
Involvement of the Innate Immune System in the Pathogenesis of Chronic Obstructive Pulmonary Disease


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
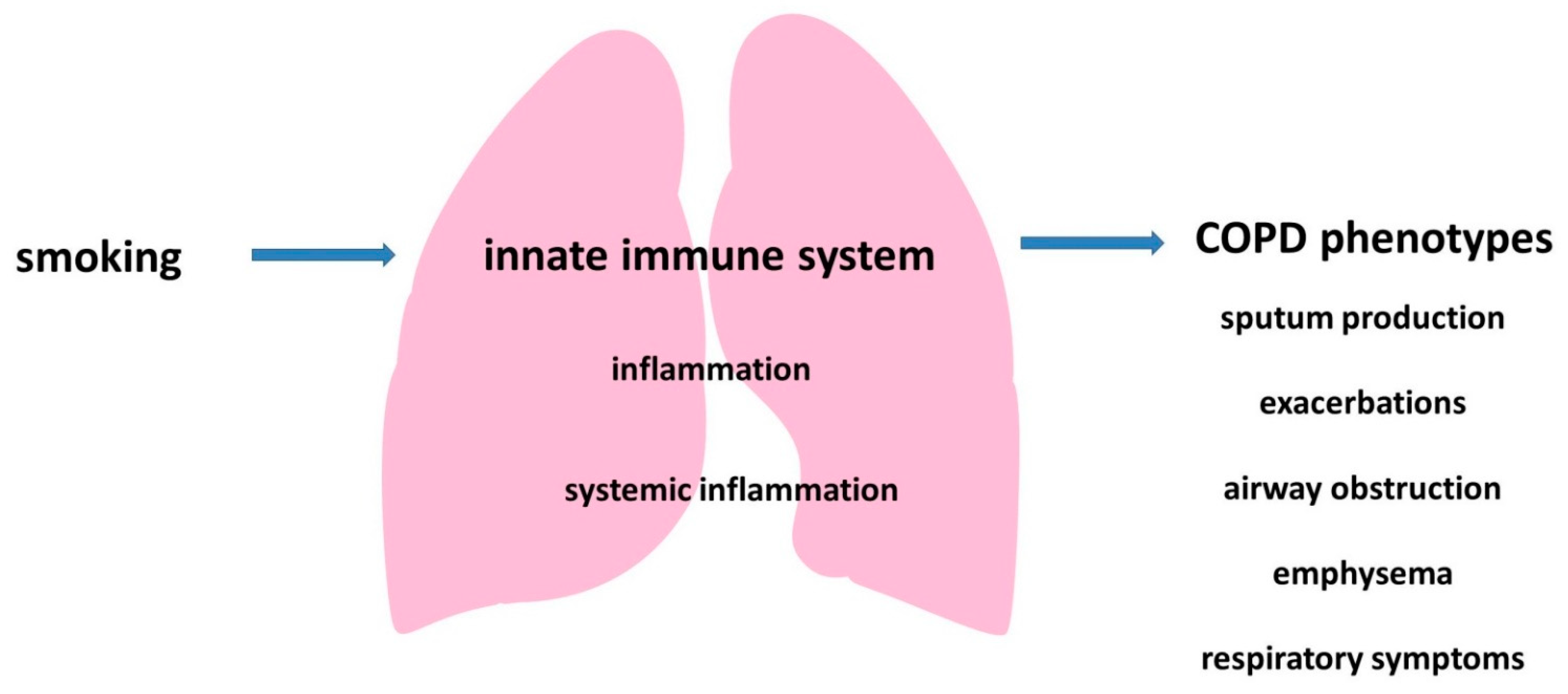
2. The Innate Immune System
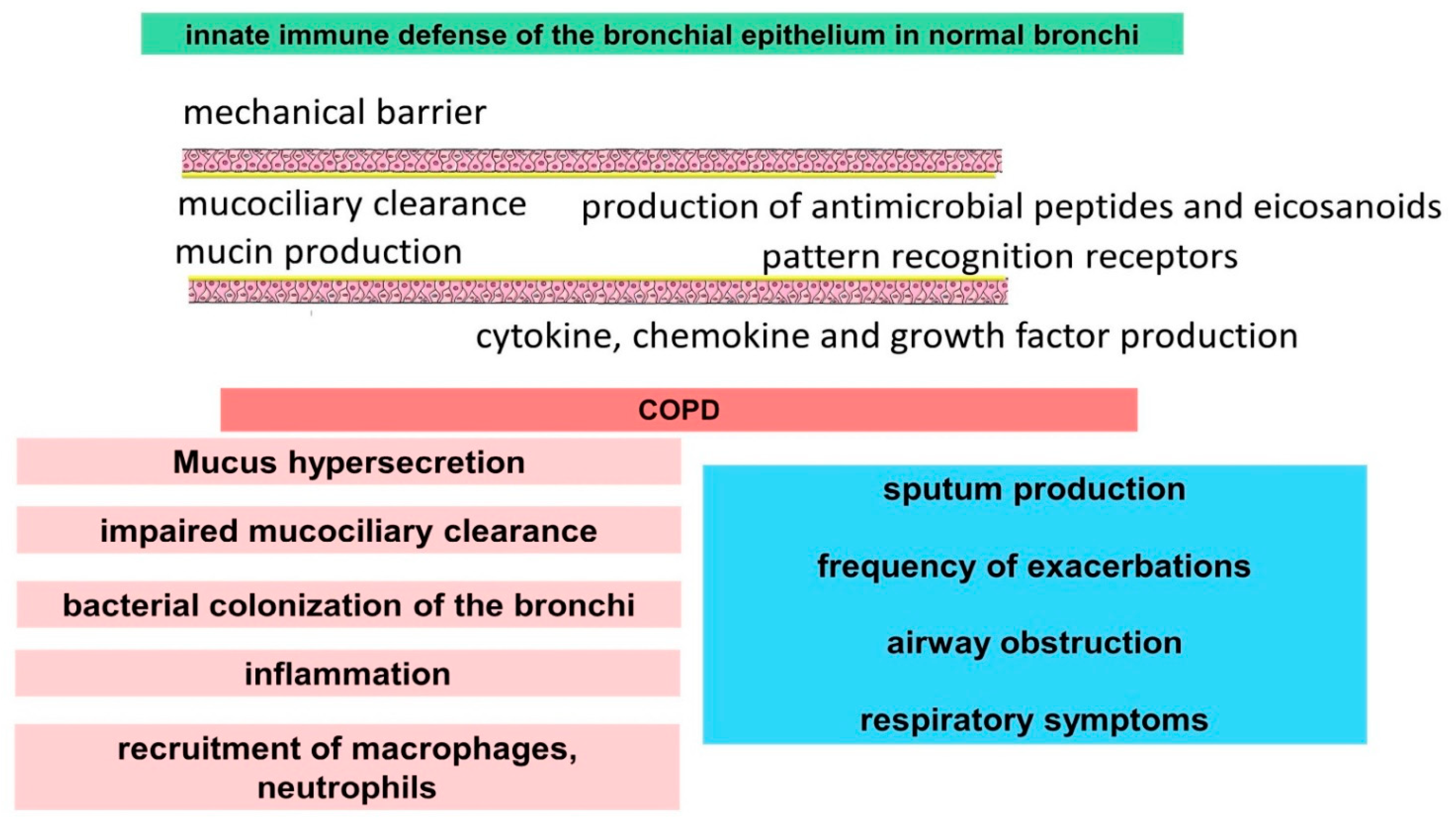
2.1. The Innate Immune Function of Airway Epithelial Cells
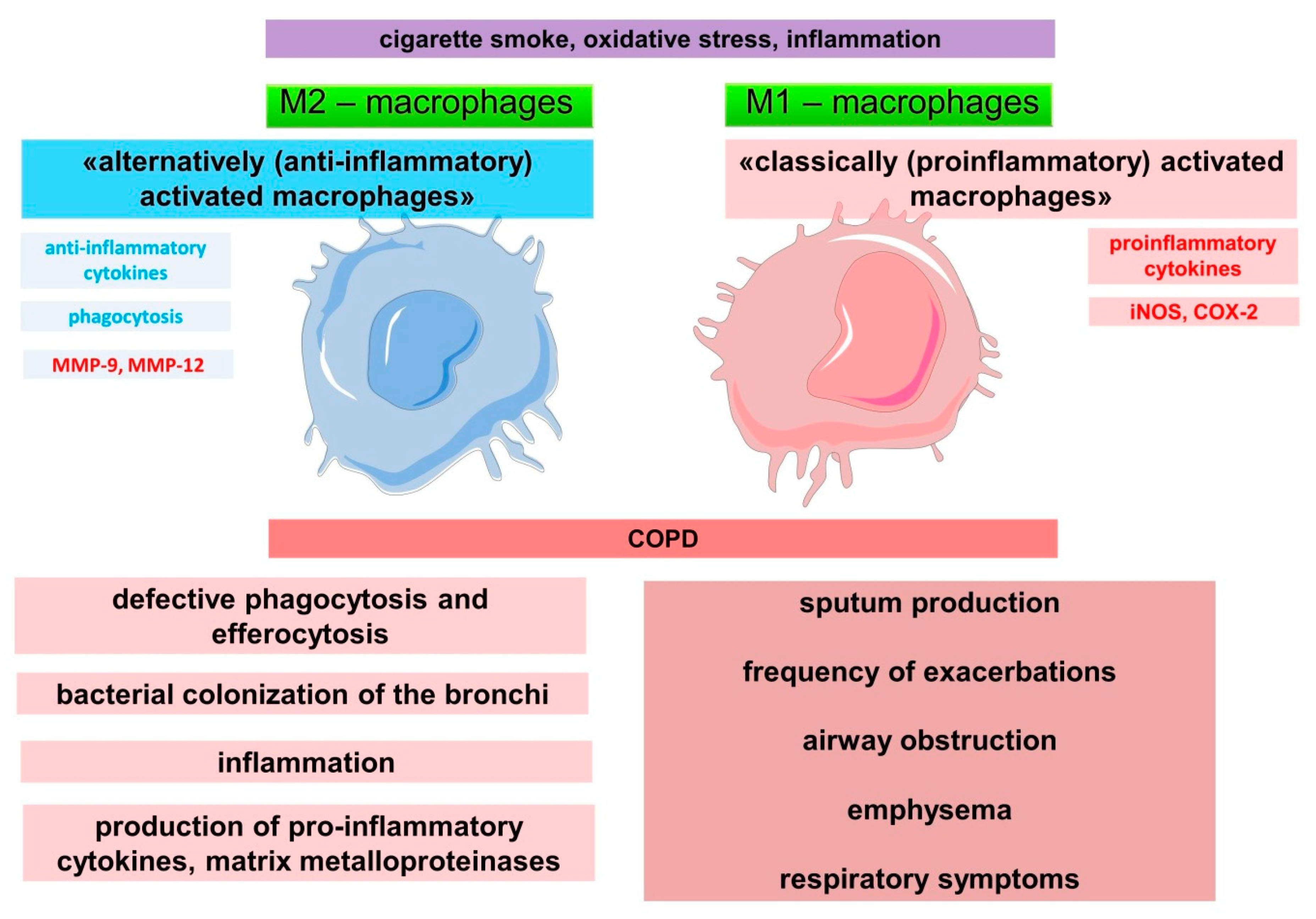
2.2. Role of Macrophages in the Innate Immune System and Pathogenesis of COPD
2.3. Role of Other Cells in the Regulation of the Innate Immune System and the Pathogenesis of COPD
3. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Soriano, J.B.; Kendrick, P.J.; Paulson, K.R.; Gupta, V.; Abrams, E.M.; Adedoyin, R.A.; Adhikari, T.B.; Advani, S.M.; Agrawal, A.; Ahmadian, E.; et al. Prevalence and attributable health burden of chronic respiratory diseases, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Respir. Med. 2020, 8, 585–596. [Google Scholar] [CrossRef]
- Çolak, Y.; Afzal, S.; Nordestgaard, B.G.; Vestbo, J.; Lange, P. Prevalence, Characteristics, and Prognosis of Early Chronic Obstructive Pulmonary Disease. The Copenhagen General Population Study. Am. J. Respir. Crit. Care Med. 2020, 201, 671–680. [Google Scholar] [CrossRef]
- Hashemi, S.Y.; Momenabadi, V.; Faramarzi, A.; Kiani, A. Trends in burden of chronic obstructive pulmonary disease in Iran, 1995–2015: Findings from the global burden of disease study. Arch. Public Health 2020, 78, 45. [Google Scholar] [CrossRef]
- Blanco, I.; Diego, I.; Bueno, P.; Casas-Maldonado, F.; Miravitlles, M. Geographic distribution of COPD prevalence in the world displayed by Geographic Information System maps. Eur. Respir. J. 2019, 54, 1900610. [Google Scholar] [CrossRef] [PubMed]
- Rycroft, C.E.; Heyes, A.; Lanza, L.; Becker, K. Epidemiology of chronic obstructive pulmonary disease: A literature review. Int. J. Chronic Obstr. Pulm. Dis. 2012, 7, 457–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quaderi, S.A.; Hurst, J.R. The unmet global burden of COPD. Glob. Health Epidemiol. Genom. 2018, 3, e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laniado-Laborín, R. Smoking and chronic obstructive pulmonary disease (COPD). Parallel epidemics of the 21 century. Int. J. Environ. Res. Public Health 2009, 6, 209–224. [Google Scholar] [CrossRef] [Green Version]
