
Tissue Proteome of 2-Hydroxyacyl-CoA Lyase Deficient Mice Reveals Peroxisome Proliferation and Activation of ω-Oxidation

 , , , , ,
, , , , ,
Abstract
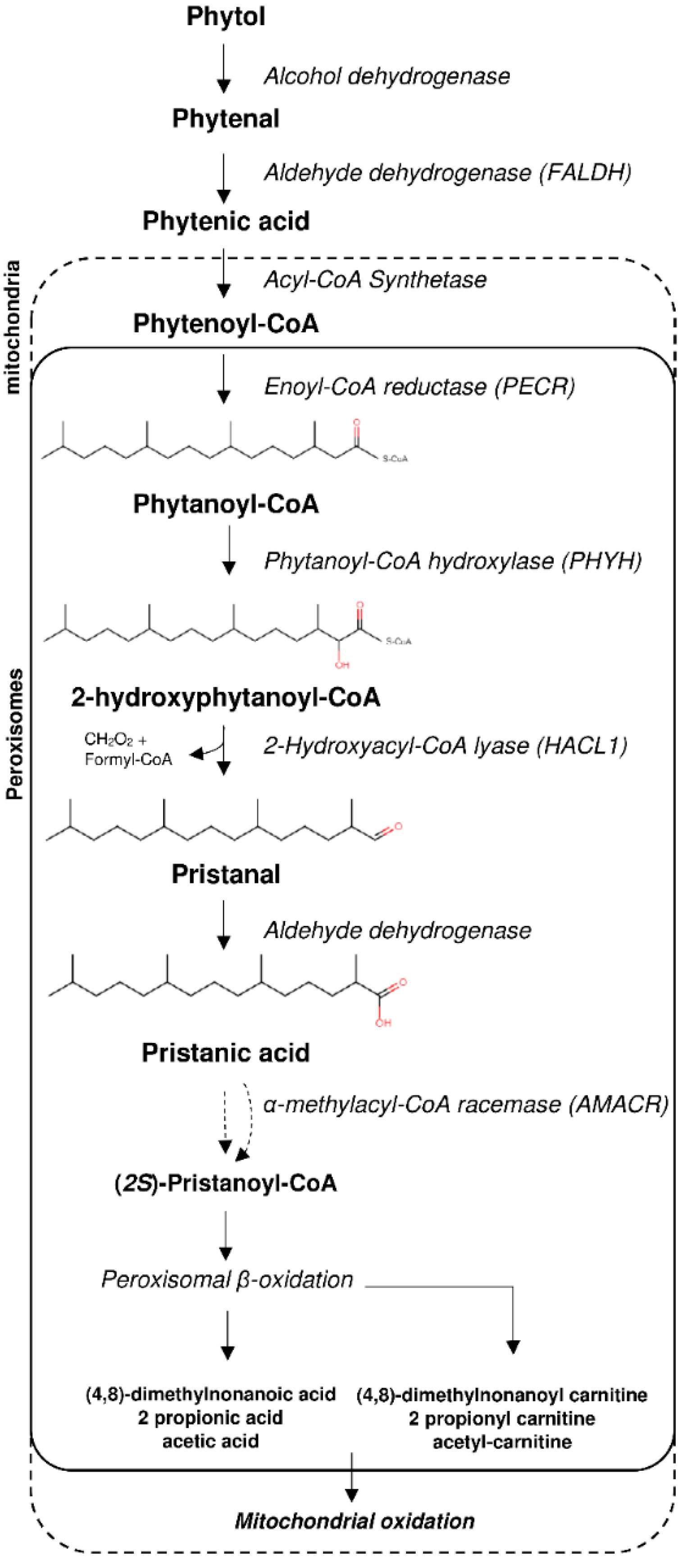
