
Rewired Cellular Metabolic Profiles in Response to Metformin under Different Oxygen and Nutrient Conditions

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
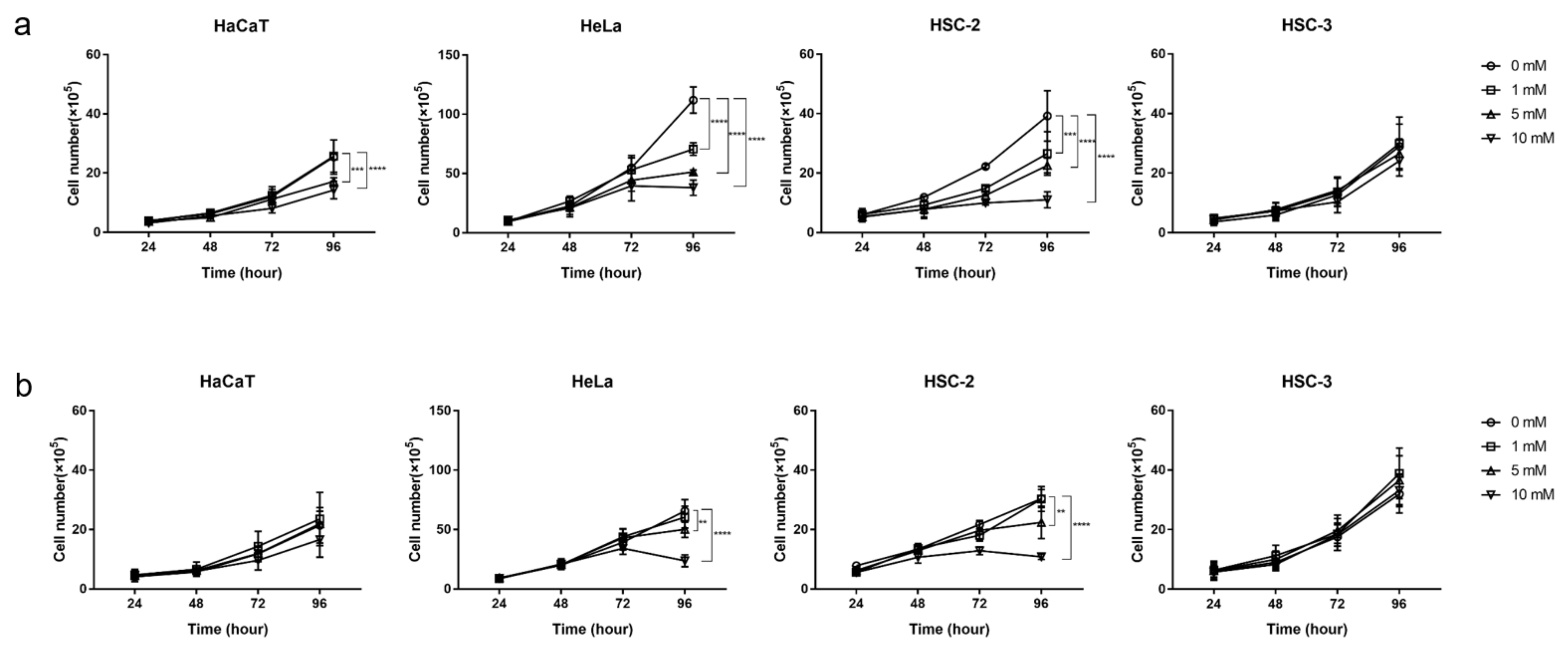
2.1. Inhibitory Effects of Metformin on the Growth of Cancer and Normal Cells
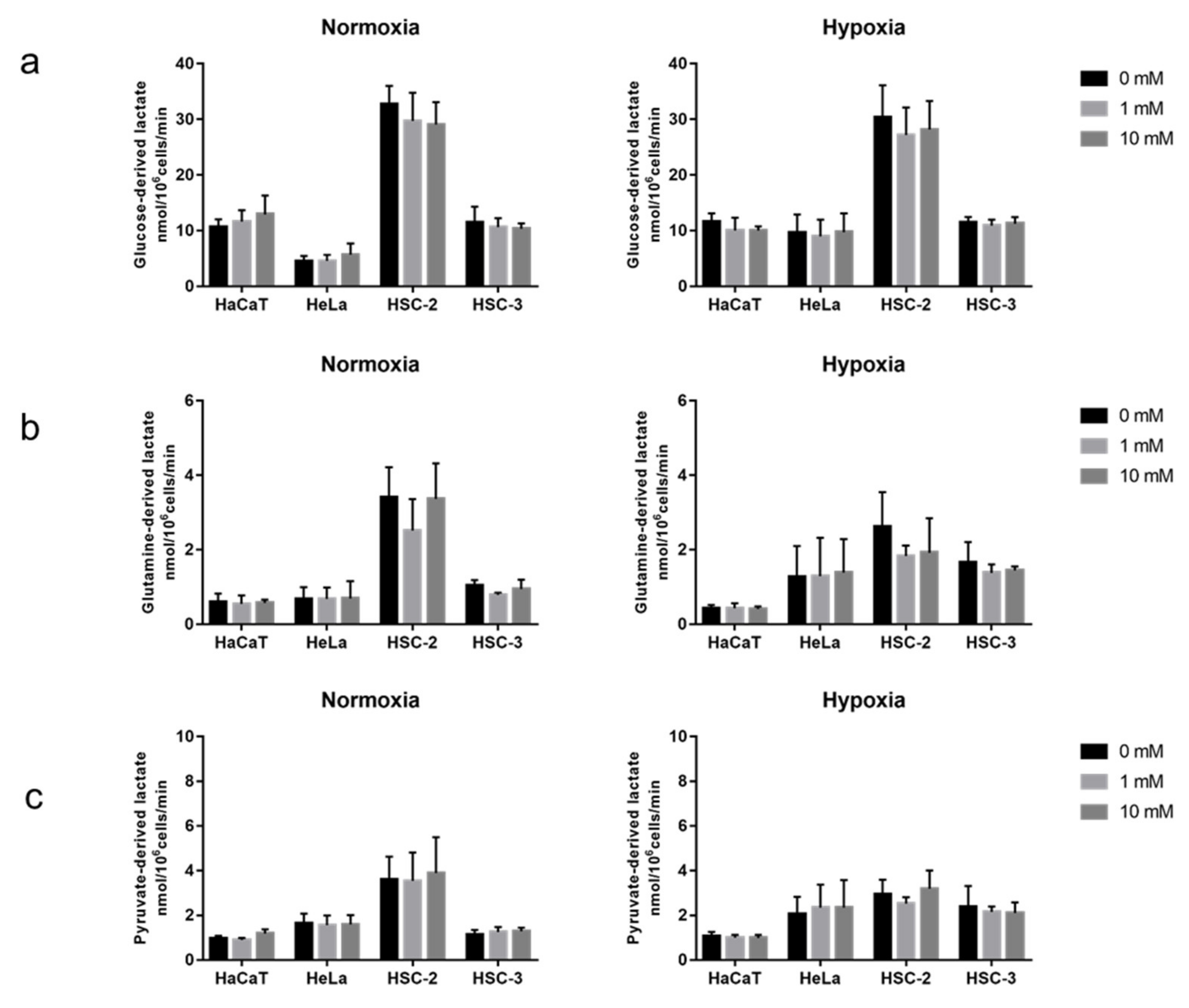
2.2. Acute Effects of Metformin on Cellular Metabolic Activity
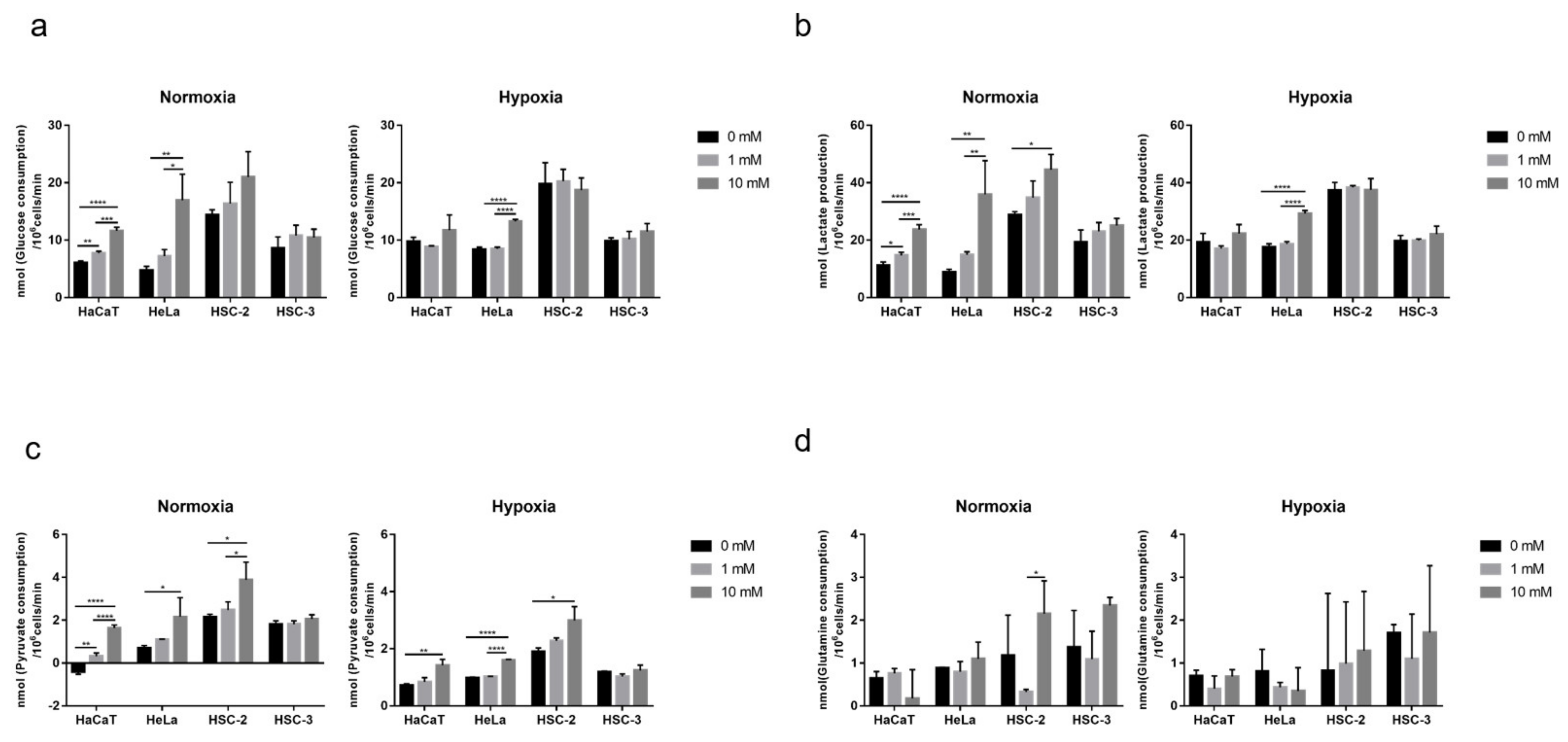
2.3. Effects of Long-Term Exposure to Metformin on Cellular Nutrient Utilization and Organic Acid Production in Culture Media
2.4. Effects of Long-Term Exposure to Metformin on Cellular Metabolic Activity
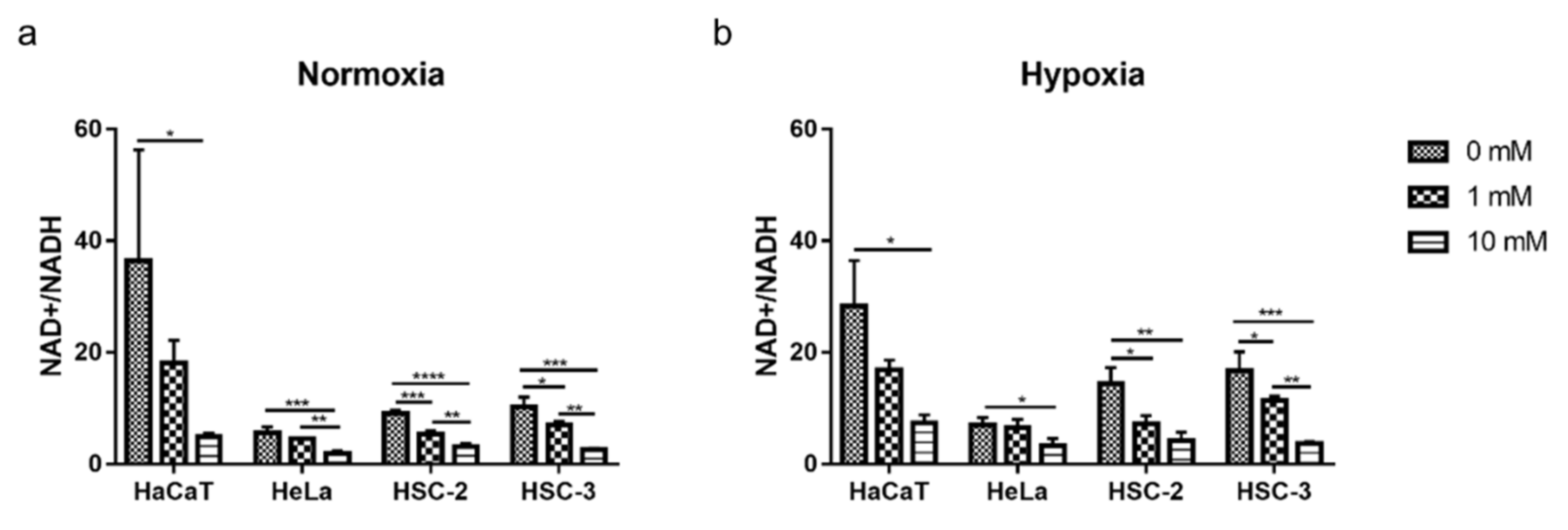
2.5. Decreased Cellular NAD+/NADH Ratio during Cell Growth with Metformin
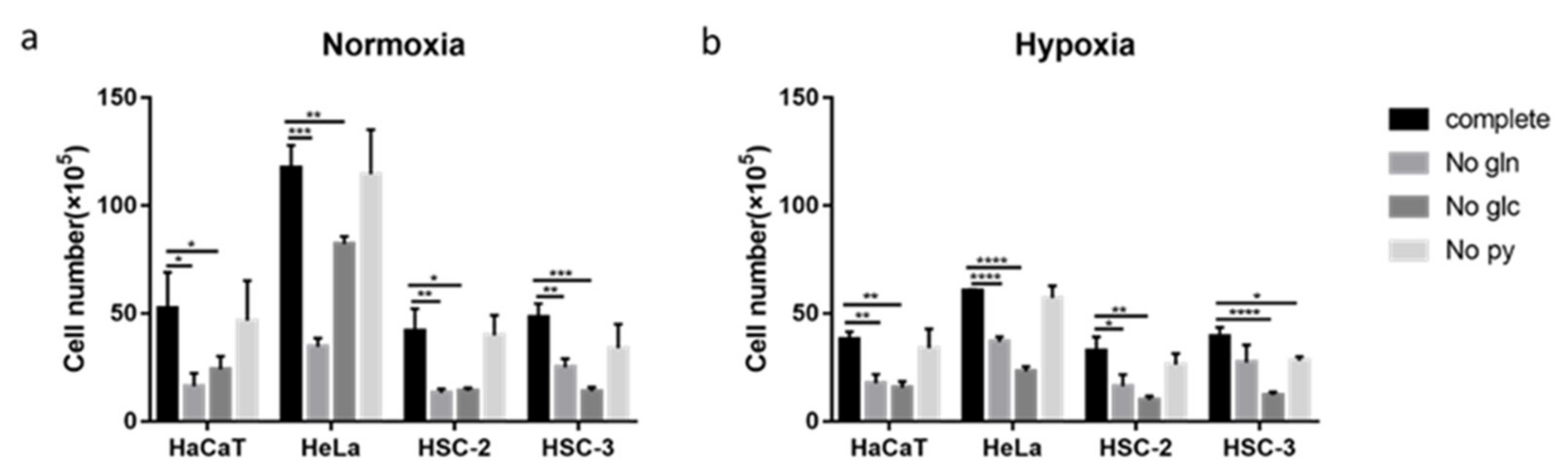
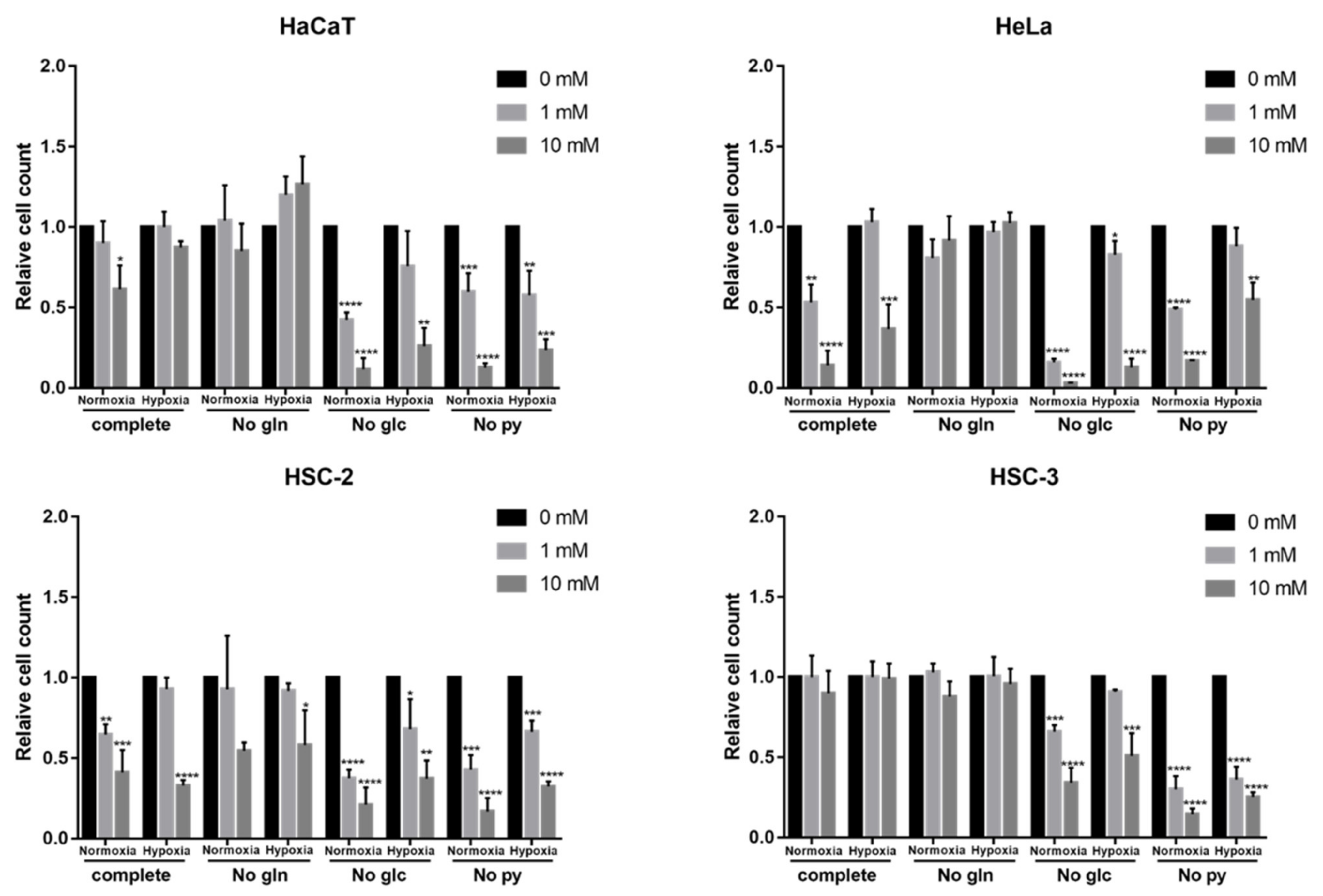
2.6. Effects of Nutritional Conditions on Metformin Efficacy
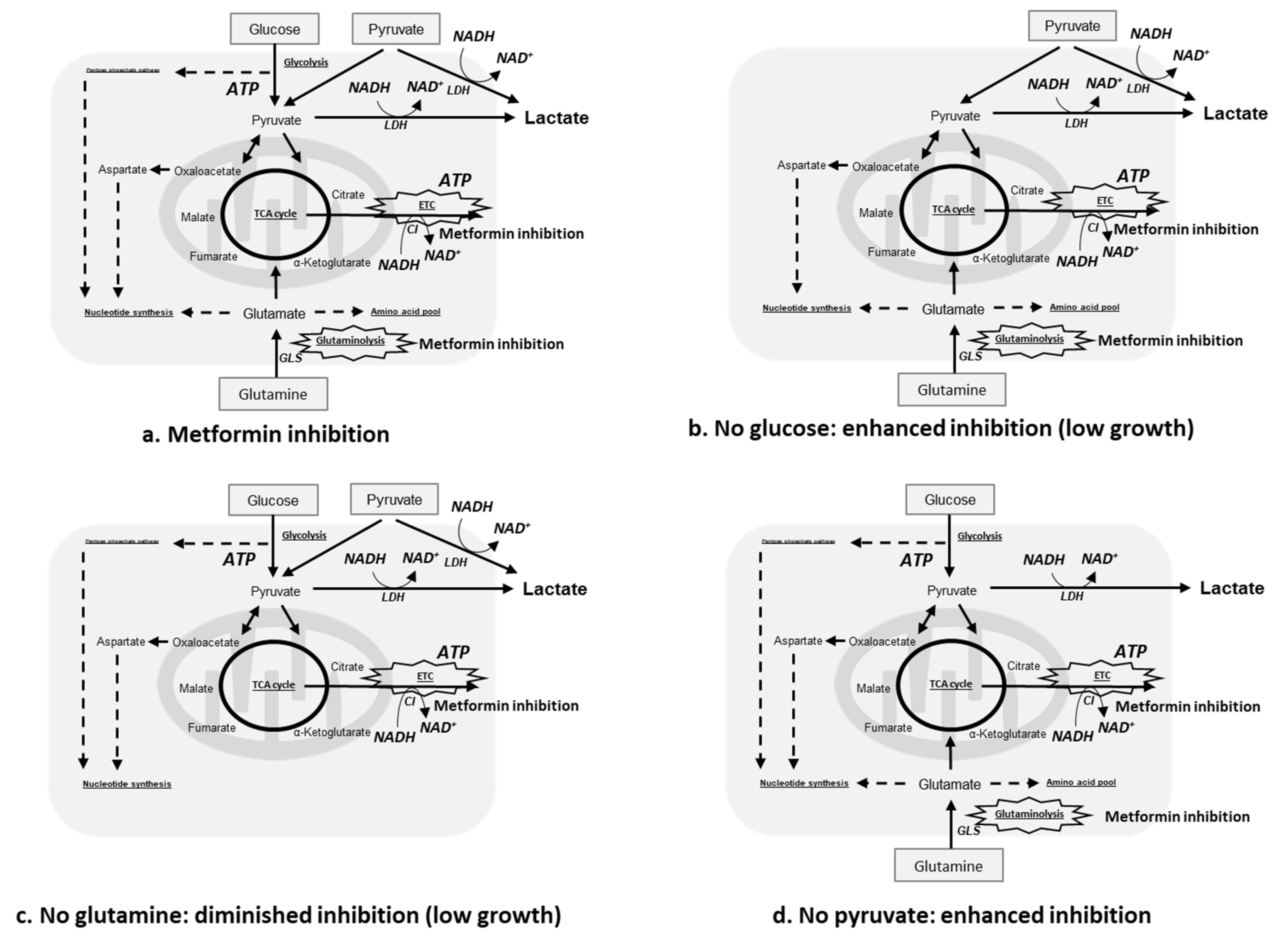
3. Discussion
4. Materials and Methods
4.1. Chemical Regents
4.2. Cell Lines and Culture Conditions
4.3. Cell Proliferation Assays
4.4. Detection of Metabolic Activities of Glucose, Glutamine, and Pyruvate in Cells with or without an Acute Exposure to Metformin
4.5. Measurement of Concentrations of Pyruvate, Glucose, and Glutamine in Culture Media
4.6. Detection of Metabolic Activities of Glucose, Glutamine, and Pyruvate in Cells Grown with or without Long-Term Exposure to Metformin
4.7. Analysis of Organic Acids with HPLC
4.8. NAD+/NADH Ratio Assay
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McKeown, S.R. Defining normoxia, physoxia and hypoxia in tumours-implications for treatment response. Br. J. Radiol. 2014, 87, 20130676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belisario, D.C.; Kopecka, J.; Pasino, M.; Akman, M.; De Smaele, E.; Donadelli, M.; Riganti, C. Hypoxia Dictates Metabolic Rewiring of Tumors: Implications for Chemoresistance. Cells 2020, 9, 2598. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, V.; Hoey, C.; Liu, L.Y.; Lalonde, E.; Ray, J.; Livingstone, J.; Lesurf, R.; Shiah, Y.J.; Vujcic, T.; Huang, X.; et al. Molecular landmarks of tumor hypoxia across cancer types. Nat. Genet. 2019, 51, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Wilson, W.R. Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer 2004, 4, 437–447. [Google Scholar] [CrossRef]
