A. The predator becomes the prey: Regulating the ubiquitin system by ubiquitylation and degradation. Nat. Rev. Mol. Cell. Biol. 2011, 12, 605–620. [Google Scholar] [CrossRef]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef]
- Groll, M.; Berkers, C.R.; Ploegh, H.L.; Ovaa, H. Crystal structure of the boronic acid-based proteasome inhibitor bortezomib in complex with the yeast 20S proteasome. Structure 2006, 14, 451–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, J.; Behnke, M.; Chen, S.; Cruickshank, A.A.; Dick, L.R.; Grenier, L.; Klunder, J.M.; Ma, Y.T.; Plamondon, L.; Stein, R.L. Potent and selective inhibitors of the proteasome: Dipeptidyl boronic acids. Bioorg. Med. Chem. Lett. 1998, 8, 333–338. [Google Scholar] [CrossRef]
- Dou, Q.P.; Zonder, J.A. Overview of proteasome inhibitor-based anti-cancer therapies: Perspective on bortezomib and second generation proteasome inhibitors versus future generation inhibitors of ubiquitin-proteasome system. Curr. Cancer Drug Targets 2014, 14, 517–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Frezza, M.; Schmitt, S.; Kanwar, J.; Dou, Q.P. Bortezomib as the first proteasome inhibitor anticancer drug: Current status and future perspectives. Curr. Cancer Drug Targets 2011, 11, 239–253. [Google Scholar] [CrossRef] [Green Version]
- Britton, M.; Lucas, M.M.; Downey, S.L.; Screen, M.; Pletnev, A.A.; Verdoes, M.; Tokhunts, R.A.; Amir, O.; Goddard, A.L.; Pelphrey, P.M.; et al. Selective inhibitor of proteasome’s caspase-like sites sensitizes cells to specific inhibition of chymotrypsin-like sites. Chem. Biol. 2009, 16, 1278–1289. [Google Scholar] [CrossRef] [Green Version]
- Screen, M.; Britton, M.; Downey, S.L.; Verdoes, M.; Voges, M.J.; Blom, A.E.; Geurink, P.P.; Risseeuw, M.D.; Florea, B.I.; van der Linden, W.A.; et al. Nature of pharmacophore influences active site specificity of proteasome inhibitors. J. Biol. Chem. 2010, 285, 40125–40134. [Google Scholar] [CrossRef] [Green Version]
- Mirabella, A.C.; Pletnev, A.A.; Downey, S.L.; Florea, B.I.; Shabaneh, T.B.; Britton, M.; Verdoes, M.; Filippov, D.V.; Overkleeft, H.S.; Kisselev, A.F. Specific cell-permeable inhibitor of proteasome trypsin-like sites selectively sensitizes myeloma cells to bortezomib and carfilzomib. Chem. Biol. 2011, 18, 608–618. [Google Scholar] [CrossRef]
- Parlati, F.; Lee, S.J.; Aujay, M.; Suzuki, E.; Levitsky, K.; Lorens, J.B.; Micklem, D.R.; Ruurs, P.; Sylvain, C.; Lu, Y.; et al. Carfilzomib can induce tumor cell death through selective inhibition of the chymotrypsin-like activity of the proteasome. Blood 2009, 114, 3439–3447. [Google Scholar] [CrossRef] [Green Version]
- Beck, P.; Dubiella, C.; Groll, M. Covalent and non-covalent reversible proteasome inhibition. Biol. Chem. 2012, 393, 1101–1120. [Google Scholar] [CrossRef]
- Rock, K.L.; York, I.A.; Saric, T.; Goldberg, A.L. Protein degradation and the generation of MHC class I-presented peptides. Adv. Immunol. 2002, 80, 1–70. [Google Scholar]
- Micale, N.; Scarbaci, K.; Troiano, V.; Ettari, R.; Grasso, S.; Zappala, M. Peptide-based proteasome inhibitors in anticancer drug design. Med. Res. Rev. 2014, 34, 1001–1069. [Google Scholar] [CrossRef] [PubMed]
- Harer, S.L.; Bhatia, M.S.; Bhatia, N.M. Proteasome inhibitors mechanism; source for design of newer therapeutic agents. J. Antibiot. 2012, 65, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Borissenko, L.; Groll, M. 20S proteasome and its inhibitors: Crystallographic knowledge for drug development. Chem. Rev. 2007, 107, 687–717. [Google Scholar] [CrossRef] [PubMed]