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Materials
2.3. Preparation of Mouse Liver for Fatty Acid Analysis
2.4. Fatty Acid Extraction and Derivatisation
2.5. GC-MS Fatty Acid Analysis
2.6. Preparation of Mouse Liver, Kidney, and Brain Tissues for Proteomic Analysis
2.7. Protein Precipitation
2.8. Protein Digestion
2.9. LC-MS Proteomic Analysis
2.10. Proteomic MS Data Analysis
2.11. Statistical Analysis and Data Plotting
3. Results
3.1. Hacl1−/− Mouse Phenotype
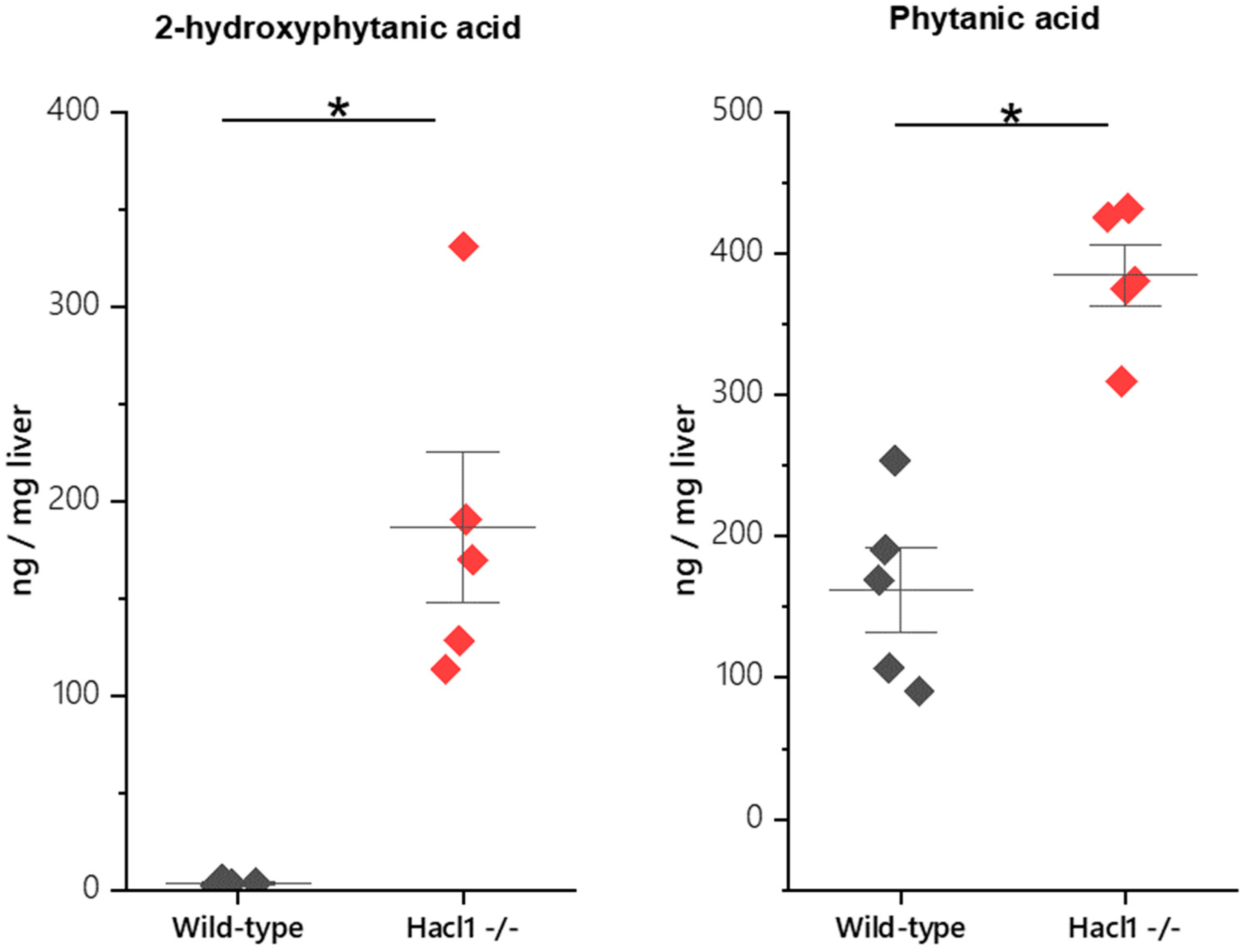
3.2. Fatty Acid Profile in Hacl1−/− Mouse Liver
