
Maternal Fructose Intake Causes Developmental Reprogramming of Hepatic Mitochondrial Catalytic Activity and Lipid Metabolism in Weanling and Young Adult Offspring

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics
2.2. Experimental Diets
2.3. Animal Model
2.4. Determination of Body Composition
2.5. Offspring Oral Glucose Tolerance Tests and Blood Glucose Analysis
2.6. Offspring Whole Blood Free Fatty Acids Analysis
2.7. Offspring Visceral Adipose and Liver Tissues Collection
2.8. Fixation of Visceral Adipose and Liver Tissues with Formalin
2.9. Plasma Metabolite Analysis
2.10. Determination of Hepatic Triglycerides
2.11. Protein Extraction from Liver Tissues
2.12. Protein Precipitation Digestion and Peptide Purification
2.13. LC–MS/MS
2.14. Protein Identification
2.15. Label-Free Quantitation of Proteins
2.16. Ingenuity Pathway Analysis
2.17. Western Blot
2.18. Statistical Analysis
3. Results
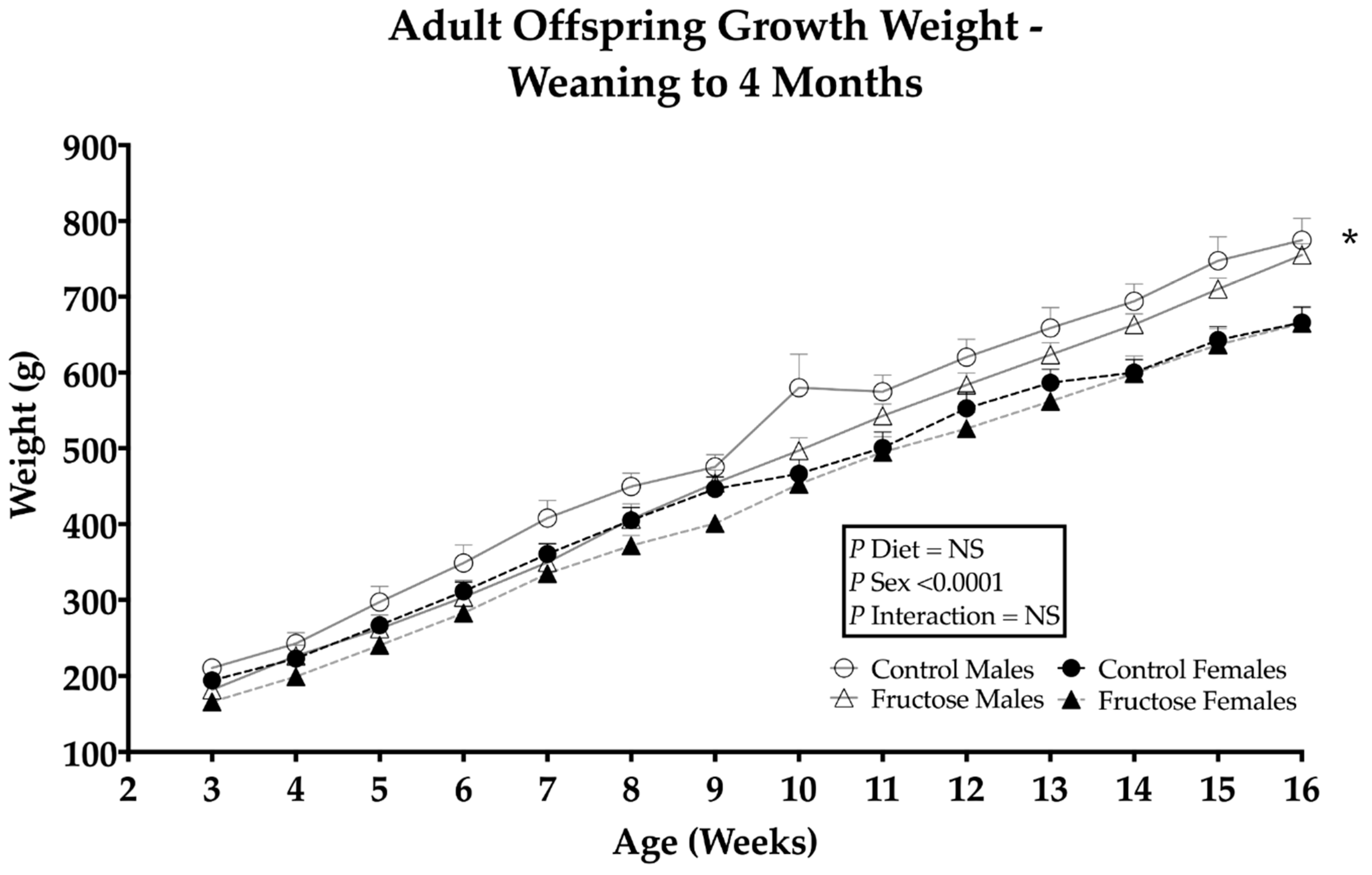
3.1. Effects of Maternal Fructose Intake on Adult Offspring Growth and Weight Gain
3.2. Effects of Excess Maternal Fructose Intake on Adult DXA Body Composition
3.3. The Effects of Excess Maternal Fructose Intake on Adult OGTT Glucose and Insulin
3.4. Effects of Excess Maternal Fructose Intake on Adult Plasma Metabolites
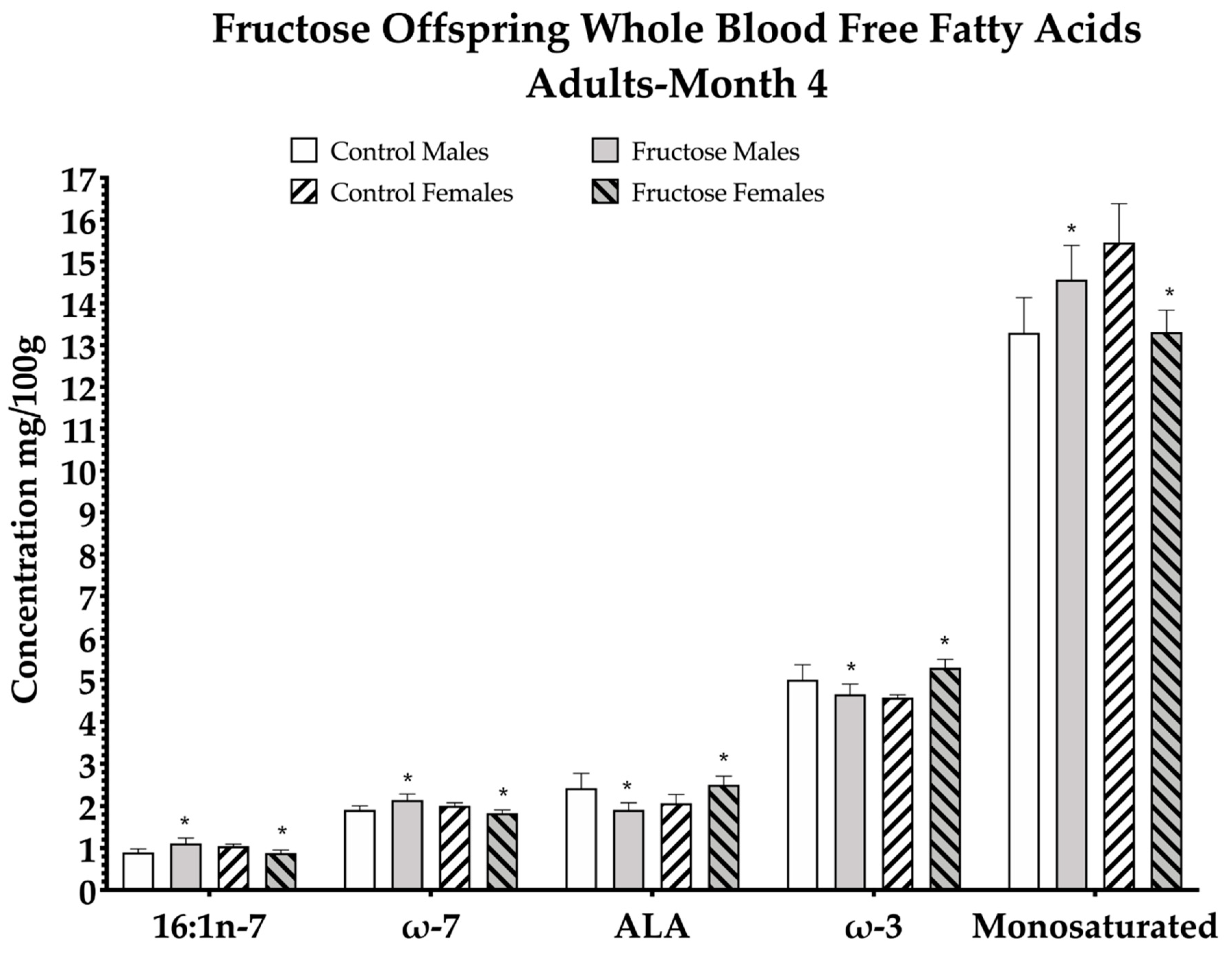
3.5. Effects of Excess Maternal Fructose Intake on Adult Whole Blood Fatty Acid Composition
3.6. Effects of Excess Maternal Fructose Intake on Offspring Hepatic Lipid Deposition
3.7. Adult Offspring Visceral Fat Morphology
3.8. Proteomics and Ingenuity Pathways Analysis
3.8.1. Weanling Males
3.8.2. Weanling Females
3.8.3. Adult Males
3.8.4. Adult Females
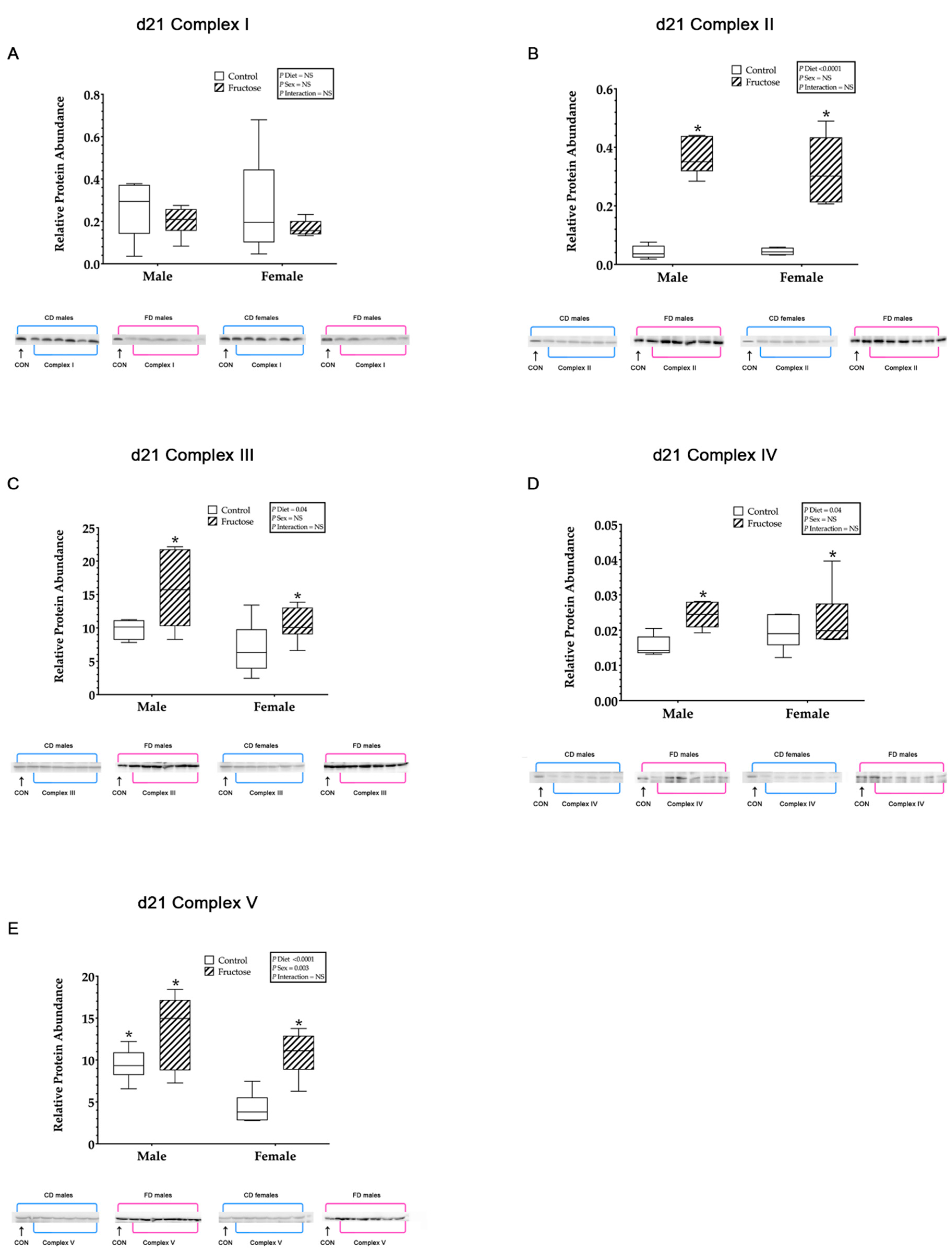
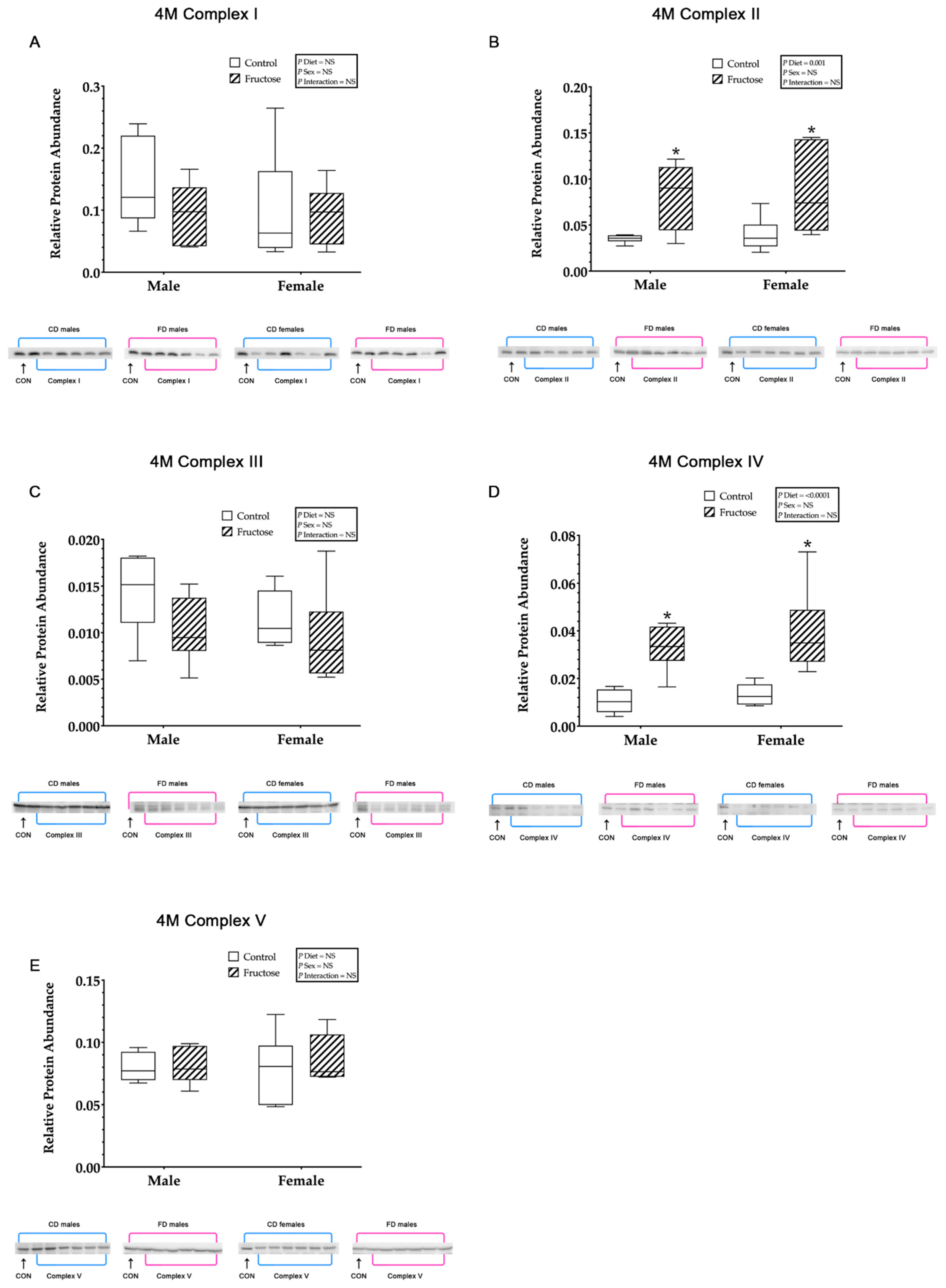
3.9. Western Blot Analysis
3.9.1. Effects of Excess Maternal Fructose Intake on Key Hepatic Proteins
3.9.2. Mitochondrial ETC Complex I
3.9.3. Mitochondrial ETC Complex II
3.9.4. Mitochondrial ETC Complex III
3.9.5. Mitochondrial ETC Complex IV
3.9.6. Mitochondrial ATP Synthase
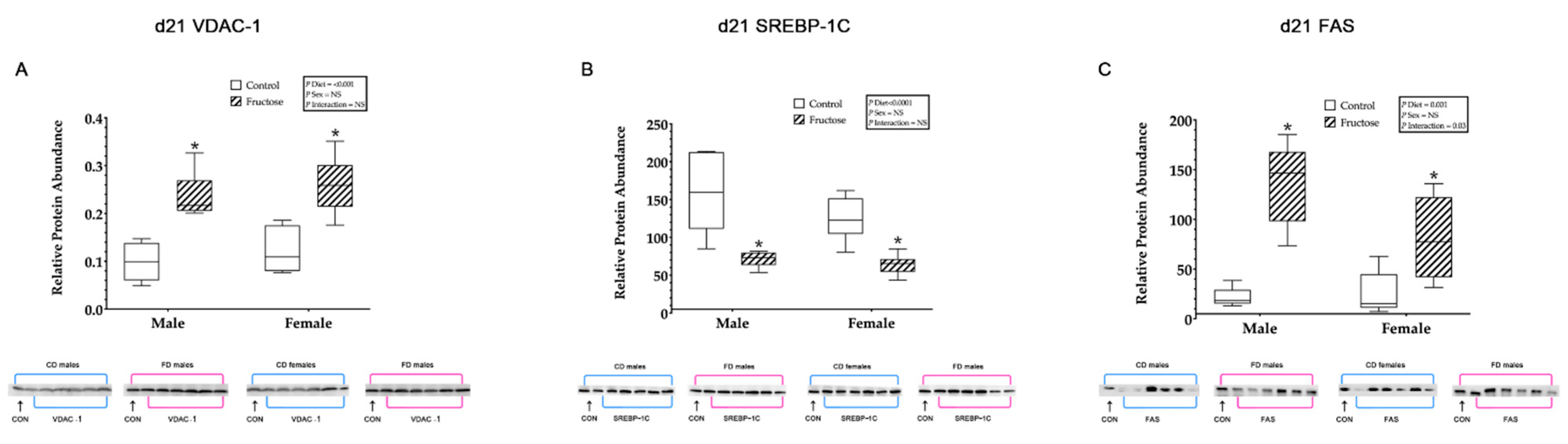
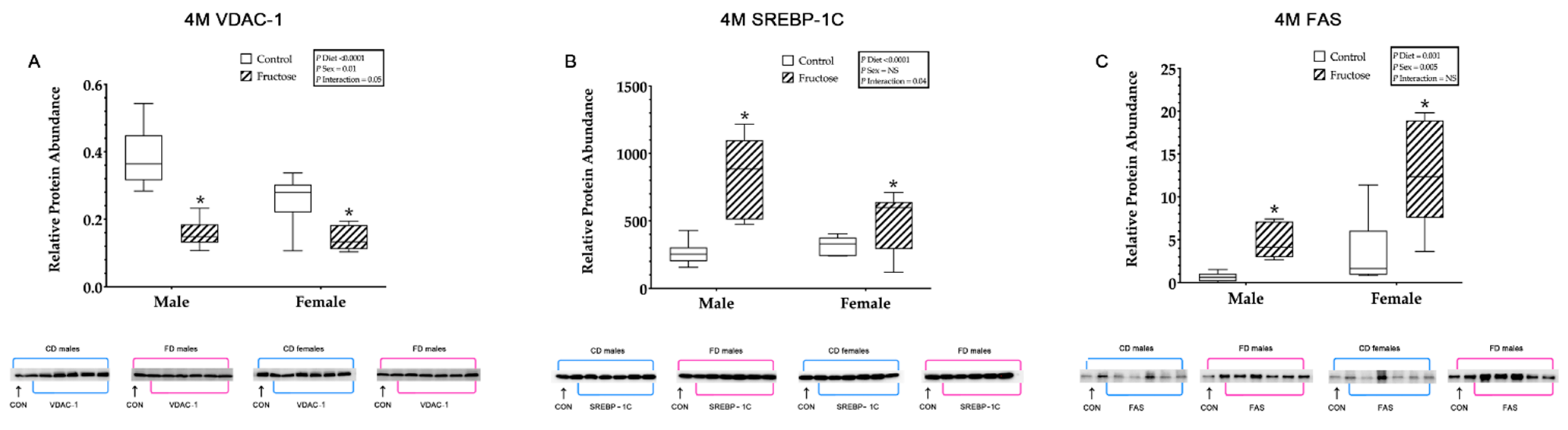
3.9.7. Hepatic VDAC1, SREBP-1c and FAS
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, D.-M.; Jiao, R.-Q.; Kong, L.-D. High Dietary Fructose: Direct or Indirect Dangerous Factors Disturbing Tissue and Organ Functions. Nutrients 2017, 9, 335. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.-C.; Wu, K.L.H.; Leu, S.; Tain, Y.-L. Translational insights on developmental origins of metabolic syndrome: Focus on fructose consumption. Biomed. J. 2018, 41, 96–101. [Google Scholar] [CrossRef]