- Scharping, N.E.; Menk, A.V.; Whetstone, R.D.; Zeng, X.; Delgoffe, G.M. Efficacy of PD-1 Blockade Is Potentiated by Metformin-Induced Reduction of Tumor Hypoxia. Cancer Immunol. Res. 2017, 5, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Hassan Venkatesh, G.; Bravo, P.; Shaaban Moustafa Elsayed, W.; Amirtharaj, F.; Wojtas, B.; Abou Khouzam, R.; Hussein Nawafleh, H.; Mallya, S.; Satyamoorthy, K.; Dessen, P.; et al. Hypoxia increases mutational load of breast cancer cells through frameshift mutations. Oncoimmunology 2020, 9, 1750750. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, A.R.; Glazer, P.M. Impact of hypoxia on DNA repair and genome integrity. Mutagenesis 2020, 35, 61–68. [Google Scholar] [CrossRef]
- Luoto, K.R.; Kumareswaran, R.; Bristow, R.G. Tumor hypoxia as a driving force in genetic instability. Genome Integr. 2013, 4, 5. [Google Scholar] [CrossRef] [Green Version]
- Shinohara, Y.; Washio, J.; Kobayashi, Y.; Abiko, Y.; Sasaki, K.; Takahashi, N. Hypoxically cultured cells of oral squamous cell carcinoma increased their glucose metabolic activity under normoxic conditions. PLoS ONE 2021, 16, e0254966. [Google Scholar] [CrossRef]
- Gilkes, D.M.; Semenza, G.L.; Wirtz, D. Hypoxia and the extracellular matrix: Drivers of tumour metastasis. Nat. Rev. Cancer 2014, 14, 430–439. [Google Scholar] [CrossRef] [Green Version]
- Qi, L.; Chen, J.; Yang, Y.; Hu, W. Hypoxia Correlates with Poor Survival and M2 Macrophage Infiltration in Colorectal Cancer. Front. Oncol. 2020, 10, 566430. [Google Scholar] [CrossRef]
- Dekervel, J.; Hompes, D.; van Malenstein, H.; Popovic, D.; Sagaert, X.; De Moor, B.; Van Cutsem, E.; D’Hoore, A.; Verslype, C.; van Pelt, J. Hypoxia-driven gene expression is an independent prognostic factor in stage II and III colon cancer patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 2159–2168. [Google Scholar] [CrossRef] [Green Version]
- Walsh, J.C.; Lebedev, A.; Aten, E.; Madsen, K.; Marciano, L.; Kolb, H.C. The clinical importance of assessing tumor hypoxia: Relationship of tumor hypoxia to prognosis and therapeutic opportunities. Antioxid. Redox Signal. 2014, 21, 1516–1554. [Google Scholar] [CrossRef]