- Huber, E.M.; Groll, M. Inhibitors for the immuno- and constitutive proteasome: Current and future trends in drug development. Angew. Chem. Int. Ed. Engl. 2012, 51, 8708–8720. [Google Scholar] [CrossRef]
- Han, L.Q.; Yuan, X.; Wu, X.Y.; Li, R.D.; Xu, B.; Cheng, Q.; Liu, Z.M.; Zhou, T.Y.; An, H.Y.; Wang, X.; et al. Urea-containing peptide boronic acids as potent proteasome inhibitors. Eur. J. Med. Chem. 2017, 125, 925–939. [Google Scholar] [CrossRef] [PubMed]
- Ruschak, A.M.; Slassi, M.; Kay, L.E.; Schimmer, A.D. Novel proteasome inhibitors to overcome bortezomib resistance. J. Natl. Cancer Inst. 2011, 103, 1007–1017. [Google Scholar] [CrossRef] [Green Version]
- Schrader, J.; Henneberg, F.; Mata, R.A.; Tittmann, K.; Schneider, T.R.; Stark, H.; Bourenkov, G.; Chari, A. The inhibition mechanism of human 20S proteasomes enables next-generation inhibitor design. Science 2016, 353, 594–598. [Google Scholar] [CrossRef] [Green Version]
- Stein, M.L.; Cui, H.; Beck, P.; Dubiella, C.; Voss, C.; Kruger, A.; Schmidt, B.; Groll, M. Systematic comparison of peptidic proteasome inhibitors highlights the alpha-ketoamide electrophile as an auspicious reversible lead motif. Angew. Chem. Int. Ed. Engl. 2014, 53, 1679–1683. [Google Scholar] [CrossRef]
- Grawert, M.A.; Gallastegui, N.; Stein, M.; Schmidt, B.; Kloetzel, P.M.; Huber, R.; Groll, M. Elucidation of the alpha-keto-aldehyde binding mechanism: A lead structure motif for proteasome inhibition. Angew. Chem. Int. Ed. Engl. 2011, 50, 542–544. [Google Scholar] [CrossRef]
- Groll, M.; Nazif, T.; Huber, R.; Bogyo, M. Probing structural determinants distal to the site of hydrolysis that control substrate specificity of the 20S proteasome. Chem. Biol. 2002, 9, 655–662. [Google Scholar] [CrossRef] [Green Version]
- Bota, D.A.; Mason, W.; Kesari, S.; Magge, R.; Winograd, B.; Elias, I.; Reich, S.D.; Levin, N.; Trikha, M.; Desjardins, A. Marizomib alone or in combination with bevacizumab in patients with recurrent glioblastoma: Phase I/II clinical trial data. Neurooncol. Adv. 2021, 3, vdab142. [Google Scholar] [CrossRef] [PubMed]
- Imbach, P.; Lang, M.; Garcia-Echeverria, C.; Guagnano, V.; Noorani, M.; Roesel, J.; Bitsch, F.; Rihs, G.; Furet, P. Novel beta-lactam derivatives: Potent and selective inhibitors of the chymotrypsin-like activity of the human 20S proteasome. Bioorg. Med. Chem. Lett. 2007, 17, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Rydzewski, R.M.; Burrill, L.; Mendonca, R.; Palmer, J.T.; Rice, M.; Tahilramani, R.; Bass, K.E.; Leung, L.; Gjerstad, E.; Janc, J.W.; et al. Optimization of subsite binding to the beta5 subunit of the human 20S proteasome using vinyl sulfones and 2-keto-1,3,4-oxadiazoles: Syntheses and cellular properties of potent, selective proteasome inhibitors. J. Med. Chem. 2006, 49, 2953–2968. [Google Scholar] [CrossRef] [PubMed]
- Ettari, R.; Bonaccorso, C.; Micale, N.; Heindl, C.; Schirmeister, T.; Calabro, M.L.; Grasso, S.; Zappala, M. Development of novel peptidomimetics containing a vinyl sulfone moiety as proteasome inhibitors. ChemMedChem 2011, 6, 1228–1237. [Google Scholar] [CrossRef] [PubMed]
- Van der Linden, W.A.; Willems, L.I.; Shabaneh, T.B.; Li, N.; Ruben, M.; Florea, B.I.; van der Marel, G.A.; Kaiser, M.; Kisselev, A.F.; Overkleeft, H.S. Discovery of a potent and highly beta1 specific proteasome inhibitor from a focused library of urea-containing peptide vinyl sulfones and peptide epoxyketones. Org. Biomol. Chem. 2012, 10, 181–194. [Google Scholar] [CrossRef] [Green Version]