- Bidwell, A.J. Chronic Fructose Ingestion as a Major Health Concern: Is a Sedentary Lifestyle Making It Worse? A Review. Nutrients 2017, 9, 549. [Google Scholar] [CrossRef] [Green Version]
- Lustig, R.H.; Schmidt, L.A.; Brindis, C.D. The toxic truth about sugar. Nature 2012, 482, 27–29. [Google Scholar] [CrossRef]
- Dekker, M.J.; Qiaozhu, S.; Baker, C.; Rutledge, A.C.; Adeli, K. Fructose: A highly lipogenic nutrient implicated in insulin resistance, hepatic steatosis, and the metabolic syndrome Mechanism of liponecrosis, a distinct mode of programmed cell death View project. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E685–E694. [Google Scholar] [CrossRef] [Green Version]
- Basciano, H.; Federico, L.; Adeli, K. Fructose, insulin resistance, and metabolic dyslipidemia. Nutr. Metab. 2005, 2, 5. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.V.L.; Dyson, R.M.; Berry, M.J.; Gray, C. Fructose Consumption during Pregnancy Influences Milk Lipid Composition and Offspring Lipid Profiles in Guinea Pigs. Front. Endocrinol. 2020, 11, 550. [Google Scholar] [CrossRef]
- Havel, P.J. Dietary fructose: Implications for dysregulation of energy homeostasis and lipid/carbohydrate metabolism. Nutr. Rev. 2005, 63, 133–157. [Google Scholar] [CrossRef]
- Gyllenhammer, L.E.; Entringer, S.; Buss, C.; Wadhwa, P.D. Developmental programming of mitochondrial biology: A conceptual framework and review. Proc. R. Soc. B 2020, 287, 20192713. [Google Scholar] [CrossRef]
- Pileggi, C.A.; Hedges, C.P.; Segovia, S.A.; Markworth, J.F.; Durainayagam, B.R.; Gray, C.; Cameron-Smith, D. Maternal high fat diet alters skeletal muscle mitochondrial catalytic activity in adult male rat offspring. Front. Physiol. 2016, 7, 546. [Google Scholar] [CrossRef]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef] [Green Version]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef]
- Aiken, C.E.; Tarry-Adkins, J.L.; Penfold, N.C.; Dearden, L.; Ozanne, S.E. Decreased ovarian reserve, dysregulation of mitochondrial biogenesis, and increased lipid peroxidation in female mouse offspring exposed to an obesogenic maternal diet. FASEB J. 2016, 30, 1548–1556. [Google Scholar] [CrossRef] [Green Version]
- Bruin, J.E.; Petre, M.A.; Raha, S.; Morrison, K.M.; Gerstein, H.C.; Holloway, A.C. Fetal and neonatal nicotine exposure in wistar rats causes progressive pancreatic mitochondrial damage and beta cell dysfunction. PLoS ONE 2008, 3, e3371. [Google Scholar] [CrossRef]
- Tarry-Adkins, J.L.; Fernandez-Twinn, D.S.; Chen, J.H.; Hargreaves, I.P.; Neergheen, V.; Aiken, C.E.; Ozanne, S.E. Poor maternal nutrition and accelerated postnatal growth induces an accelerated aging phenotype and oxidative stress in skeletal muscle of male rats. Dis. Models Mech. 2016, 9, 1221–1229. [Google Scholar] [CrossRef] [Green Version]