- Mamo, M.; Ye, I.C.; DiGiacomo, J.W.; Park, J.Y.; Downs, B.; Gilkes, D.M. Hypoxia Alters the Response to Anti-EGFR Therapy by Regulating EGFR Expression and Downstream Signaling in a DNA Methylation-Specific and HIF-Dependent Manner. Cancer Res. 2020, 80, 4998–5010. [Google Scholar] [CrossRef]
- Daniel, S.K.; Sullivan, K.M.; Labadie, K.P.; Pillarisetty, V.G. Hypoxia as a barrier to immunotherapy in pancreatic adenocarcinoma. Clin. Transl. Med. 2019, 8, 10. [Google Scholar] [CrossRef]
- Jayaprakash, P.; Ai, M.; Liu, A.; Budhani, P.; Bartkowiak, T.; Sheng, J.; Ager, C.; Nicholas, C.; Jaiswal, A.R.; Sun, Y.; et al. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J. Clin. Investig. 2018, 128, 5137–5149. [Google Scholar] [CrossRef]
- Sullivan, M.R.; Vander Heiden, M.G. Determinants of nutrient limitation in cancer. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 193–207. [Google Scholar] [CrossRef]
- Ahmadiankia, N. In vitro and in vivo studies of cancer cell behavior under nutrient deprivation. Cell Biol. Int. 2020, 44, 1588–1597. [Google Scholar] [CrossRef]
- Onodera, T.; Momose, I.; Adachi, H.; Yamazaki, Y.; Sawa, R.; Ohba, S.I.; Kawada, M. Human pancreatic cancer cells under nutrient deprivation are vulnerable to redox system inhibition. J. Biol. Chem. 2020, 295, 16678–16690. [Google Scholar] [CrossRef]
- Prasad, P.; Ghosh, S.; Roy, S.S. Glutamine deficiency promotes stemness and chemoresistance in tumor cells through DRP1-induced mitochondrial fragmentation. Cell. Mol. Life Sci. CMLS 2021, 78, 4821–4845. [Google Scholar] [CrossRef]
- Ledoux, S.; Yang, R.; Friedlander, G.; Laouari, D. Glucose depletion enhances P-glycoprotein expression in hepatoma cells: Role of endoplasmic reticulum stress response. Cancer Res. 2003, 63, 7284–7290. [Google Scholar]
- Romero, R.; Erez, O.; Hüttemann, M.; Maymon, E.; Panaitescu, B.; Conde-Agudelo, A.; Pacora, P.; Yoon, B.H.; Grossman, L.I. Metformin, the aspirin of the 21st century: Its role in gestational diabetes mellitus, prevention of preeclampsia and cancer, and the promotion of longevity. Am. J. Obstet. Gynecol. 2017, 217, 282–302. [Google Scholar] [CrossRef]