- Tkacova, R. Systemic inflammation in chronic obstructive pulmonary disease: May adipose tissue play a role? Review of the literature and future perspectives. Mediators Inflamm. 2010, 2010, 585989. [Google Scholar] [CrossRef]
- Agustí, A.G.N. Systemic Effects of Chronic Obstructive Pulmonary Disease. Proc. Am. Thorac. Soc. 2005, 2, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Johns, D.P.; Walters, J.A.E.; Walters, E.H. Diagnosis and early detection of COPD using spirometry. J. Thorac. Dis. 2014, 6, 1557–1569. [Google Scholar] [CrossRef]
- Ruppel, G.L.; Carlin, B.W.; Hart, M.; Doherty, D.E. Office Spirometry in Primary Care for the Diagnosis and Management of COPD: National Lung Health Education Program Update. Respir. Care 2018, 63, 242–252. [Google Scholar] [CrossRef] [Green Version]
- Kotlyarov, S.N. Spirometry screening in evaluation of chronic obstructive pulmonary disease at primary care. I.P. Pavlov. Russ. Med. Biol. Her. 2011, 19, 91–95. [Google Scholar] [CrossRef]
- Viniol, C.; Vogelmeier, C.F. Exacerbations of COPD. Eur. Respir. Rev. 2018, 27, 170103. [Google Scholar] [CrossRef] [Green Version]
- Sethi, S.; Maloney, J.; Grove, L.; Wrona, C.; Berenson, C.S. Airway inflammation and bronchial bacterial colonization in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2006, 173, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Hogea, S.-P.; Tudorache, E.; Fildan, A.P.; Fira-Mladinescu, O.; Marc, M.; Oancea, C. Risk factors of chronic obstructive pulmonary disease exacerbations. Clin. Respir. J. 2020, 14, 183–197. [Google Scholar] [CrossRef]
- Wilkinson, T.M.; Patel, I.S.; Wilks, M.; Donaldson, G.C.; Wedzicha, J.A. Airway bacterial load and FEV1 decline in patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2003, 167, 1090–1095. [Google Scholar] [CrossRef] [PubMed]
- Buchmann, K. Evolution of Innate Immunity: Clues from Invertebrates via Fish to Mammals. Front. Immunol. 2014, 5, 459. [Google Scholar] [CrossRef] [Green Version]
- Alberts, B.; Johnson, A.; Lewis, J.; Morgan, D.; Raff, M.; Roberts, K.; Walter, P.; Wilson, J.; Hunt, T. Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002; Innate Immunity. Available online: https://www.ncbi.nlm.nih.gov/books/NBK26846/ (accessed on 26 December 2021).