- Pitkänen, S.; Robinson, B.H. Mitochondrial complex I deficiency leads to increased production of superoxide radicals and induction of superoxide dismutase. J. Clin. Investig. 1996, 98, 345–351. [Google Scholar] [CrossRef] [Green Version]
- Leary, C.; Leese, H.J.; Sturmey, R.G. Human embryos from overweight and obese women display phenotypic and metabolic abnormalities. Hum. Reprod. 2015, 30, 122–132. [Google Scholar] [CrossRef] [Green Version]
- Gruzieva, O.; Xu, C.J.; Breton, C.V.; Annesi-Maesano, I.; Antó, J.M.; Auffray, C.; Melén, E. Epigenome-wide meta-analysis of methylation in children related to prenatal NO2 air pollution exposure. Environ. Health Perspect. 2017, 125, 104–110. [Google Scholar] [CrossRef] [Green Version]
- Hastie, R.; Lappas, M. The effect of pre-existing maternal obesity and diabetes on placental mitochondrial content and electron transport chain activity. Placenta 2014, 35, 673–683. [Google Scholar] [CrossRef]
- Brunst, K.J.; Sanchez-Guerra, M.; Chiu, Y.H.M.; Wilson, A.; Coull, B.A.; Kloog, I.; Wright, R.J. Prenatal particulate matter exposure and mitochondrial dysfunction at the maternal-fetal interface: Effect modification by maternal lifetime trauma and child sex. Environ. Int. 2018, 112, 49–58. [Google Scholar] [CrossRef]
- Clemente, D.B.P.; Casas, M.; Vilahur, N.; Begiristain, H.; Bustamante, M.; Carsin, A.E.; Nawrot, T.S. Prenatal ambient air pollution, placental mitochondrial DNA content, and birth weight in the INMA (Spain) and ENVIRONAGE (Belgium) birth Cohorts. Environ. Health Perspect. 2016, 124, 659–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janssen, B.G.; Byun, H.M.; Gyselaers, W.; Lefebvre, W.; Baccarelli, A.A.; Nawrot, T.S. Placental mitochondrial methylation and exposure to airborne particulate matter in the early life environment: An ENVIRONAGE birth cohort study. Epigenetics 2015, 10, 536–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janssen, B.G.; Munters, E.; Pieters, N.; Smeets, K.; Cox, B.; Cuypers, A.; Nawrot, T.S. Placental mitochondrial DNA content and particulate air pollution during in utero life. Environ. Health Perspect. 2012, 120, 1346–1352. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Feng, Q.; Zhang, Q.; Wang, T.; Xiao, X. Early life fructose exposure and its implications for long-term cardiometabolic health in offspring. Nutrients 2016, 8, 685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloboda, D.M.; Li, M.; Patel, R.; Clayton, Z.E.; Yap, C.; Vickers, M.H. Early life exposure to fructose and offspring phenotype: Implications for long term metabolic homeostasis. J. Obes. 2014, 2014, 203474. [Google Scholar] [CrossRef]