- Flory, J.; Lipska, K. Metformin in 2019. JAMA 2019, 321, 1926–1927. [Google Scholar] [CrossRef]
- Salvatore, T.; Pafundi, P.C.; Morgillo, F.; Di Liello, R.; Galiero, R.; Nevola, R.; Marfella, R.; Monaco, L.; Rinaldi, L.; Adinolfi, L.E.; et al. Metformin: An old drug against old age and associated morbidities. Diabetes Res. Clin. Pract. 2020, 160, 108025. [Google Scholar] [CrossRef]
- Salvatore, T.; Pafundi, P.C.; Galiero, R.; Rinaldi, L.; Caturano, A.; Vetrano, E.; Aprea, C.; Albanese, G.; Di Martino, A.; Ricozzi, C.; et al. Can Metformin Exert as an Active Drug on Endothelial Dysfunction in Diabetic Subjects? Biomedicines 2020, 9, 3. [Google Scholar] [CrossRef]
- Emami Riedmaier, A.; Fisel, P.; Nies, A.T.; Schaeffeler, E.; Schwab, M. Metformin and cancer: From the old medicine cabinet to pharmacological pitfalls and prospects. Trends Pharmacol. Sci. 2013, 34, 126–135. [Google Scholar] [CrossRef]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [Green Version]
- Parikh, A.B.; Marrone, K.A.; Becker, D.J.; Brahmer, J.R.; Ettinger, D.S.; Levy, B.P. A pooled analysis of two phase II trials evaluating metformin plus platinum-based chemotherapy in advanced non-small cell lung cancer. Cancer Treat. Res. Commun. 2019, 20, 100150. [Google Scholar] [CrossRef]
- Morgillo, F.; Fasano, M.; Della Corte, C.M.; Sasso, F.C.; Papaccio, F.; Viscardi, G.; Esposito, G.; Di Liello, R.; Normanno, N.; Capuano, A.; et al. Results of the safety run-in part of the METAL (METformin in Advanced Lung cancer) study: A multicentre, open-label phase I-II study of metformin with erlotinib in second-line therapy of patients with stage IV non-small-cell lung cancer. ESMO Open 2017, 2, e000132. [Google Scholar] [CrossRef] [Green Version]
- Chun, S.G.; Liao, Z.; Jeter, M.D.; Chang, J.Y.; Lin, S.H.; Komaki, R.U.; Guerrero, T.M.; Mayo, R.C.; Korah, B.M.; Koshy, S.M.; et al. Metabolic Responses to Metformin in Inoperable Early-stage Non-Small Cell Lung Cancer Treated With Stereotactic Radiotherapy: Results of a Randomized Phase II Clinical Trial. Am. J. Clin. Oncol. 2020, 43, 231–235. [Google Scholar] [CrossRef]