- Kotlyarov, S.; Kotlyarova, A. Molecular Mechanisms of Lipid Metabolism Disorders in Infectious Exacerbations of Chronic Obstructive Pulmonary Disease. Int. J. Mol. Sci. 2021, 22, 7634. [Google Scholar] [CrossRef] [PubMed]
- Töpfer, E.; Boraschi, D.; Italiani, P. Innate Immune Memory: The Latest Frontier of Adjuvanticity. J. Immunol. Res. 2015, 2015, 478408. [Google Scholar] [CrossRef] [Green Version]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, K. Tobacco smoke: Involvement of reactive oxygen species and stable free radicals in mechanisms of oxidative damage, carcinogenesis and synergistic effects with other respirable particles. Int. J. Environ. Res. Public Health 2009, 6, 445–462. [Google Scholar] [CrossRef] [PubMed]
- Bals, R.; Hiemstra, P.S. Innate immunity in the lung: How epithelial cells fight against respiratory pathogens. Eur. Respir. J. 2004, 23, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Parker, D.; Prince, A. Innate immunity in the respiratory epithelium. Am. J. Respir. Cell Mol. Biol. 2011, 45, 189–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, S.L.; Goldblatt, D.L.; Evans, S.E.; Tuvim, M.J.; Dickey, B.F. Airway Epithelial Innate Immunity. Front. Physiol. 2021, 12, 749077. [Google Scholar] [CrossRef]
- Knowles, M.R.; Boucher, R.C. Mucus clearance as a primary innate defense mechanism for mammalian airways. J. Clin. Investig. 2002, 109, 571–577. [Google Scholar] [CrossRef]
- Hariri, B.M.; Cohen, N.A. New insights into upper airway innate immunity. Am. J. Rhinol. Allergy 2016, 30, 319–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahy, J.V.; Dickey, B.F. Airway mucus function and dysfunction. N. Engl. J. Med. 2010, 363, 2233–2247. [Google Scholar] [CrossRef] [Green Version]
- Hiemstra, P.S. Antimicrobial peptides in the real world: Implications for cystic fibrosis. Eur. Respir. J. 2007, 29, 617–618. [Google Scholar] [CrossRef] [PubMed]
- Thornton, D.J.; Rousseau, K.; McGuckin, M.A. Structure and function of the polymeric mucins in airways mucus. Annu. Rev. Physiol. 2008, 70, 459–486. [Google Scholar] [CrossRef]
- Rose, M.C.; Voynow, J.A. Respiratory tract mucin genes and mucin glycoproteins in health and disease. Physiol. Rev. 2006, 86, 245–278. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Ye, Z. The Potential Role and Regulatory Mechanisms of MUC5AC in Chronic Obstructive Pulmonary Disease. Molecules 2020, 25, 4437. [Google Scholar] [CrossRef]
- Saco, T.V.; Breitzig, M.T.; Lockey, R.F.; Kolliputi, N. Epigenetics of Mucus Hypersecretion in Chronic Respiratory Diseases. Am. J. Respir. Cell Mol. Biol. 2018, 58, 299–309. [Google Scholar] [CrossRef]
- Buisine, M.P.; Devisme, L.; Copin, M.C.; Durand-Réville, M.; Gosselin, B.; Aubert, J.P.; Porchet, N. Developmental mucin gene expression in the human respiratory tract. Am. J. Respir. Cell Mol. Biol. 1999, 20, 209–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radicioni, G.; Ceppe, A.; Ford, A.A.; Alexis, N.E.; Barr, R.G.; Bleecker, E.R.; Christenson, S.A.; Cooper, C.B.; Han, M.K.; Hansel, N.N.; et al. Airway mucin MUC5AC and MUC5B concentrations and the initiation and progression of chronic obstructive pulmonary disease: An analysis of the SPIROMICS cohort. Lancet Respir. Med. 2021, 9, 1241–1254. [Google Scholar] [CrossRef]
- Leopold, P.L.; O’Mahony, M.J.; Lian, X.J.; Tilley, A.E.; Harvey, B.G.; Crystal, R.G. Smoking is associated with shortened airway cilia. PLoS ONE 2009, 4, e8157. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Defensins: Antimicrobial peptides of innate immunity. Nat. Rev. Immunol. 2003, 3, 710–720. [Google Scholar] [CrossRef]
- Herr, C.; Beisswenger, C.; Hess, C.; Kandler, K.; Suttorp, N.; Welte, T.; Schroeder, J.-M.; Vogelmeier, C.; R Bals for the CAPNETZ Study Group. Suppression of pulmonary innate host defence in smokers. Thorax 2009, 64, 144–149. [Google Scholar] [CrossRef] [Green Version]
- Bals, R.; Wang, X.; Wu, Z.; Freeman, T.; Bafna, V.; Zasloff, M.; Wilson, J.M. Human beta-defensin 2 is a salt-sensitive peptide antibiotic expressed in human lung. J. Clin. Investig. 1998, 102, 874–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCray, P.B., Jr.; Bentley, L. Human airway epithelia express a beta-defensin. Am. J. Respir. Cell Mol. Biol. 1997, 16, 343–349. [Google Scholar] [CrossRef]
- Feng, Z.; Jiang, B.; Chandra, J.; Ghannoum, M.; Nelson, S.; Weinberg, A. Human beta-defensins: Differential activity against candidal species and regulation by Candida albicans. J. Dent. Res. 2005, 84, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.S.; Wiens, M.E.; Smith, J.G. Antiviral mechanisms of human defensins. J. Mol. Biol. 2013, 425, 4965–4980. [Google Scholar] [CrossRef]
- Proud, D.; Sanders, S.P.; Wiehler, S. Human rhinovirus infection induces airway epithelial cell production of human beta-defensin 2 both in vitro and in vivo. J. Immunol. 2004, 172, 4637–4645. [Google Scholar] [CrossRef] [PubMed]
- Harder, J.; Meyer-Hoffert, U.; Teran, L.M.; Schwichtenberg, L.; Bartels, J.; Maune, S.; Schröder, J.M. Mucoid Pseudomonas aeruginosa, TNF-alpha, and IL-1beta, but not IL-6, induce human beta-defensin-2 in respiratory epithelia. Am. J. Respir. Cell Mol. Biol. 2000, 22, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Baines, K.J.; Wright, T.K.; Simpson, J.L.; McDonald, V.M.; Wood, L.G.; Parsons, K.S.; Wark, P.A.; Gibson, P.G. Airway β-Defensin-1 Protein Is Elevated in COPD and Severe Asthma. Mediators Inflamm. 2015, 2015, 407271. [Google Scholar] [CrossRef] [Green Version]
- Cane, J.; Tregidgo, L.; Thulborn, S.; Finch, D.; Bafadhel, M. Antimicrobial Peptides SLPI and Beta Defensin-1 in Sputum are Negatively Correlated with FEV(1). Int. J. Chronic Obstr. Pulm. Dis. 2021, 16, 1437–1447. [Google Scholar] [CrossRef]
- Liao, Z.; Dong, J.; Hu, X.; Wang, T.; Wan, C.; Li, X.o.; Li, L.; Guo, L.; Xu, D.; Wen, F. Enhanced expression of human β-defensin 2 in peripheral lungs of patients with chronic obstructive pulmonary disease. Peptides 2012, 38, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.; Cho, Y.; Dinh, N.N.; Waring, A.J.; Lehrer, R.I. Activities of LL-37, a cathelin-associated antimicrobial peptide of human neutrophils. Antimicrob. Agents Chemother. 1998, 42, 2206–2214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frohm, M.; Agerberth, B.; Ahangari, G.; Stâhle-Bäckdahl, M.; Lidén, S.; Wigzell, H.; Gudmundsson, G.H. The expression of the gene coding for the antibacterial peptide LL-37 is induced in human keratinocytes during inflammatory disorders. J. Biol. Chem. 1997, 272, 15258–15263. [Google Scholar] [CrossRef] [Green Version]