- Regnault, T.R.; Gentili, S.; Sarr, O.; Toop, C.R.; Sloboda, D.M. Fructose, pregnancy and later life impacts. Clin. Exp. Pharmacol. Physiol. 2013, 40, 824–837. [Google Scholar] [CrossRef]
- Clayton, Z.E.; Vickers, M.H.; Bernal, A.; Yap, C.; Sloboda, D.M. Early Life Exposure to Fructose Alters Maternal, Fetal and Neonatal Hepatic Gene Expression and Leads to Sex-Dependent Changes in Lipid Metabolism in Rat Offspring. PLoS ONE 2015, 10, e0141962. [Google Scholar] [CrossRef]
- Carta, G.; Murru, E.; Banni, S.; Manca, C. Palmitic acid: Physiological role, metabolism and nutritional implications. Front. Physiol. 2017, 8, 902. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, Y.; Imajo, K.; Honda, Y.; Kessoku, T.; Tomeno, W.; Kato, S.; Nakajima, A. Palmitate-induced lipotoxicity is crucial for the pathogenesis of nonalcoholic fatty liver disease in cooperation with gut-derived endotoxin. Sci. Rep. 2018, 8, 1–14. [Google Scholar]
- Okada, T.; Furuhashi, N.; Kuromori, Y.; Miyashita, M.; Iwata, F.; Harada, K. Plasma palmitoleic acid content and obesity in children. Am. J. Clin. Nutr. 2005, 82, 747–750. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, J.R.; Ferreira, C.F.; Senter, G.; Krolow, R.; de Aguiar, B.W.; Portella, A.K.; Silveira, P.P. Early Life Stress Interacts with the Diet Deficiency of Omega-3 Fatty Acids during the Life Course Increasing the Metabolic Vulnerability in Adult Rats. PLoS ONE 2013, 8, e62031. [Google Scholar] [CrossRef] [Green Version]
- Bolsoni-Lopes, A.; Festuccia, W.T.; Farias, T.S.M.; Chimin, P.; Torres-Leal, F.L.; Derogis, P.B.M.; Alonso-Vale, M.I.C. Palmitoleic acid (n-7) increases white adipocyte lipolysis and lipase content in a PPARα-dependent manner. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E1093–E1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, M.M.; Lopes, A.B.; Crisma, A.R.; de Sá, R.C.C.; Kuwabara, W.M.T.; Curi, R.; Alonso-Vale, M.I.C. Palmitoleic acid (16:1n7) increases oxygen consumption, fatty acid oxidation and ATP content in white adipocytes. Lipids Health Dis. 2018, 17, 55. [Google Scholar] [CrossRef] [Green Version]
- Tricò, D.; Mengozzi, A.; Nesti, L.; Hatunic, M.; Gabriel Sanchez, R.; Konrad, T.; Natali, A. Circulating palmitoleic acid is an independent determinant of insulin sensitivity, beta cell function and glucose tolerance in non-diabetic individuals: A longitudinal analysis. Diabetologia 2020, 63, 206–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Softic, S.; Cohen, D.E.; Kahn, C.R. Role of Dietary Fructose