- Brown, J.R.; Chan, D.K.; Shank, J.J.; Griffith, K.A.; Fan, H.; Szulawski, R.; Yang, K.; Reynolds, R.K.; Johnston, C.; McLean, K.; et al. Phase II clinical trial of metformin as a cancer stem cell-targeting agent in ovarian cancer. JCI Insight 2020, 5, e133247. [Google Scholar] [CrossRef] [PubMed]
- Miranda, V.C.; Braghiroli, M.I.; Faria, L.D.; Bariani, G.; Alex, A.; Bezerra Neto, J.E.; Capareli, F.C.; Sabbaga, J.; Lobo Dos Santos, J.F.; Hoff, P.M.; et al. Phase 2 Trial of Metformin Combined With 5-Fluorouracil in Patients With Refractory Metastatic Colorectal Cancer. Clin. Colorectal Cancer 2016, 15, 321–328.e321. [Google Scholar] [CrossRef] [PubMed]
- Bragagnoli, A.C.; Araujo, R.L.C.; Ferraz, M.W.; Dos Santos, L.V.; Abdalla, K.C.; Comar, F.; Santos, F.A.; Oliveira, M.A.; Carvalheira, J.B.C.; Cárcano, F.M.; et al. Metformin plus lrinotecan in patients with refractory colorectal cancer: A phase 2 clinical trial. Br. J. Cancer 2021, 124, 1072–1078. [Google Scholar] [CrossRef] [PubMed]
- Fenn, K.; Maurer, M.; Lee, S.M.; Crew, K.D.; Trivedi, M.S.; Accordino, M.K.; Hershman, D.L.; Kalinsky, K. Phase 1 Study of Erlotinib and Metformin in Metastatic Triple-Negative Breast Cancer. Clin. Breast Cancer 2020, 20, 80–86. [Google Scholar] [CrossRef]
- Gulati, S.; Desai, J.; Palackdharry, S.M.; Morris, J.C.; Zhu, Z.; Jandarov, R.; Riaz, M.K.; Takiar, V.; Mierzwa, M.; Gutkind, J.S.; et al. Phase 1 dose-finding study of metformin in combination with concurrent cisplatin and radiotherapy in patients with locally advanced head and neck squamous cell cancer. Cancer 2020, 126, 354–362. [Google Scholar] [CrossRef]
- Curry, J.; Johnson, J.; Tassone, P.; Vidal, M.D.; Menezes, D.W.; Sprandio, J.; Mollaee, M.; Cotzia, P.; Birbe, R.; Lin, Z.; et al. Metformin effects on head and neck squamous carcinoma microenvironment: Window of opportunity trial. Laryngoscope 2017, 127, 1808–1815. [Google Scholar] [CrossRef]
- Morales, D.R.; Morris, A.D. Metformin in cancer treatment and prevention. Annu. Rev. Med. 2015, 66, 17–29. [Google Scholar] [CrossRef]
- Farmer, R.E.; Ford, D.; Forbes, H.J.; Chaturvedi, N.; Kaplan, R.; Smeeth, L.; Bhaskaran, K. Metformin and cancer in type 2 diabetes: A systematic review and comprehensive bias evaluation. Int. J. Epidemiol. 2017, 46, 728–744. [Google Scholar] [CrossRef]