- Bals, R.; Weiner, D.J.; Moscioni, A.D.; Meegalla, R.L.; Wilson, J.M. Augmentation of innate host defense by expression of a cathelicidin antimicrobial peptide. Infect. Immun. 1999, 67, 6084–6089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.-Y.; Xiao, W.; Zhu, M.-X.; Yang, Z.-H.; Pan, X.-J.; Zhang, Y.; Sun, C.-C.; Xing, Y. The effect of human antibacterial peptide LL-37 in the pathogenesis of chronic obstructive pulmonary disease. Respir. Med. 2012, 106, 1680–1689. [Google Scholar] [CrossRef] [Green Version]
- De Smet, K.; Contreras, R. Human Antimicrobial Peptides: Defensins, Cathelicidins and Histatins. Biotechnol. Lett. 2005, 27, 1337–1347. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.G.; Davidson, D.J.; Gold, M.R.; Bowdish, D.; Hancock, R.E.W. The Human Antimicrobial Peptide LL-37 Is a Multifunctional Modulator of Innate Immune Responses. J. Immunol. 2002, 169, 3883–3891. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, K.; Okumura, K.; Isogai, H.; Isogai, E. The Human Cathelicidin Antimicrobial Peptide LL-37 and Mimics are Potential Anticancer Drugs. Front. Oncol. 2015, 5, 144. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Sunkara, L.T.; Zeng, X.; Deng, Z.; Myers, S.M.; Zhang, G. Differential regulation of human cathelicidin LL-37 by free fatty acids and their analogs. Peptides 2013, 50, 129–138. [Google Scholar] [CrossRef]
- Schauber, J.; Iffland, K.; Frisch, S.; Kudlich, T.; Schmausser, B.; Eck, M.; Menzel, T.; Gostner, A.; Lührs, H.; Scheppach, W. Histone-deacetylase inhibitors induce the cathelicidin LL-37 in gastrointestinal cells. Mol. Immunol. 2004, 41, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Jiao, D.; Wong, C.-K.; Tsang, M.S.-M.; Chu, I.M.-T.; Liu, D.; Zhu, J.; Chu, M.; Lam, C.W.-K. Activation of Eosinophils Interacting with Bronchial Epithelial Cells by Antimicrobial Peptide LL-37: Implications in Allergic Asthma. Sci. Rep. 2017, 7, 1848. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhu, M.; Yang, Z.; Pan, X.; Jiang, Y.; Sun, C.; Wang, Q.; Xiao, W. The human Cathelicidin LL-37 induces MUC5AC mucin production by airway epithelial cells via TACE-TGF-α-EGFR pathway. Exp. Lung Res. 2014, 40, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Parameswaran, G.I.; Sethi, S.; Murphy, T.F. Effects of bacterial infection on airway antimicrobial peptides and proteins in COPD. Chest 2011, 140, 611–617. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.-M.; Guo, Y.-F.; Zhang, H.-S.; Sun, T.-Y. Antimicrobial peptide LL-37 circulating levels in chronic obstructive pulmonary disease patients with high risk of frequent exacerbations. J. Thorac. Dis. 2015, 7, 740–745. [Google Scholar] [CrossRef]
- Tatsuta, M.; Kan, O.K.; Ishii, Y.; Yamamoto, N.; Ogawa, T.; Fukuyama, S.; Ogawa, A.; Fujita, A.; Nakanishi, Y.; Matsumoto, K. Effects of cigarette smoke on barrier function and tight junction proteins in the bronchial epithelium: Protective role of cathelicidin LL-37. Respir. Res. 2019, 20, 251. [Google Scholar] [CrossRef]
- Kotlyarov, S.; Kotlyarova, A. Anti-Inflammatory Function of Fatty Acids and Involvement of Their Metabolites in the Resolution of Inflammation in Chronic Obstructive Pulmonary Disease. Int. J. Mol. Sci. 2021, 22, 12803. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, T.; Shimokawaji, T.; Kanoh, S.; Rubin, B.K. Secretory phospholipases A2 are secreted from ciliated cells and increase mucin and eicosanoid secretion from goblet cells. Chest 2015, 147, 1599–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buczynski, M.W.; Dumlao, D.S.; Dennis, E.A. Thematic Review Series: Proteomics. An integrated omics analysis of eicosanoid biology. J. Lipid Res. 2009, 50, 1015–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakiela, B.; Gielicz, A.; Plutecka, H.; Hubalewska, M.; Mastalerz, L.; Bochenek, G.; Soja, J.; Januszek, R.; Musial, J.; Sanak, M. Eicosanoid biosynthesis during mucociliary and mucous metaplastic differentiation of bronchial epithelial cells. Prostaglandins Other Lipid Mediat. 2013, 106, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Profita, M.; Sala, A.; Riccobono, L.; Pace, E.; Paternò, A.; Zarini, S.; Siena, L.; Mirabella, A.; Bonsignore, G.; Vignola, A.M. 15(S)-HETE modulates LTB4 production and neutrophil chemotaxis in chronic bronchitis. Am. J. Physiol. -Cell Physiol. 2000, 279, C1249–C1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, C.K.W.; Polosa, R.; Holgate, S.T. Effect of 15-(s)-Hydroxyeicosatetraenoic Acid on Allergen-induced Asthmatic Responses. Am. Rev. Respir. Dis. 1990, 141, 1423–1427. [Google Scholar] [CrossRef] [PubMed]
- Nieves, D.; Moreno, J.J. Enantioselective effect of 12(S)-hydroxyeicosatetraenoic acid on 3T6 fibroblast growth through ERK 1/2 and p38 MAPK pathways and cyclin D1 activation. Biochem. Pharmacol. 2008, 76, 654–661. [Google Scholar] [CrossRef]
- Chavis, C.; Godard, P.; Crastes de Paulet, A.; Damon, M. Formation of lipoxins and leukotrienes by human alveolar macrophages incubated with 15(S)-HETE: A model for cellular cooperation between macrophages and airway epithelial cells. Eicosanoids 1992, 5, 203–211. [Google Scholar]
- Markovič, T.; Jakopin, Ž.; Dolenc, M.S.; Mlinarič-Raščan, I. Structural features of subtype-selective EP receptor modulators. Drug Discov. Today 2017, 22, 57–71. [Google Scholar] [CrossRef] [Green Version]
- Sastre, B.; del Pozo, V. Role of PGE2 in asthma and nonasthmatic eosinophilic bronchitis. Mediat. Inflamm. 2012, 2012, 645383. [Google Scholar] [CrossRef] [Green Version]
- Pavord, I.D.; Tattersfield, A.E. Bronchoprotective role for endogenous prostaglandin E2. Lancet 1995, 345, 436–438. [Google Scholar] [CrossRef]
- Gauvreau, G.M.; Watson, R.M.; O’Byrne, P.M. Protective effects of inhaled PGE2 on allergen-induced airway responses and airway inflammation. Am. J. Respir. Crit. Care Med. 1999, 159, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Birrell, M.A.; Maher, S.A.; Dekkak, B.; Jones, V.; Wong, S.; Brook, P.; Belvisi, M.G. Anti-inflammatory effects of PGE2 in the lung: Role of the EP4 receptor subtype. Thorax 2015, 70, 740–747. [Google Scholar] [CrossRef] [Green Version]
- Vancheri, C.; Mastruzzo, C.; Sortino, M.A.; Crimi, N. The lung as a privileged site for the beneficial actions of PGE2. Trends Immunol. 2004, 25, 40–46. [Google Scholar] [CrossRef]
- van der Pouw Kraan, T.C.; Boeije, L.C.; Smeenk, R.J.; Wijdenes, J.; Aarden, L.A. Prostaglandin-E2 is a potent inhibitor of human interleukin 12 production. J. Exp. Med. 1995, 181, 775–779. [Google Scholar] [CrossRef] [PubMed]
- Standiford, T.J.; Kunkel, S.L.; Rolfe, M.W.; Evanoff, H.L.; Allen, R.M.; Strieter, R.M. Regulation of human alveolar macrophage- and blood monocyte-derived interleukin-8 by prostaglandin E2 and dexamethasone. Am. J. Respir. Cell Mol. Biol. 1992, 6, 75–81. [Google Scholar] [CrossRef]