and Hepatic De Novo Lipogenesis in Fatty Liver Disease. Dig. Dis. Sci. 2016, 61, 1282–1293. [Google Scholar] [CrossRef] [Green Version]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Moon, Y.A.; Shah, N.A.; Mohapatra, S.; Warrington, J.A.; Horton, J.D. Identification of a mammalian long chain fatty acyl elongase regulated by sterol regulatory element-binding proteins. J. Biol. Chem. 2001, 276, 45358–45366. [Google Scholar] [CrossRef] [Green Version]
- Edwards, P.A.; Tabor, D.; Kast, H.R.; Venkateswaran, A. Regulation of gene expression by SREBP and SCAP. Biochim. Biophys. Acta 2000, 1529, 103–113. [Google Scholar] [CrossRef]
- Crescenzo, R.; Bianco, F.; Falcone, I.; Coppola, P.; Liverini, G.; Lossa, S. Increased hepatic de novo lipogenesis and mitochondrial effeciency in a model of obesity induced by diet rich in fructose. Eur. J. Nutr. 2013, 52, 537–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cioffi, F.; Senese, R.; Lasala, P.; Ziello, A.; Mazzoli, A.; Crescenzo, R.; Iossa, S. Fructose-Rich Diet Affects Mitochondrial DNA Damage and Repair in Rats. Nutrients 2017, 9, 323. [Google Scholar] [CrossRef] [Green Version]
- Huizing, M.; Ruitenbeek, W.; Thinnes, F.P.; DePinto, V.; Wendel, U.; Trijbels, F.J.M.; Van Den Heuvel, L.P. Deficiency of the voltage-dependent anion channel: A novel cause of mitochondriopathy. Pediatr. Res. 1996, 39, 760–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camara, A.K.S.; Zhou, Y.; Wen, P.-C.; Tajkhorshid, E.; Kwok, W.-M. Mitchondrial VDAC1: A key gatekeeper as potential theraputic target. Front. Physiol. 2017, 8, 460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yakubu, D.P.; Mostyn, A.; Hyatt, M.A.; Kurlak, L.O.; Budge, H.; Stephenson, T.; Symonds, M.E. Ontogeny and nutritional programming of mitochondrial proteins in the ovine kidney, liver and lung. Reproduction 2007, 134, 823–830. [Google Scholar] [CrossRef] [Green Version]
- National Research Council; Division on Earth and Life Studies; Institute for Laboratory Animal Research; Committee for the Update of the Guide for the Care and Use of Laboratory Animals. Guide for the Care and Use of Laboratory Animals: Eighth Edition; The National Academies Press: Washington, DC, USA, 2011. [Google Scholar]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving Bioscience Research Reporting: The ARRIVE Guidelines for Reporting Animal Research. PLoS Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef]