- Chen, K.; Li, Y.; Guo, Z.; Zeng, Y.; Zhang, W.; Wang, H. Metformin: Current clinical applications in nondiabetic patients with cancer. Aging 2020, 12, 3993–4009. [Google Scholar] [CrossRef]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Gui, D.Y.; Sullivan, L.B.; Luengo, A.; Hosios, A.M.; Bush, L.N.; Gitego, N.; Davidson, S.M.; Freinkman, E.; Thomas, C.J.; Vander Heiden, M.G. Environment Dictates Dependence on Mitochondrial Complex I for NAD+ and Aspartate Production and Determines Cancer Cell Sensitivity to Metformin. Cell Metab. 2016, 24, 716–727. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Romero, I.L.; Litchfield, L.M.; Lengyel, E.; Locasale, J.W. Metformin Targets Central Carbon Metabolism and Reveals Mitochondrial Requirements in Human Cancers. Cell Metab. 2016, 24, 728–739. [Google Scholar] [CrossRef] [Green Version]
- Wahdan-Alaswad, R.S.; Edgerton, S.M.; Salem, H.S.; Thor, A.D. Metformin Targets Glucose Metabolism in Triple Negative Breast Cancer. J. Oncol. Transl. Res. 2018, 4, 129. [Google Scholar] [CrossRef]
- Hu, L.; Zeng, Z.; Xia, Q.; Liu, Z.; Feng, X.; Chen, J.; Huang, M.; Chen, L.; Fang, Z.; Liu, Q.; et al. Metformin attenuates hepatoma cell proliferation by decreasing glycolytic flux through the HIF-1α/PFKFB3/PFK1 pathway. Life Sci. 2019, 239, 116966. [Google Scholar] [CrossRef]
- Harada, K.; Ferdous, T.; Harada, T.; Ueyama, Y. Metformin in combination with 5-fluorouracil suppresses tumor growth by inhibiting the Warburg effect in human oral squamous cell carcinoma. Int. J. Oncol. 2016, 49, 276–284. [Google Scholar] [CrossRef] [Green Version]
- Ma, M.; Ma, C.; Li, P.; Ma, C.; Ping, F.; Li, W.; Xu, L.; Zhang, H.; Sun, Q.; Li, Y. Low glucose enhanced metformin’s inhibitory effect on pancreatic cancer cells by suppressing glycolysis and inducing energy stress via up-regulation of miR-210-5p. Cell Cycle 2020, 19, 2168–2181. [Google Scholar] [CrossRef]
- Saladini, S.; Aventaggiato, M.; Barreca, F.; Morgante, E.; Sansone, L.; Russo, M.A.; Tafani, M. Metformin Impairs Glutamine Metabolism and Autophagy in Tumour Cells. Cells 2019, 8, 49. [Google Scholar] [CrossRef] [Green Version]