- Lazzeri, N.; Belvisi, M.G.; Patel, H.J.; Yacoub, M.H.; Chung, K.F.; Mitchell, J.A. Effects of prostaglandin E2 and cAMP elevating drugs on GM-CSF release by cultured human airway smooth muscle cells. Relevance to asthma therapy. Am. J. Respir. Cell Mol. Biol. 2001, 24, 44–48. [Google Scholar] [CrossRef]
- N’Guessan, P.D.; Temmesfeld-Wollbrück, B.; Zahlten, J.; Eitel, J.; Zabel, S.; Schmeck, B.; Opitz, B.; Hippenstiel, S.; Suttorp, N.; Slevogt, H. Moraxella catarrhalis induces ERK- and NF-κB-dependent COX-2 and prostaglandin E2 in lung epithelium. Eur. Respir. J. 2007, 30, 443–451. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Xu, Z.; Zhang, R.; Wu, Z.; Lim, J.-H.; Koga, T.; Li, J.-D.; Shen, H. Nontypeable Haemophilus influenzae induces COX-2 and PGE2 expression in lung epithelial cells via activation of p38 MAPK and NF-kappa B. Respir. Res. 2008, 9, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- N’Guessan, P.D.; Hippenstiel, S.; Etouem, M.O.; Zahlten, J.; Beermann, W.; Lindner, D.; Opitz, B.; Witzenrath, M.; Rosseau, S.; Suttorp, N.; et al. Streptococcus pneumoniae induced p38 MAPK- and NF-κB-dependent COX-2 expression in human lung epithelium. Am. J. Physiol. -Lung Cell. Mol. Physiol. 2006, 290, L1131–L1138. [Google Scholar] [CrossRef] [Green Version]
- Montuschi, P.; Kharitonov, S.A.; Ciabattoni, G.; Barnes, P.J. Exhaled leukotrienes and prostaglandins in COPD. Thorax 2003, 58, 585–588. [Google Scholar] [CrossRef] [Green Version]
- De Keijzer, S.; Meddens, M.B.M.; Torensma, R.; Cambi, A. The Multiple Faces of Prostaglandin E2 G-Protein Coupled Receptor Signaling during the Dendritic Cell Life Cycle. Int. J. Mol. Sci. 2013, 14, 6542–6555. [Google Scholar] [CrossRef] [Green Version]
- Bayarri, M.A.; Milara, J.; Estornut, C.; Cortijo, J. Nitric Oxide System and Bronchial Epithelium: More Than a Barrier. Front. Physiol. 2021, 12, 687381. [Google Scholar] [CrossRef] [PubMed]
- Kotlyarov, S. Diversity of Lipid Function in Atherogenesis: A Focus on Endothelial Mechanobiology. Int. J. Mol. Sci. 2021, 22, 11545. [Google Scholar] [CrossRef]
- Lane, C.; Knight, D.; Burgess, S.; Franklin, P.; Horak, F.; Legg, J.; Moeller, A.; Stick, S. Epithelial inducible nitric oxide synthase activity is the major determinant of nitric oxide concentration in exhaled breath. Thorax 2004, 59, 757–760. [Google Scholar] [CrossRef] [Green Version]
- Mattila, J.T.; Thomas, A.C. Nitric Oxide Synthase: Non-Canonical Expression Patterns. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Shirakami, G.; Zhan, X.; Johns, R.A. Regulation of ciliary beat frequency by the nitric oxide-cyclic guanosine monophosphate signaling pathway in rat airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2000, 23, 175–181. [Google Scholar] [CrossRef]
- Hardiman, K.M.; McNicholas-Bevensee, C.M.; Fortenberry, J.; Myles, C.T.; Malik, B.; Eaton, D.C.; Matalon, S. Regulation of amiloride-sensitive Na(+) transport by basal nitric oxide. Am. J. Respir. Cell Mol. Biol. 2004, 30, 720–728. [Google Scholar] [CrossRef]
- Olson, N.; Greul, A.K.; Hristova, M.; Bove, P.F.; Kasahara, D.I.; van der Vliet, A. Nitric oxide and airway epithelial barrier function: Regulation of tight junction proteins and epithelial permeability. Arch. Biochem. Biophys. 2009, 484, 205–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogdan, C. Nitric oxide and the immune response. Nat. Immunol. 2001, 2, 907–916. [Google Scholar] [CrossRef] [PubMed]
- MacMicking, J.; Xie, Q.-W.; Nathan, C. Nitric oxide and macrophage function. Annu. Rev. Immunol. 1997, 15, 323–350. [Google Scholar] [CrossRef]
- Wink, D.A.; Hines, H.B.; Cheng, R.Y.S.; Switzer, C.H.; Flores-Santana, W.; Vitek, M.P.; Ridnour, L.A.; Colton, C.A. Nitric oxide and redox mechanisms in the immune response. J. Leukoc. Biol. 2011, 89, 873–891. [Google Scholar] [CrossRef] [Green Version]
- Flak, T.A.; Goldman, W.E. Autotoxicity of nitric oxide in airway disease. Am. J. Respir. Crit. Care Med. 1996, 154, S202–S206. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.H.; Springall, D.R.; Bishop, A.E.; Morgan, K.; Evans, T.J.; Habib, S.; Gruenert, D.C.; Gyi, K.M.; Hodson, M.E.; Yacoub, M.H.; et al. Lack of inducible nitric oxide synthase in bronchial epithelium: A possible mechanism of susceptibility to infection in cystic fibrosis. J. Pathol. 1998, 184, 323–331. [Google Scholar] [CrossRef]
- Robbins, R.A.; Barnes, P.J.; Springall, D.R.; Warren, J.B.; Kwon, O.J.; Buttery, L.D.; Wilson, A.J.; Geller, D.A.; Polak, J.M. Expression of inducible nitric oxide in human lung epithelial cells. Biochem. Biophys. Res. Commun. 1994, 203, 209–218. [Google Scholar] [CrossRef]
- Asano, K.; Chee, C.B.; Gaston, B.; Lilly, C.M.; Gerard, C.; Drazen, J.M.; Stamler, J.S. Constitutive and inducible nitric oxide synthase gene expression, regulation, and activity in human lung epithelial cells. Proc. Natl. Acad. Sci. USA 1994, 91, 10089–10093. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.-T.; Liu, X.-S.; Xu, Y.-J.; Ni, W.; Chen, S.-X. Expression of Nitric Oxide Synthase Isoenzyme in Lung Tissue of Smokers with and without Chronic Obstructive Pulmonary Disease. Chin. Med. J. 2015, 128, 1584–1589. [Google Scholar] [CrossRef]
- Mio, T.; Romberger, D.J.; Thompson, A.B.; Robbins, R.A.; Heires, A.; Rennard, S.I. Cigarette smoke induces interleukin-8 release from human bronchial epithelial cells. Am. J. Respir. Crit. Care Med. 1997, 155, 1770–1776. [Google Scholar] [CrossRef] [PubMed]
- Hellermann, G.R.; Nagy, S.B.; Kong, X.; Lockey, R.F.; Mohapatra, S.S. Mechanism of cigarette smoke condensate-induced acute inflammatory response in human bronchial epithelial cells. Respir. Res. 2002, 3, 22. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.-J.; Sun, Y.-H.; Jiang, J.-X.; Dong, X.-W.; Zhou, J.-Y.; Xie, Q.-M. Epoxyeicosatrienoic acids attenuate cigarette smoke extract-induced interleukin-8 production in bronchial epithelial cells. Prostaglandins Leukot. Essent. Fat. Acids 2015, 94, 13–19. [Google Scholar] [CrossRef]
- Keatings, V.M.; Collins, P.D.; Scott, D.M.; Barnes, P.J. Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am. J. Respir. Crit. Care Med. 1996, 153, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [Green Version]
- Mayer, A.K.; Muehmer, M.; Mages, J.; Gueinzius, K.; Hess, C.; Heeg, K.; Bals, R.; Lang, R.; Dalpke, A.H. Differential Recognition of TLR-Dependent Microbial Ligands in Human Bronchial Epithelial Cells. J. Immunol. 2007, 178, 3134–3142. [Google Scholar] [CrossRef]
- de C Ventura, G.M.; Le Goffic, R.; Balloy, V.; Plotkowski, M.-C.; Chignard, M.; Si-Tahar, M. TLR 5, but neither TLR2 nor TLR4, is involved in lung epithelial cell response to Burkholderia cenocepacia. FEMS Immunol. Med. Microbiol. 2008, 54, 37–44. [Google Scholar] [CrossRef] [Green Version]