- Matsuda, M.; DeFronzo, R.A. Insulin sensitivity indices obtained from oral glucose tolerance testing: Comparison with the euglycemic insulin clamp. Diabetes Care 1999, 22, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, C.; Haffner, S.M.; Stančáková, A.; Kuusisto, J.; Laakso, M. Fasting and OGTT-derived Measures of Insulin Resistance as Compared With the Euglycemic-Hyperinsulinemic Clamp in Nondiabetic Finnish Offspring of Type 2 Diabetic Individuals. J. Clin. Endocrinol. Metab. 2015, 100, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Lepage, G.; Roy, C.C. Direct transesterification of all classes of lipids in a one-step reaction. J. Lipid. Res. 1986, 27, 114–120. [Google Scholar] [CrossRef]
- Hübner, N.F.; Horch, R.E.; Polykandriotis, E.; Rau, T.T.; Dragu, A. A Histopathologic and Immunohistochemical Study on Liquification of Human Adipose Tissue Ex Vivo. Aesthet. Plast. Surg. 2014, 38, 976–984. [Google Scholar] [CrossRef] [PubMed]
- Sarr, O.; Thompson, J.A.; Zhao, L.; Lee, T.-Y.; Regnault, T.R.H. Low Birth Weight Male Guinea Pig Offspring Display Increased Visceral Adiposity in Early Adulthood. PLoS ONE 2014, 9, e98433. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; VerBerkmoes, N.C.; Langston, M.A.; Uberbacher, E.; Hettich, R.L.; Samatova, N.F. Detecting differential and correlated protein expression in label-free shotgun proteomics. J. Proteome Res. 2006, 5, 2909–2918. [Google Scholar] [CrossRef]
- Bosch, P.J.; Peng, L.; Kivell, B.M. Proteomics Analysis of Dorsal Striatum Reveals Changes in Synaptosomal Proteins following Methamphetamine Self-Administration in Rats. PLoS ONE 2015, 10, e0139829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meissner, F.; Mann, M. Quantitative shotgun proteomics: Considerations for a high-quality workflow in immunology. Nat. Immunol. 2014, 15, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Nesvizhskii, A.I.; Keller, A.; Kolker, E.; Aebersold, R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003, 75, 4646–4658. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.; Nesvizhskii, A.I.; Kolker, E.; Aebersold, R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 2002, 74, 5383–5392. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adult Offspring Plasma Metabolites | Sex | Month 4 | |
|---|---|---|---|
| Control | Fructose | ||
| TAG (mmol/L) | Male | 0.55 ± 0.05 | 0.62 ± 0.05 * |
| Female | 0.69 ± 0.05 * | 1.05 ± 0.05 ** | |
| CHOL (mmol/L) | Male | 0.72 ± 0.05 | 0.98 ± 0.05 |
| Female | 0.82 ± 0.05 * | 1.12 ± 0.05 * | |
| LDL (mmol/L) | Male | 0.67 ± 0.05 | 0.71 ± 0.05 |
| Female | 0.88 ± 0.05 * | 0.96 ± 0.05 * | |
| FRUC (umol/L) | Male | 240.16 ± 6.57 | 222.57 ± 6.57 |
| Female | 239.00 ± 6.57 | 270.80 ± 6.57 * | |
| ALP (U/L) | Male | 13.33 ± 1.37 | 4.88 ± 1.37 * |
| Female | 5.16 ± 1.37 | 3.50 ± 1.37 * | |
| HDL (mmol/L) | Male | 0.03 ± 0.00 | 0.41 ± 0.00 |