- Baldisserotto, A.; Destro, F.; Vertuani, G.; Marastoni, M.; Gavioli, R.; Tomatis, R. N-terminal-prolonged vinyl ester-based peptides as selective proteasome beta1 subunit inhibitors. Bioorg. Med. Chem. 2009, 17, 5535–5540. [Google Scholar] [CrossRef]
- Clerc, J.; Schellenberg, B.; Groll, M.; Bachmann, A.S.; Huber, R.; Dudler, R.; Kaiser, M. Convergent Synthesis and Biological Evaluation of Syringolin A and Derivatives as Eukaryotic 20S Proteasome Inhibitors. Eur. J. Org. Chem. 2010, 2010, 3991–4003. [Google Scholar] [CrossRef]
- Groll, M.; Gallastegui, N.; Marechal, X.; Le Ravalec, V.; Basse, N.; Richy, N.; Genin, E.; Huber, R.; Moroder, L.; Vidal, J.; et al. 20S proteasome inhibition: Designing noncovalent linear peptide mimics of the natural product TMC-95A. ChemMedChem 2010, 5, 1701–1705. [Google Scholar] [CrossRef]
- Genin, E.; Reboud-Ravaux, M.; Vidal, J. Proteasome inhibitors: Recent advances and new perspectives in medicinal chemistry. Curr. Top. Med. Chem. 2010, 10, 232–256. [Google Scholar] [CrossRef] [Green Version]
- Mishto, M.; Bellavista, E.; Santoro, A.; Stolzing, A.; Ligorio, C.; Nacmias, B.; Spazzafumo, L.; Chiappelli, M.; Licastro, F.; Sorbi, S.; et al. Immunoproteasome and LMP2 polymorphism in aged and Alzheimer’s disease brains. Neurobiol. Aging 2006, 27, 54–66. [Google Scholar] [CrossRef]
- Kohno, J.; Koguchi, Y.; Nishio, M.; Nakao, K.; Kuroda, M.; Shimizu, R.; Ohnuki, T.; Komatsubara, S. Structures of TMC-95A-D: Novel proteasome inhibitors from Apiospora montagnei sacc. TC 1093. J. Org. Chem. 2000, 65, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, M.; Groll, M.; Renner, C.; Huber, R.; Moroder, L. The core structure of TMC-95A is a promising lead for reversible proteasome inhibition. Angew. Chem. Int. Ed. Engl. 2002, 41, 780–783. [Google Scholar] [CrossRef]
- Song, R.; Qiao, W.; He, J.; Huang, J.; Luo, Y.; Yang, T. Proteases and Their Modulators in Cancer Therapy: Challenges and Opportunities. J. Med. Chem. 2021, 64, 2851–2877. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.S. Synthetic Aziridines in Medicinal Chemistry: A Mini-Review. Mini Rev. Med. Chem. 2016, 16, 892–904. [Google Scholar] [CrossRef] [PubMed]
- Vega-Perez, J.M.; Palo-Nieto, C.; Vega-Holm, M.; Gongora-Vargas, P.; Calderon-Montano, J.M.; Burgos-Moron, E.; Lopez-Lazaro, M.; Iglesias-Guerra, F. Aziridines from alkenyl-beta-D-galactopyranoside derivatives: Stereoselective synthesis and in vitro selective anticancer activity. Eur. J. Med. Chem. 2013, 70, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Ielo, L.; Touqeer, S.; Roller, A.; Langer, T.; Holzer, W.; Pace, V. Telescoped, Divergent, Chemoselective C1 and C1-C1 Homologation of Imine Surrogates: Access to Quaternary Chloro- and Halomethyl-Trifluoromethyl Aziridines. Angew. Chem. Int. Edit. 2019, 58, 2479–2484. [Google Scholar] [CrossRef]
- Miele, M.; Citarella, A.; Langer, T.; Urban, E.; Zehl, M.; Holzer, W.; Ielo, L.; Pace, V. Chemoselective Homologation-Deoxygenation Strategy Enabling the Direct Conversion of Carbonyls into (n+1)-Halomethyl-Alkanes. Org. Lett. 2020, 22, 7629–7634. [Google Scholar] [CrossRef]
- Citarella, A.; Gentile, D.; Rescifina, A.; Piperno, A.; Mognetti, B.; Gribaudo, G.; Sciortino, M.T.; Holzer, W.; Pace, V.; Micale, N. Pseudo-Dipeptide Bearing α,α-Difluoromethyl Ketone Moiety as Electrophilic Warhead with Activity against Coronaviruses. Int. J. Mol. Sci. 2021, 22, 1398. [Google Scholar] [CrossRef]