- Go, H.; Koh, J.; Kim, H.S.; Jeon, Y.K.; Chung, D.H. Expression of toll-like receptor 2 and 4 is increased in the respiratory epithelial cells of chronic idiopathic interstitial pneumonia patients. Respir. Med. 2014, 108, 783–792. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- MacRedmond, R.E.; Greene, C.M.; Dorscheid, D.R.; McElvaney, N.G.; O’Neill, S.J. Epithelial expression of TLR4 is modulated in COPD and by steroids, salmeterol and cigarette smoke. Respir. Res. 2007, 8, 84. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.W.; Kim, D.R.; Kim, T.J.; Paik, J.H.; Chung, J.-H.; Jheon, S.; Huh, J.W.; Lee, J.-H.; Lee, C.-T. The association of down-regulated toll-like receptor 4 expression with airflow limitation and emphysema in smokers. Respir. Res. 2012, 13, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Stefano, A.; Ricciardolo, F.L.M.; Caramori, G.; Adcock, I.M.; Chung, K.F.; Barnes, P.J.; Brun, P.; Leonardi, A.; Andò, F.; Vallese, D.; et al. Bronchial inflammation and bacterial load in stable COPD is associated with TLR4 overexpression. Eur. Respir. J. 2017, 49, 1602006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, H.W.; Jeyaseelan, S.; Rino, J.G.; Voelker, D.R.; Wexler, R.B.; Campbell, K.; Harbeck, R.J.; Martin, R.J. TLR2 signaling is critical for Mycoplasma pneumoniae-induced airway mucin expression. J. Immunol. 2005, 174, 5713–5719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, D.; Conway, S. Bacterial modulation of mucosal innate immunity. Mol. Immunol. 2005, 42, 895–901. [Google Scholar] [CrossRef]
- Abreu, M.T.; Vora, P.; Faure, E.; Thomas, L.S.; Arnold, E.T.; Arditi, M. Decreased Expression of Toll-Like Receptor-4 and MD-2 Correlates with Intestinal Epithelial Cell Protection against Dysregulated Proinflammatory Gene Expression in Response to Bacterial Lipopolysaccharide. J. Immunol. 2001, 167, 1609–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sand, J.; Rønnow, S.R.; Langholm, L.; Karsdal, M.A.; Manon-Jensen, T.; Tal-Singer, R.; Miller, B.E.; Vestbo, J.; Leeming, D.J. A combination of biomarkers for fibrinolysis and basement membrane destruction predicts mortality in the ECLIPSE COPD cohort. Eur. Respir. J. 2020, 56, 4716. [Google Scholar] [CrossRef]
- Kieliszek, M.; Lipinski, B. Pathophysiological significance of protein hydrophobic interactions: An emerging hypothesis. Med. Hypotheses 2018, 110, 15–22. [Google Scholar] [CrossRef]
- Vlahos, R.; Bozinovski, S. Role of Alveolar Macrophages in Chronic Obstructive Pulmonary Disease. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapellos, T.S.; Bassler, K.; Aschenbrenner, A.C.; Fujii, W.; Schultze, J.L. Dysregulated Functions of Lung Macrophage Populations in COPD. J. Immunol. Res. 2018, 2018, 2349045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarling, J.D.; Lin, H.-S.; Hsu, S. Self-Renewal of Pulmonary Alveolar Macrophages: Evidence from Radiation Chimera Studies. J. Leukoc. Biol. 1987, 42, 443–446. [Google Scholar] [CrossRef]
- SHAPIRO, S.D. The Macrophage in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 1999, 160, S29–S32. [Google Scholar] [CrossRef] [PubMed]
- Pesci, A.; Balbi, B.; Majori, M.; Cacciani, G.; Bertacco, S.; Alciato, P.; Donner, C. Inflammatory cells and mediators in bronchial lavage of patients with chronic obstructive pulmonary disease. Eur. Respir. J. 1998, 12, 380–386. [Google Scholar] [CrossRef] [Green Version]
- Di Stefano, A.; Capelli, A.; Lusuardi, M.; Balbo, P.; Vecchio, C.; Maestrelli, P.; Mapp, C.E.; Fabbri, L.M.; Donner, C.F.; Saetta, M. Severity of Airflow Limitation Is Associated with Severity of Airway Inflammation in Smokers. Am. J. Respir. Crit. Care Med. 1998, 158, 1277–1285. [Google Scholar] [CrossRef]
- Retamales, I.; Elliott, W.M.; Meshi, B.; Coxson, H.O.; Pare, P.D.; Sciurba, F.C.; Rogers, R.M.; Hayashi, S.; Hogg, J.C. Amplification of Inflammation in emphysema and Its Association with Latent Adenoviral Infection. Am. J. Respir. Crit. Care Med. 2001, 164, 469–473. [Google Scholar] [CrossRef]
- Beckett, E.L.; Stevens, R.L.; Jarnicki, A.G.; Kim, R.Y.; Hanish, I.; Hansbro, N.G.; Deane, A.; Keely, S.; Horvat, J.C.; Yang, M.; et al. A new short-term mouse model of chronic obstructive pulmonary disease identifies a role for mast cell tryptase in pathogenesis. J. Allergy Clin. Immunol. 2013, 131, 752–762.e757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS–) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef]
- Saqib, U.; Sarkar, S.; Suk, K.; Mohammad, O.; Baig, M.S.; Savai, R. Phytochemicals as modulators of M1-M2 macrophages in inflammation. Oncotarget 2018, 9, 17937–17950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.; Liang, H.; Zen, K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef] [Green Version]
- Brown, B.N.; Valentin, J.E.; Stewart-Akers, A.M.; McCabe, G.P.; Badylak, S.F. Macrophage phenotype and remodeling outcomes in response to biologic scaffolds with and without a cellular component. Biomaterials 2009, 30, 1482–1491. [Google Scholar] [CrossRef] [Green Version]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V. Macrophages: The Potent Immunoregulatory Innate Immune Cells. Macrophage Act. -Biol. Dis. 2019. [Google Scholar] [CrossRef] [Green Version]
- Bazzan, E.; Turato, G.; Tinè, M.; Radu, C.M.; Balestro, E.; Rigobello, C.; Biondini, D.; Schiavon, M.; Lunardi, F.; Baraldo, S.; et al. Dual polarization of human alveolar macrophages progressively increases with smoking and COPD severity. Respir. Res. 2017, 18, 40. [Google Scholar] [CrossRef] [PubMed]
- Hodge, S.; Hodge, G.; Ahern, J.; Jersmann, H.; Holmes, M.; Reynolds, P.N. Smoking alters alveolar macrophage recognition and phagocytic ability: Implications in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2007, 37, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Berenson, C.S.; Kruzel, R.L.; Eberhardt, E.; Sethi, S. Phagocytic dysfunction of human alveolar macrophages and severity of chronic obstructive pulmonary disease. J. Infect. Dis. 2013, 208, 2036–2045. [Google Scholar] [CrossRef] [Green Version]
- Han, C.Z.; Juncadella, I.J.; Kinchen, J.M.; Buckley, M.W.; Klibanov, A.L.; Dryden, K.; Onengut-Gumuscu, S.; Erdbrügger, U.; Turner, S.D.; Shim, Y.M.; et al. Macrophages redirect phagocytosis by non-professional phagocytes and influence inflammation. Nature 2016, 539, 570–574. [Google Scholar] [CrossRef] [Green Version]
- Uribe-Querol, E.; Rosales, C. Phagocytosis: Our Current Understanding of a Universal Biological Process. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Aderem, A.; Underhill, D.M. Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol 1999, 17, 593–623. [Google Scholar] [CrossRef] [PubMed]
- Grassin-Delyle, S.; Abrial, C.; Salvator, H.; Brollo, M.; Naline, E.; Devillier, P. The Role of Toll-Like Receptors in the Production of Cytokines by Human Lung Macrophages. J. Innate Immun. 2020, 12, 63–73. [Google Scholar] [CrossRef]