- Fendt, S.M.; Bell, E.L.; Keibler, M.A.; Davidson, S.M.; Wirth, G.J.; Fiske, B.; Mayers, J.R.; Schwab, M.; Bellinger, G.; Csibi, A.; et al. Metformin decreases glucose oxidation and increases the dependency of prostate cancer cells on reductive glutamine metabolism. Cancer Res. 2013, 73, 4429–4438. [Google Scholar] [CrossRef] [Green Version]
- Sivalingam, V.N.; Latif, A.; Kitson, S.; McVey, R.; Finegan, K.G.; Marshall, K.; Lisanti, M.P.; Sotgia, F.; Stratford, I.J.; Crosbie, E.J. Hypoxia and hyperglycaemia determine why some endometrial tumours fail to respond to metformin. Br. J. Cancer 2020, 122, 62–71. [Google Scholar] [CrossRef]
- Khan, H.; Anshu, A.; Prasad, A.; Roy, S.; Jeffery, J.; Kittipongdaja, W.; Yang, D.T.; Schieke, S.M. Metabolic Rewiring in Response to Biguanides Is Mediated by mROS/HIF-1a in Malignant Lymphocytes. Cell Rep. 2019, 29, 3009–3018.e4. [Google Scholar] [CrossRef] [Green Version]
- Lord, S.R.; Cheng, W.C.; Liu, D.; Gaude, E.; Haider, S.; Metcalf, T.; Patel, N.; Teoh, E.J.; Gleeson, F.; Bradley, K.; et al. Integrated Pharmacodynamic Analysis Identifies Two Metabolic Adaption Pathways to Metformin in Breast Cancer. Cell Metab. 2018, 28, 679–688.e4. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Bermudez, J.; Baudrier, L.; La, K.; Zhu, X.G.; Fidelin, J.; Sviderskiy, V.O.; Papagiannakopoulos, T.; Molina, H.; Snuderl, M.; Lewis, C.A.; et al. Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat. Cell Biol. 2018, 20, 775–781. [Google Scholar] [CrossRef]
- Liu, Y.; He, C.; Huang, X. Metformin partially reverses the carboplatin-resistance in NSCLC by inhibiting glucose metabolism. Oncotarget 2017, 8, 75206–75216. [Google Scholar] [CrossRef] [Green Version]
- Salani, B.; Del Rio, A.; Marini, C.; Sambuceti, G.; Cordera, R.; Maggi, D. Metformin, cancer and glucose metabolism. Endocr. Relat. Cancer 2014, 21, R461–R471. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Hang, C.; Jiang, T.; Yi, S.; Shao, W.; Li, W.; Lin, D. Nuclear Magnetic Resonance-Based Metabolomic Analysis of the Anticancer Effect of Metformin Treatment on Cholangiocarcinoma Cells. Front. Oncol. 2020, 10, 570516. [Google Scholar] [CrossRef]