| Female | 0.03 ± 0.00 | 0.05 ± 0.00 | |
| UA (µmol/L | Male | 16.33 ± 2.18 | 24.88 ± 2.18 |
| Female | 21.50 ± 2.18 | 26.83 ± 2.18 | |
| LIP (U/L) | Male | 6.20 ± 0.81 | 10.18 ± 0.81 |
| Female | 6.88 ± 0.81 | 07.31 ± 0.81 | |
| hs-CRP (mg/L) | Male | 0.10 ± 0.01 | 0.09 ± 0.01 |
| Female | 0.14 ± 0.01 | 0.09 ± 0.01 | |
| ALT (U/L) | Male | 72.82 ± 9.13 | 99.33 ± 9.13 |
| Female | 66.50 ± 9.13 | 91.51 ± 9.13 | |
| GGT (U/L) | Male | 171.80 ± 15.93 | 168.85 ± 15.93 |
| Female | 89.40 ± 15.93 | 156.75 ± 15.93 | |
| Leptin (ng/nL) | Male | 4.85 ± 0.41 | 3.98 ± 0.41 |
| Female | 3.93 ± 0.41 | 4.34 ± 0.41 | |
| Adult Offspring Whole Blood FFA (mg/100 g) | Sex | Month 4 | |
|---|---|---|---|
| Control | Fructose | ||
| Palmitoleic acid (16:1n-7) | Male | 0.89 ± 0.04 | 1.11 ± 0.04 * |
| Female | 1.04 ± 0.04 | 0.88 ± 0.04 * | |
| Total Omega-7 | Male | 1.90 ± 0.05 | 2.14 ± 0.05 * |
| Female | 2.00 ± 0.05 | 1.83 ± 0.05 * | |
| ALA (18:3n-3) | Male | 2.41 ± 0.11 | 1.91 ± 0.11 * |
| Female | 2.06 ± 0.11 | 2.50 ± 0.11 * | |
| Total Omega-3 | Male | 5.00 ± 0.12 | 4.66 ± 0.12 * |
| Female | 4.58 ± 0.12 | 5.28 ± 0.12 * | |
| Total Monosaturated fats | Male | 13.28 ± 0.42 | 14.57 ± 0.42 * |
| Female | 15.45 ± 0.42 | 13.31 ± 0.42 * | |
| EPA (20:5n-3) | Male | 0.14 ± 0.01 | 0.15 ± 0.01 |
| Female | 0.22 ± 0.01 * | 0.20 ± 0.01 * | |
| LA (18:2n-6) | Male | 19.86 ± 0.40 | 19.31 ± 0.40 |
| Female | 18.05 ± 0.40 * | 21.04 ± 0.40 * | |
| Total Saturates | Male | 48.09 ± 0.32 * | 48.52 ± 0.32 * |
| Female | 46.92 ± 0.32 | 46.65 ± 0.32 | |
| Myristic acid (14:0) | Male | 1.04 ± 0.04 | 0.17 ± 0.04 * |
| Female | 0.95 ± 0.04 | 0.80 ± 0.04 * | |
| Arachidic acid (20:0) | Male | 0.68 ± 0.19 | 0.76 ± 0.19 * |
| Female | 0.79 ± 0.19 | 0.62 ± 0.19 * | |
| Total Trans Fatty acids | Male | 0.65 ± 0.09 | 1.01 ± 0.09 * |
| Female | 0.68 ± 0.09 | 1.15 ± 0.09 * | |
| Gondoic Acid (20:1n-9) | Male | 0.48 ± 0.08 | 1.14 ± 0.08 * |
| Female | 0.83 ± 0.08 | 0.52 ± 0.08 * | |
| GLA (18:3n-6) | Male | 0.22 ± 0.01 | 0.13 ± 0.01 * |
| Female | 0.18 ± 0.01 | 0.13 ± 0.01 * | |
| DGLA (20:3n-6) | Male | 0.36 ± 0.01 | 0.37 ± 0.01 |
| Female | 0.43 ± 0.01 * | 0.43 ± 0.01 * | |
| Margaric acid (17:0) | Male | 1.45 ± 0.02 | 1.45 ± 0.02 |
| Female | 1.36 ± 0.02 | 1.37 ± 0.02 | |
| DHA (22:6n-3) | Male | 0.45 ± 0.02 | 0.54 ± 0.02 |
| Female | 0.56 ± 0.02 | 0.56 ± 0.02 | |
| Pentadecanoic acid (15:0) | Male | 0.84 ± 0.04 | 0.90 ± 0.04 |
| Female | 0.83 ± 0.04 | 0.69 ± 0.04 | |
| dma16:0 | Male | 0.40 ± 0.06 | 0.72 ± 0.06 |
| Female | 0.50 ± 0.06 | 0.44 ± 0.06 | |
| Total Omega-6 | Male | 31.32 ± 0.49 | 31.07 ± 0.49 |
| Female | 30.83 ± 0.49 | 33.58 ± 0.49 | |