- Karimi, K.; Sarir, H.; Mortaz, E.; Smit, J.J.; Hosseini, H.; De Kimpe, S.J.; Nijkamp, F.P.; Folkerts, G. Toll-like receptor-4 mediates cigarette smoke-induced cytokine production by human macrophages. Respir. Res. 2006, 7, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Nardo, D.; De Nardo, C.M.; Latz, E. New insights into mechanisms controlling the NLRP3 inflammasome and its role in lung disease. Am. J. Pathol. 2014, 184, 42–54. [Google Scholar] [CrossRef] [Green Version]
- Colarusso, C.; Terlizzi, M.; Molino, A.; Pinto, A.; Sorrentino, R. Role of the inflammasome in chronic obstructive pulmonary disease (COPD). Oncotarget 2017, 8, 81813–81824. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Xu, Q.; Sun, W.; Zhou, X.; Fu, D.; Mao, L. New Insights into the Role of NLRP3 Inflammasome in Pathogenesis and Treatment of Chronic Obstructive Pulmonary Disease. J. Inflamm. Res. 2021, 14, 4155–4168. [Google Scholar] [CrossRef]
- Faner, R.; Sobradillo, P.; Noguera, A.; Gomez, C.; Cruz, T.; López-Giraldo, A.; Ballester, E.; Soler, N.; Arostegui, J.I.; Pelegrín, P.; et al. The inflammasome pathway in stable COPD and acute exacerbations. ERJ Open Res. 2016, 2, 00002–02016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Lv, C.e.; Wang, S.; Ying, H.; Weng, Y.; Yu, W. NLRP3 Inflammasome Involves in the Acute Exacerbation of Patients with Chronic Obstructive Pulmonary Disease. Inflammation 2018, 41, 1321–1333. [Google Scholar] [CrossRef]
- Tran, H.B.; Lewis, M.D.; Tan, L.W.; Lester, S.E.; Baker, L.M.; Ng, J.; Hamilton-Bruce, M.A.; Hill, C.L.; Koblar, S.A.; Rischmueller, M.; et al. Immunolocalization of NLRP3 Inflammasome in Normal Murine Airway Epithelium and Changes following Induction of Ovalbumin-Induced Airway Inflammation. J. Allergy 2012, 2012, 819176. [Google Scholar] [CrossRef] [Green Version]
- Nachmias, N.; Langier, S.; Brzezinski, R.Y.; Siterman, M.; Stark, M.; Etkin, S.; Avriel, A.; Schwarz, Y.; Shenhar-Tsarfaty, S.; Bar-Shai, A. NLRP3 inflammasome activity is upregulated in an in-vitro model of COPD exacerbation. PLoS ONE 2019, 14, e0214622. [Google Scholar] [CrossRef] [Green Version]
- Hassane, M.; Demon, D.; Soulard, D.; Fontaine, J.; Keller, L.E.; Patin, E.C.; Porte, R.; Prinz, I.; Ryffel, B.; Kadioglu, A.; et al. Neutrophilic NLRP3 inflammasome-dependent IL-1β secretion regulates the γδT17 cell response in respiratory bacterial infections. Mucosal Immunol. 2017, 10, 1056–1068. [Google Scholar] [CrossRef] [Green Version]
- Peiró, T.; Patel, D.F.; Akthar, S.; Gregory, L.G.; Pyle, C.J.; Harker, J.A.; Birrell, M.A.; Lloyd, C.M.; Snelgrove, R.J. Neutrophils drive alveolar macrophage IL-1β release during respiratory viral infection. Thorax 2018, 73, 546–556. [Google Scholar] [CrossRef] [Green Version]
- Lea, S.R.; Reynolds, S.L.; Kaur, M.; Simpson, K.D.; Hall, S.R.; Hessel, E.M.; Singh, D. The effects of repeated Toll-like receptors 2 and 4 stimulation in COPD alveolar macrophages. Int. J. Chronic Obstr. Pulm. Dis. 2018, 13, 771–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soler, N.; Ewig, S.; Torres, A.; Filella, X.; Gonzalez, J.; Zaubet, A. Airway inflammation and bronchial microbial patterns in patients with stable chronic obstructive pulmonary disease. Eur. Respir. J. 1999, 14, 1015–1022. [Google Scholar] [CrossRef] [Green Version]
- Foronjy, R.; Nkyimbeng, T.; Wallace, A.; Thankachen, J.; Okada, Y.; Lemaitre, V.; D’Armiento, J. Transgenic expression of matrix metalloproteinase-9 causes adult-onset emphysema in mice associated with the loss of alveolar elastin. Am. J. Physiol. -Lung Cell. Mol. Physiol. 2008, 294, L1149–L1157. [Google Scholar] [CrossRef] [Green Version]
- Russell, R.E.K.; Thorley, A.; Culpitt, S.V.; Dodd, S.; Donnelly, L.E.; Demattos, C.; Fitzgerald, M.; Barnes, P.J. Alveolar macrophage-mediated elastolysis: Roles of matrix metalloproteinases, cysteine, and serine proteases. Am. J. Physiol. -Lung Cell. Mol. Physiol. 2002, 283, L867–L873. [Google Scholar] [CrossRef] [Green Version]
- Wallace, A.M.; Sandford, A.J.; English, J.C.; Burkett, K.M.; Li, H.; Finley, R.J.; Müller, N.L.; Coxson, H.O.; Paré, P.D.; Abboud, R.T. Matrix Metalloproteinase Expression by Human Alveolar Macrophages in Relation to Emphysema. COPD J. Chronic Obstr. Pulm. Dis. 2008, 5, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Finlay, G.A.; O’Driscoll, L.R.; Russell, K.J.; D’Arcy, E.M.; Masterson, J.B.; Fitzgerald, M.X.; O’Connor, C.M. Matrix Metalloproteinase Expression and Production by Alveolar Macrophages in Emphysema. Am. J. Respir. Crit. Care Med. 1997, 156, 240–247. [Google Scholar] [CrossRef] [Green Version]
- Kasahara, Y.; Tuder, R.M.; Cool, C.D.; Lynch, D.A.; Flores, S.C.; Voelkel, N.F. Endothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysema. Am. J. Respir. Crit. Care Med. 2001, 163, 737–744. [Google Scholar] [CrossRef] [Green Version]
- Konradt, C.; Hunter, C.A. Pathogen interactions with endothelial cells and the induction of innate and adaptive immunity. Eur. J. Immunol. 2018, 48, 1607–1620. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Shan, P.; Jiang, G.; Cohn, L.; Lee, P.J. Toll-like receptor 4 deficiency causes pulmonary emphysema. J. Clin. Investig. 2006, 116, 3050–3059. [Google Scholar] [CrossRef] [PubMed]
- Lacoste, J.Y.; Bousquet, J.; Chanez, P.; Van Vyve, T.; Simony-Lafontaine, J.; Lequeu, N.; Vic, P.; Enander, I.; Godard, P.; Michel, F.B. Eosinophilic and neutrophilic inflammation in asthma, chronic bronchitis, and chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 1993, 92, 537–548. [Google Scholar] [CrossRef]
- Barnes, P.J. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2016, 138, 16–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stănescu, D.; Sanna, A.; Veriter, C.; Kostianev, S.; Calcagni, P.G.; Fabbri, L.M.; Maestrelli, P. Airways obstruction, chronic expectoration, and rapid decline of FEV1 in smokers are associated with increased levels of sputum neutrophils. Thorax 1996, 51, 267–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagon, J.Y.; Chastre, J.; Trouillet, J.L.; Domart, Y.; Dombret, M.C.; Bornet, M.; Gibert, C. Characterization of distal bronchial microflora during acute exacerbation of chronic bronchitis. Use of the protected specimen brush technique in 54 mechanically ventilated patients. Am. Rev. Respir. Dis. 1990, 142, 1004–1008. [Google Scholar] [CrossRef]
- Jasper, A.E.; McIver, W.J.; Sapey, E.; Walton, G.M. Understanding the role of neutrophils in chronic inflammatory airway disease. F1000Research 2019, 8, F1000 Faculty Rev-557. [Google Scholar] [CrossRef]