- Citarella, A.; Micale, N. Peptidyl Fluoromethyl Ketones and Their Applications in Medicinal Chemistry. Molecules 2020, 25, 4031. [Google Scholar] [CrossRef]
- Miele, M.; Citarella, A.; Micale, N.; Holzer, W.; Pace, V. Direct and Chemoselective Synthesis of Tertiary Difluoroketones via Weinreb Amide Homologation with a CHF2-Carbene Equivalent. Org. Lett. 2019, 21, 8261–8265. [Google Scholar] [CrossRef]
- Trivella, D.B.; Pereira, A.R.; Stein, M.L.; Kasai, Y.; Byrum, T.; Valeriote, F.A.; Tantillo, D.J.; Groll, M.; Gerwick, W.H.; Moore, B.S. Enzyme inhibition by hydroamination: Design and mechanism of a hybrid carmaphycin-syringolin enone proteasome inhibitor. Chem. Biol. 2014, 21, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Scarbaci, K.; Troiano, V.; Micale, N.; Ettari, R.; Tamborini, L.; Di Giovanni, C.; Cerchia, C.; Grasso, S.; Novellino, E.; Schirmeister, T.; et al. Identification of a new series of amides as non-covalent proteasome inhibitors. Eur. J. Med. Chem. 2014, 76, 1–9. [Google Scholar] [CrossRef]
- Troiano, V.; Scarbaci, K.; Ettari, R.; Micale, N.; Cerchia, C.; Pinto, A.; Schirmeister, T.; Novellino, E.; Grasso, S.; Lavecchia, A.; et al. Optimization of peptidomimetic boronates bearing a P3 bicyclic scaffold as proteasome inhibitors. Eur. J. Med. Chem. 2014, 83, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Micale, N.; Schirmeister, T.; Ettari, R.; Cinellu, M.A.; Maiore, L.; Serratrice, M.; Gabbiani, C.; Massai, L.; Messori, L. Selected cytotoxic gold compounds cause significant inhibition of 20S proteasome catalytic activities. J. Inorg. Biochem. 2014, 141, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Scarbaci, K.; Troiano, V.; Ettari, R.; Pinto, A.; Micale, N.; Di Giovanni, C.; Cerchia, C.; Schirmeister, T.; Novellino, E.; Lavecchia, A.; et al. Development of novel selective peptidomimetics containing a boronic acid moiety, targeting the 20S proteasome as anticancer agents. ChemMedChem 2014, 9, 1801–1816. [Google Scholar] [CrossRef] [PubMed]
- Micale, N.; Ettari, R.; Lavecchia, A.; Di Giovanni, C.; Scarbaci, K.; Troiano, V.; Grasso, S.; Novellino, E.; Schirmeister, T.; Zappala, M. Development of peptidomimetic boronates as proteasome inhibitors. Eur. J. Med. Chem. 2013, 64, 23–34. [Google Scholar] [CrossRef]
- Kisselev, A.F.; Goldberg, A.L. Monitoring activity and inhibition of 26S proteasomes with fluorogenic peptide substrates. Methods Enzymol. 2005, 398, 364–378. [Google Scholar]
- Abdelfatah, S.; Bockers, M.; Asensio, M.; Kadioglu, O.; Klinger, A.; Fleischer, E.; Efferth, T. Isopetasin and S-isopetasin as novel P-glycoprotein inhibitors against multidrug-resistant cancer cells. Phytomedicine 2021, 86, 153196. [Google Scholar] [CrossRef]
- Mahmoud, N.; Saeed, M.E.M.; Sugimoto, Y.; Klinger, A.; Fleischer, E.; Efferth, T. Putative molecular determinants mediating sensitivity or resistance towards carnosic acid tumor cell responses. Phytomedicine 2020, 77, 153271. [Google Scholar] [CrossRef]
- O’Brien, J.; Wilson, I.; Orton, T.; Pognan, F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 2000, 267, 5421–5426. [Google Scholar] [CrossRef]
- Kimmig, A.; Gekeler, V.; Neumann, M.; Frese, G.; Handgretinger, R.; Kardos, G.; Diddens, H.; Niethammer, D. Susceptibility of Multidrug-Resistant Human Leukemia-Cell Lines to Human Interleukin 2-Activated Killer-Cells. Cancer Res. 1990, 50, 6793–6799. [Google Scholar]