| Vaccenic acid (t18:1n-7) | Male | 0.28 ± 0.01 | 0.36 ± 0.01 |
| Female | 0.28 ± 0.01 | 0.31 ± 0.01 | |
| Palmitic acid (16:0) | Male | 15.50 ± 0.19 | 15.26 ± 0.19 |
| Female | 15.20 ± 0.19 | 15.64 ± 0.19 | |
| Elaidic acid (t18:1n-9) | Male | 0.21 ± 0.01 | 0.19 ± 0.01 |
| Female | 0.24 ± 0.01 | 0.20 ± 0.01 | |
| dma18:0 | Male | 0.42 ± 0.08 | 0.85 ± 0.08 |
| Female | 0.45 ± 0.08 | 0.54 ± 0.08 | |
| Behenic acid (22:0) | Male | 0.94 ± 0.04 | 0.94 ± 0.04 |
| Female | 1.04 ± 0.04 | 0.92 ± 0.04 | |
| Adrenic acid (22:4n-6) + Docosatrienoate acid (22:3n-3) | Male | 2.32 ± 0.05 | 2.27 ± 0.05 |
| AA (20:4n-6) | Male | 8.62 ± 0.15 | 8.47 ± 0.15 |
| Female | 8.57 ± 0.15 | 8.97 ± 0.15 | |
| DPA (22:5n-3) | Male | 2.15 ± 0.06 | 1.91 ± 0.06 |
| Female | 1.97 ± 0.06 | 1.97 ± 0.06 | |
| Cis-Vaccenic acid (18:1n-7) | Male | 0.97 ± 0.01 | 0.98 ± 0.01 |
| Female | 0.97 ± 0.01 | 0.97 ± 0.01 | |
| Eicosadienoic acid (20:2n-6) | Male | 0.77 ± 0.02 | 0.74 ± 0.02 |
| Female | 0.66 ± 0.02 | 0.71 ± 0.02 | |
| Lignoceric acid (24:0) | Male | 1.65 ± 0.06 | 1.67 ± 0.06 |
| Female | 1.79 ± 0.06 | 1.77 ± 0.06 | |
| Stearic acid (18:0) | Male | 24.75 ± 0.32 | 25.33 ± 0.32 |
| Female | 23.47 ± 0.32 | 24.17 ± 0.32 | |
| Oleic acid (18:1n-9) | Male | 9.59 ± 0.18 | 9.77 ± 0.18 |
| Female | 10.16 ± 0.18 | 10.40 ± 0.18 | |
| Total Omega-9 | Male | 10.42 ± 0.19 | 10.94 ± 0.19 |
| Female | 11.46 ± 0.19 | 10.95 ± 0.19 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, E.V.L.; Dyson, R.M.; Vanderboor, C.M.G.; Sarr, O.; Anderson, J.; Berry, M.J.; Regnault, T.R.H.; Peng, L.; Gray, C. Maternal Fructose Intake Causes Developmental Reprogramming of Hepatic Mitochondrial Catalytic Activity and Lipid Metabolism in Weanling and Young Adult Offspring. Int. J. Mol. Sci. 2022, 23, 999. https://doi.org/10.3390/ijms23020999
Smith EVL, Dyson RM, Vanderboor CMG, Sarr O, Anderson J, Berry MJ, Regnault TRH, Peng L, Gray C. Maternal Fructose Intake Causes Developmental Reprogramming of Hepatic Mitochondrial Catalytic Activity and Lipid Metabolism in Weanling and Young Adult Offspring. International Journal of Molecular Sciences. 2022; 23(2):999. https://doi.org/10.3390/ijms23020999
Chicago/Turabian StyleSmith, Erin Vanessa LaRae, Rebecca Maree Dyson, Christina M. G. Vanderboor, Ousseynou Sarr, Jane Anderson, Mary J. Berry, Timothy R. H. Regnault, Lifeng Peng, and Clint Gray. 2022. "Maternal Fructose Intake Causes Developmental Reprogramming of Hepatic Mitochondrial Catalytic Activity and Lipid Metabolism in Weanling and Young Adult Offspring" International Journal of Molecular Sciences 23, no. 2: 999. https://doi.org/10.3390/ijms23020999