- Donaldson, G.C.; Wedzicha, J.A. COPD exacerbations .1: Epidemiology. Thorax 2006, 61, 164–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traves, S.L.; Culpitt, S.V.; Russell, R.E.K.; Barnes, P.J.; Donnelly, L.E. Increased levels of the chemokines GROα and MCP-1 in sputum samples from patients with COPD. Thorax 2002, 57, 590–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rovina, N.; Koutsoukou, A.; Koulouris, N.G. Inflammation and Immune Response in COPD: Where Do We Stand? Mediat. Inflamm. 2013, 2013, 413735. [Google Scholar] [CrossRef] [Green Version]
- Knabe, L.; Petit, A.; Vernisse, C.; Charriot, J.; Pugnière, M.; Henriquet, C.; Sasorith, S.; Molinari, N.; Chanez, P.; Berthet, J.-P.; et al. CCSP counterbalances airway epithelial-driven neutrophilic chemotaxis. Eur. Respir. J. 2019, 54, 1802408. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.J.; Sapey, E.; Stockley, R. Neutrophil phenotypes in chronic lung disease. Expert Rev. Respir. Med. 2019, 13, 951–967. [Google Scholar] [CrossRef]
- Barnes, P.J.; Shapiro, S.D.; Pauwels, R.A. Chronic obstructive pulmonary disease: Molecular and cellularmechanisms. Eur. Respir. J. 2003, 22, 672–688. [Google Scholar] [CrossRef]
- Janoff, A.; Scherer, J. Mediators of inflammation in leukocyte lysosomes. IX. Elastinolytic activity in granules of human polymorphonuclear leukocytes. J. Exp. Med. 1968, 128, 1137–1155. [Google Scholar] [CrossRef]
- Kafienah, W.; Buttle, D.J.; Burnett, D.; Hollander, A.P. Cleavage of native type I collagen by human neutrophil elastase. Biochem. J. 1998, 330 Pt 2, 897–902. [Google Scholar] [CrossRef] [Green Version]
- Finkelstein, R.; Fraser, R.S.; Ghezzo, H.; Cosio, M.G. Alveolar inflammation and its relation to emphysema in smokers. Am. J. Respir. Crit. Care Med. 1995, 152, 1666–1672. [Google Scholar] [CrossRef]
- Shaykhiev, R.; Crystal, R.G. Innate immunity and chronic obstructive pulmonary disease: A mini-review. Gerontology 2013, 59, 481–489. [Google Scholar] [CrossRef] [Green Version]
- Banchereau, J.; Briere, F.; Caux, C.; Davoust, J.; Lebecque, S.; Liu, Y.J.; Pulendran, B.; Palucka, K. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000, 18, 767–811. [Google Scholar] [CrossRef]
- Holt, P.G.; Stumbles, P.A. Regulation of immunologic homeostasis in peripheral tissues by dendritic cells: The respiratory tract as a paradigm. J. Allergy Clin. Immunol. 2000, 105, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Freeman, C.M.; Curtis, J.L. Lung Dendritic Cells: Shaping Immune Responses throughout Chronic Obstructive Pulmonary Disease Progression. Am. J. Respir. Cell Mol. Biol. 2017, 56, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Tsoumakidou, M.; Koutsopoulos, A.V.; Tzanakis, N.; Dambaki, K.; Tzortzaki, E.; Zakynthinos, S.; Jeffery, P.K.; Siafakas, N.M. Decreased small airway and alveolar CD83+ dendritic cells in COPD. Chest 2009, 136, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Rogers, A.V.; Adelroth, E.; Hattotuwa, K.; Dewar, A.; Jeffery, P.K. Bronchial mucosal dendritic cells in smokers and ex-smokers with COPD: An electron microscopic study. Thorax 2008, 63, 108–114. [Google Scholar] [CrossRef] [Green Version]
- Divangahi, M.; Aaby, P.; Khader, S.A.; Barreiro, L.B.; Bekkering, S.; Chavakis, T.; van Crevel, R.; Curtis, N.; DiNardo, A.R.; Dominguez-Andres, J.; et al. Trained immunity, tolerance, priming and differentiation: Distinct immunological processes. Nat. Immunol. 2021, 22, 2–6. [Google Scholar] [CrossRef]
- Cheng, S.-C.; Quintin, J.; Cramer, R.A.; Shepardson, K.M.; Saeed, S.; Kumar, V.; Giamarellos-Bourboulis, E.J.; Martens, J.H.A.; Rao, N.A.; Aghajanirefah, A.; et al. mTOR- and HIF-1α–mediated aerobic glycolysis as metabolic basis for trained immunity. Science 2014, 345, 1250684. [Google Scholar] [CrossRef] [Green Version]
- Van Belleghem, J.D.; Bollyky, P.L. Macrophages and innate immune memory against Staphylococcus skin infections. Proc. Natl. Acad. Sci. USA 2018, 115, 11865–11867. [Google Scholar] [CrossRef] [Green Version]
- Netea, M.G.; Joosten, L.A.B.; Latz, E.; Mills, K.H.G.; Natoli, G.; Stunnenberg, H.G.; O’Neill, L.A.J.; Xavier, R.J. Trained immunity: A program of innate immune memory in health and disease. Science 2016, 352, aaf1098. [Google Scholar] [CrossRef] [Green Version]
- Netea, M.G.; Domínguez-Andrés, J.; Barreiro, L.B.; Chavakis, T.; Divangahi, M.; Fuchs, E.; Joosten, L.A.B.; van der Meer, J.W.M.; Mhlanga, M.M.; Mulder, W.J.M.; et al. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 2020, 20, 375–388. [Google Scholar] [CrossRef] [Green Version]
- Novakovic, B.; Habibi, E.; Wang, S.Y.; Arts, R.J.W.; Davar, R.; Megchelenbrink, W.; Kim, B.; Kuznetsova, T.; Kox, M.; Zwaag, J.; et al. β-Glucan Reverses the Epigenetic State of LPS-Induced Immunological Tolerance. Cell 2016, 167, 1354–1368.e1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Y.; Jeyanathan, M.; Haddadi, S.; Barra, N.G.; Vaseghi-Shanjani, M.; Damjanovic, D.; Lai, R.; Afkhami, S.; Chen, Y.; Dvorkin-Gheva, A.; et al. Induction of Autonomous Memory Alveolar Macrophages Requires T Cell Help and Is Critical to Trained Immunity. Cell 2018, 175, 1634–1650.e1617. [Google Scholar] [CrossRef] [Green Version]
- Zhong, C.; Yang, X.; Feng, Y.; Yu, J. Trained Immunity: An Underlying Driver of Inflammatory Atherosclerosis. Front. Immunol. 2020, 11, 284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bekkering, S.; Quintin, J.; Joosten, L.A.B.; Meer, J.W.M.v.d.; Netea, M.G.; Riksen, N.P. Oxidized Low-Density Lipoprotein Induces Long-Term Proinflammatory Cytokine Production and Foam Cell Formation via Epigenetic Reprogramming of Monocytes. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1731–1738. [Google Scholar] [CrossRef]
- Christ, A.; Günther, P.; Lauterbach, M.A.R.; Duewell, P.; Biswas, D.; Pelka, K.; Scholz, C.J.; Oosting, M.; Haendler, K.; Baßler, K.; et al. Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell 2018, 172, 162–175.e114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norata, G.D. Trained immunity and cardiovascular disease: Is it time for translation to humans? Cardiovasc. Res. 2018, 114, e41–e42. [Google Scholar] [CrossRef]
- Mehta, D.; Petes, C.; Gee, K.; Basta, S. The Role of Virus Infection in Deregulating the Cytokine Response to Secondary Bacterial Infection. J. Interferon Cytokine Res. 2015, 35, 925–934. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kotlyarov, S. Involvement of the Innate Immune System in the Pathogenesis of Chronic Obstructive Pulmonary Disease. Int. J. Mol. Sci. 2022, 23, 985. https://doi.org/10.3390/ijms23020985
Kotlyarov S. Involvement of the Innate Immune System in the Pathogenesis of Chronic Obstructive Pulmonary Disease. International Journal of Molecular Sciences. 2022; 23(2):985. https://doi.org/10.3390/ijms23020985
Chicago/Turabian StyleKotlyarov, Stanislav. 2022. "Involvement of the Innate Immune System in the Pathogenesis of Chronic Obstructive Pulmonary Disease" International Journal of Molecular Sciences 23, no. 2: 985. https://doi.org/10.3390/ijms23020985