- Efferth, T.; Sauerbrey, A.; Olbrich, A.; Gebhart, E.; Rauch, P.; Weber, H.O.; Hengstler, J.G.; Halatsch, M.E.; Volm, M.; Tew, K.D.; et al. Molecular modes of action of artesunate in tumor cell lines. Mol. Pharmacol. 2003, 64, 382–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efferth, T.; Konkimalla, V.B.; Wang, Y.F.; Sauerbrey, A.; Meinhardt, S.; Zintl, F.; Mattern, J.; Volm, M. Prediction of broad spectrum resistance of tumors towards anticancer drugs. Clin. Cancer Res. 2008, 14, 2405–2412. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- Castoldi, L.; Holzer, W.; Langer, T.; Pace, V. Evidence and isolation of tetrahedral intermediates formed upon the addition of lithium carbenoids to Weinreb amides and N-acylpyrroles. Chem. Commun. 2017, 53, 9498–9501. [Google Scholar] [CrossRef] [Green Version]
- Suffert, J. Simple Direct Titration of Organolithium Reagents Using N-Pivaloyl-o -toluidine and/or N-pivaloyl-o-benzylaniline. J. Org. Chem. 1989, 54, 509–510. [Google Scholar] [CrossRef]
- Uneyama, K.; Amii, H.; Katagiri, T.; Kobayashi, T.; Hosokawa, T. A rich chemistry of fluorinated imidoyl halides. J. Fluor. 2005, 126, 165–171. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | ΔG Vina | Ki Calc. (μM) | ChT-L Activity (% Inhibition at 20 μM) 1 | IC50/Ki (μM) ChT-L 2 |
|---|---|---|---|---|
| 15 | −6.91 | 8.56 | <20 | - |
| 16 | −7.18 | 5.42 | 25 ± 1.1 | - |
| 17 | −7.07 | 6.53 | <20 | - |
| 18 | −7.64 | 2.49 | 37 ± 0.5 | - |
| 19 | −7.40 | 3.74 | <20 | - |
| 20 | −7.89 | 1.64 | <20 | - |
| 21 | −8.31 | 0.80 | 68 ± 0.4 | 13.6 ± 1.1/1.6 ± 0.13 |
| 22 | −8.59 | 0.50 | 67 ± 0.1 | 14.1 ± 0.7/1.6 ± 0.08 |
| 23 | −7.88 | 1.66 | 41 ± 1.2 | - |
| 24 | −7.85 | 1.75 | 27 ± 0.3 | - |
| 25 | −8.11 | 1.31 | 34 ± 1.4 | - |
| 26 | −8.75 | 0.38 | - | - |
| Compound | CCRF-CEM IC50 (µM) | CEM/ADR5000 IC50 (µM) | PBMCs CC50 (µM) | Degree of Resistance |
|---|---|---|---|---|
| 15 | >100 | >100 | - | - |
| 16 | 55.03 ± 1.33 | >100 | - | - |
| 17 | >100 | >100 | - | - |
| 18 | >100 | >100 | - | - |
| 19 | >100 | >100 | - | - |
| 20 | >100 | >100 | - | - |
| 21 | 25.45 ± 4.08 | 24.08 ± 4.05 | >100 | 0.95 |
| 22 | 32.82 ± 1.28 | 67.72 ± 8.80 | >100 | 2.06 |
| 23 | 56.95 ± 2.20 | >100 | - | - |
| 24 | 42.15 ± 1.86 | >100 | - | - |
| 25 | 82.41 ± 3.50 | 67.97 ± 3.20 | - | 0.82 |
| Doxorubicin | 0.044 ± 0.01 | 20.50 ± 2.59 | - | 463.90 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ielo, L.; Patamia, V.; Citarella, A.; Efferth, T.; Shahhamzehei, N.; Schirmeister, T.; Stagno, C.; Langer, T.; Rescifina, A.; Micale, N.; et al. Novel Class of Proteasome Inhibitors: In Silico and In Vitro Evaluation of Diverse Chloro(trifluoromethyl)aziridines. Int. J. Mol. Sci. 2022, 23, 12363. https://doi.org/10.3390/ijms232012363
Ielo L, Patamia V, Citarella A, Efferth T, Shahhamzehei N, Schirmeister T, Stagno C, Langer T, Rescifina A, Micale N, et al. Novel Class of Proteasome Inhibitors: In Silico and In Vitro Evaluation of Diverse Chloro(trifluoromethyl)aziridines. International Journal of Molecular Sciences. 2022; 23(20):12363. https://doi.org/10.3390/ijms232012363
Chicago/Turabian StyleIelo, Laura, Vincenzo Patamia, Andrea Citarella, Thomas Efferth, Nasim Shahhamzehei, Tanja Schirmeister, Claudio Stagno, Thierry Langer, Antonio Rescifina, Nicola Micale, and et al. 2022. "Novel Class of Proteasome Inhibitors: In Silico and In Vitro Evaluation of Diverse Chloro(trifluoromethyl)aziridines" International Journal of Molecular Sciences 23, no. 20: 12363. https://doi.org/10.3390/ijms232012363