- Salvatore, T.; Pafundi, P.C.; Marfella, R.; Sardu, C.; Rinaldi, L.; Monaco, L.; Ricozzi, C.; Imbriani, S.; Nevola, R.; Adinolfi, L.E.; et al. Metformin lactic acidosis: Should we still be afraid? Diabetes Res. Clin. Pract. 2019, 157, 107879. [Google Scholar] [CrossRef]
- Vincent, E.E.; Sergushichev, A.; Griss, T.; Gingras, M.C.; Samborska, B.; Ntimbane, T.; Coelho, P.P.; Blagih, J.; Raissi, T.C.; Choinière, L.; et al. Mitochondrial Phosphoenolpyruvate Carboxykinase Regulates Metabolic Adaptation and Enables Glucose-Independent Tumor Growth. Mol. Cell 2015, 60, 195–207. [Google Scholar] [CrossRef] [Green Version]
- Javeshghani, S.; Zakikhani, M.; Austin, S.; Bazile, M.; Blouin, M.J.; Topisirovic, I.; St-Pierre, J.; Pollak, M.N. Carbon source and myc expression influence the antiproliferative actions of metformin. Cancer Res. 2012, 72, 6257–6267. [Google Scholar] [CrossRef] [Green Version]
- Birsoy, K.; Wang, T.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 2015, 162, 540–551. [Google Scholar] [CrossRef] [Green Version]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Kim, J.; DeBerardinis, R.J. Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metab. 2019, 30, 434–446. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Washio, J.; Sato, S.; Abiko, Y.; Shinohara, Y.; Kobayashi, Y.; Otani, H.; Sasaki, S.; Wang, X.; Takahashi, N. Rewired Cellular Metabolic Profiles in Response to Metformin under Different Oxygen and Nutrient Conditions. Int. J. Mol. Sci. 2022, 23, 989. https://doi.org/10.3390/ijms23020989
Liu S, Washio J, Sato S, Abiko Y, Shinohara Y, Kobayashi Y, Otani H, Sasaki S, Wang X, Takahashi N. Rewired Cellular Metabolic Profiles in Response to Metformin under Different Oxygen and Nutrient Conditions. International Journal of Molecular Sciences. 2022; 23(2):989. https://doi.org/10.3390/ijms23020989
Chicago/Turabian StyleLiu, Shan, Jumpei Washio, Satoko Sato, Yuki Abiko, Yuta Shinohara, Yuri Kobayashi, Haruki Otani, Shiori Sasaki, Xiaoyi Wang, and Nobuhiro Takahashi. 2022. "Rewired Cellular Metabolic Profiles in Response to Metformin under Different Oxygen and Nutrient Conditions" International Journal of Molecular Sciences 23, no. 2: 989. https://doi.org/10.3390/ijms23020989