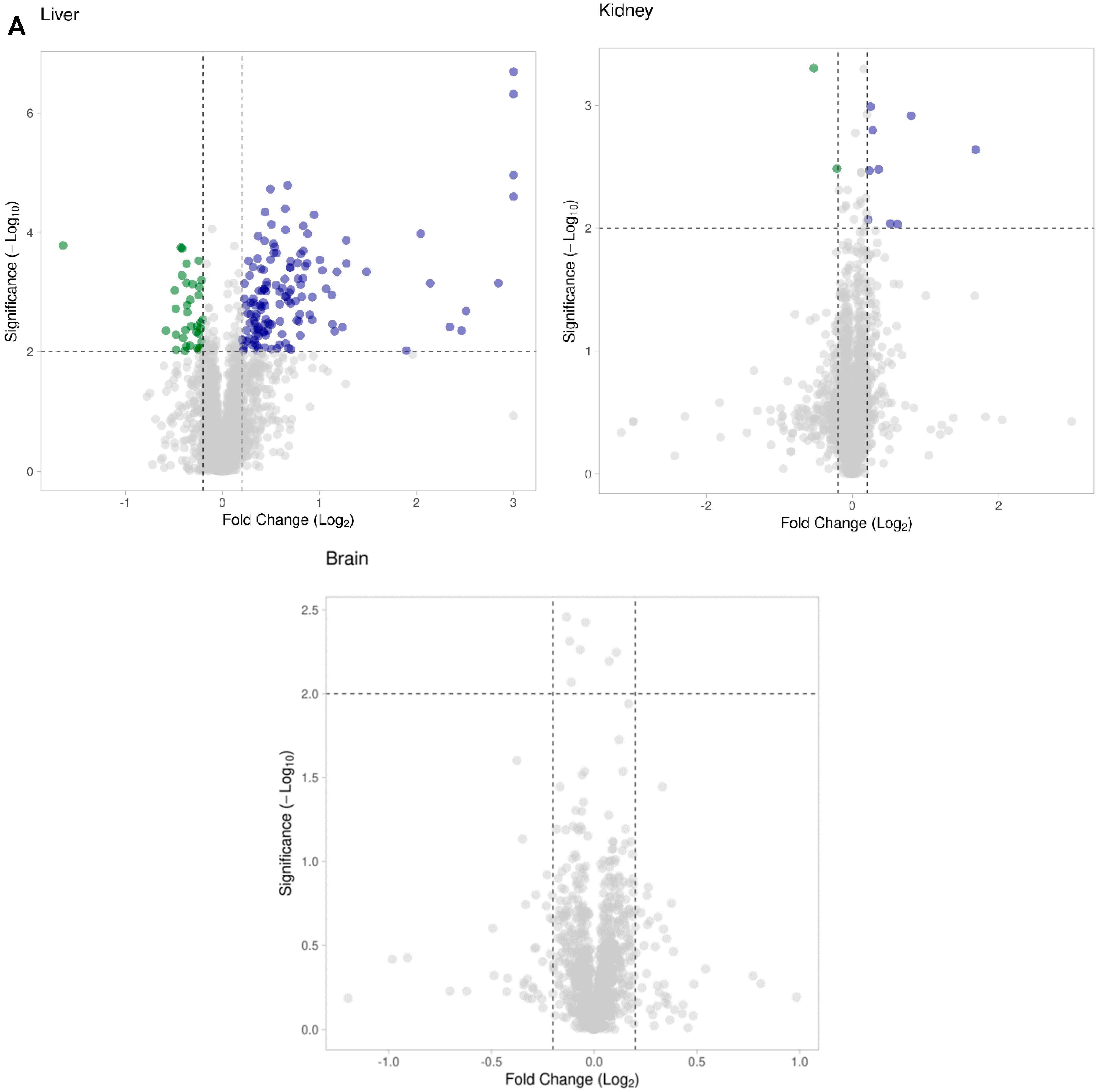
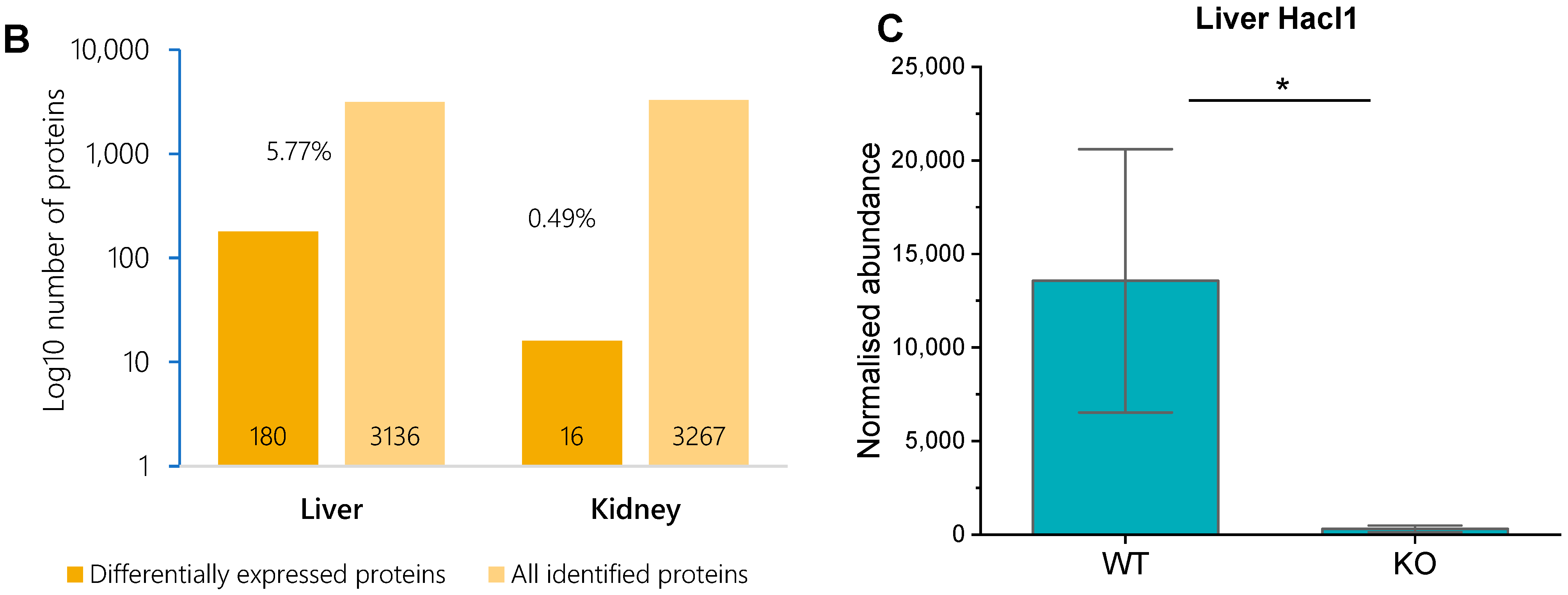
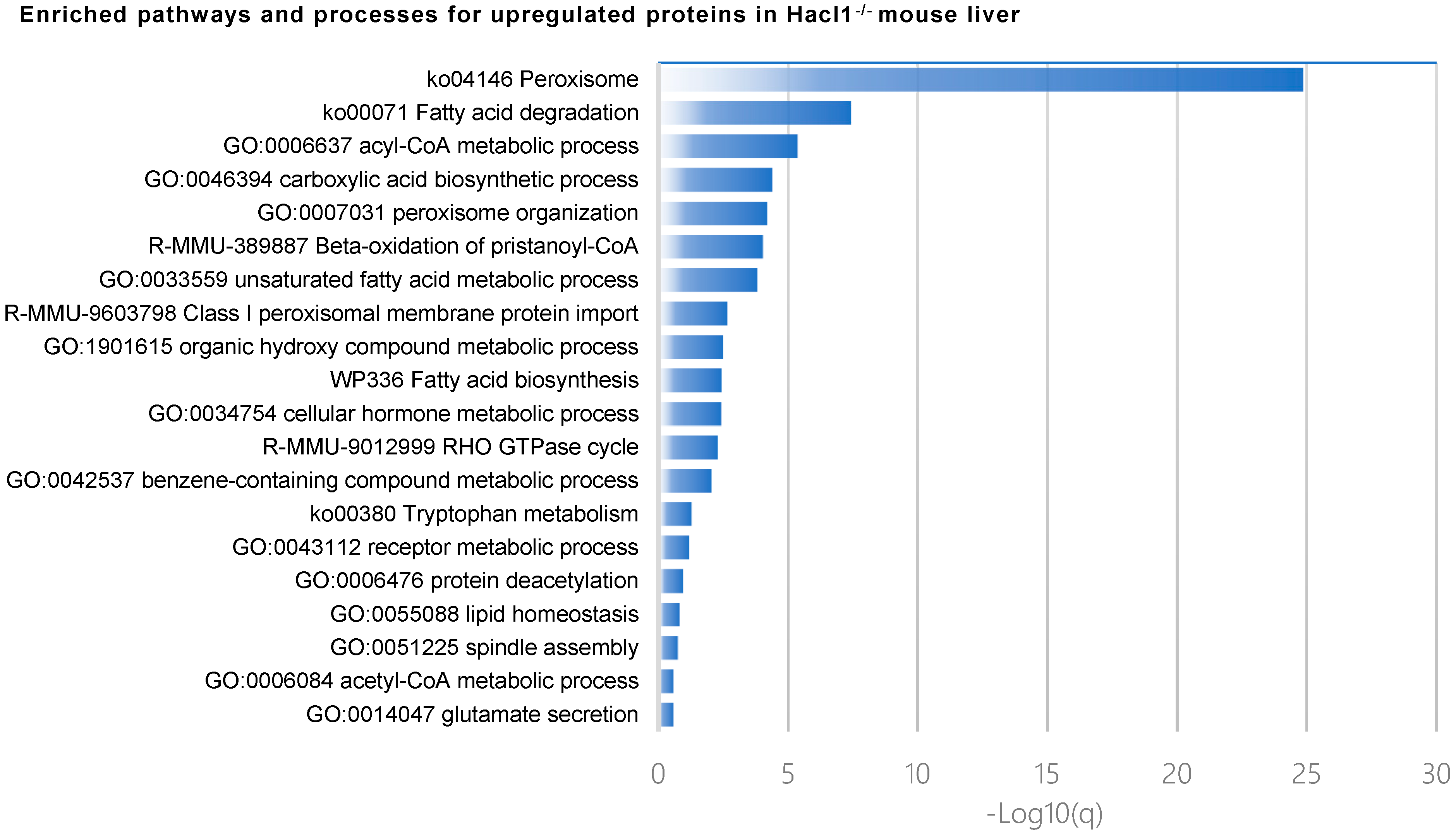
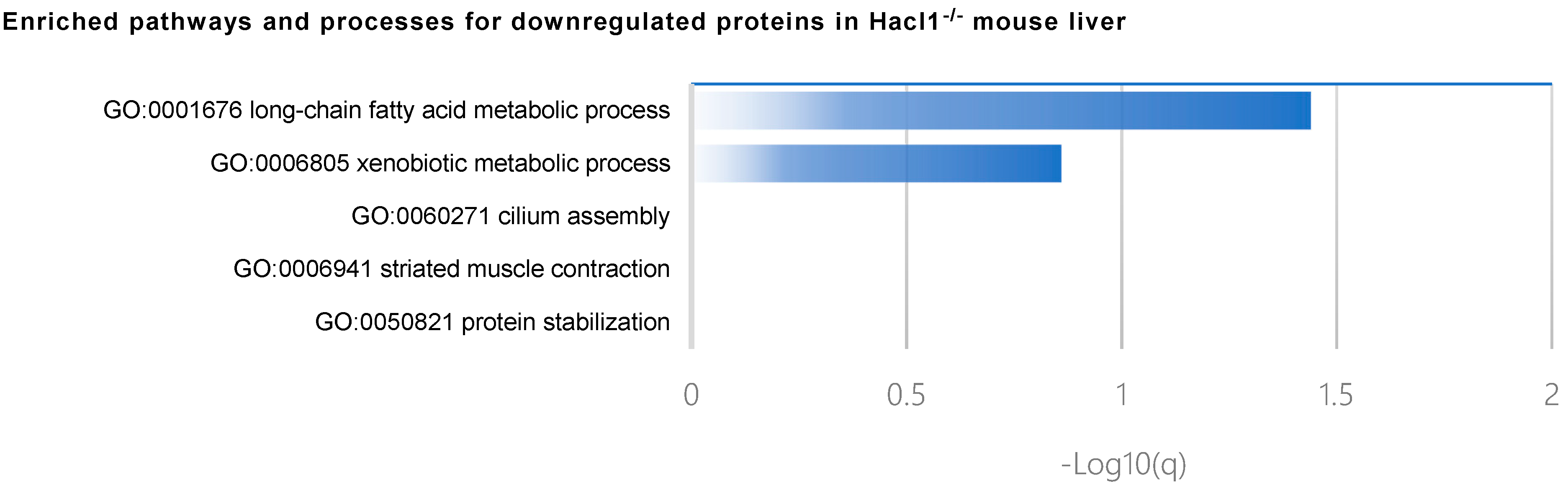
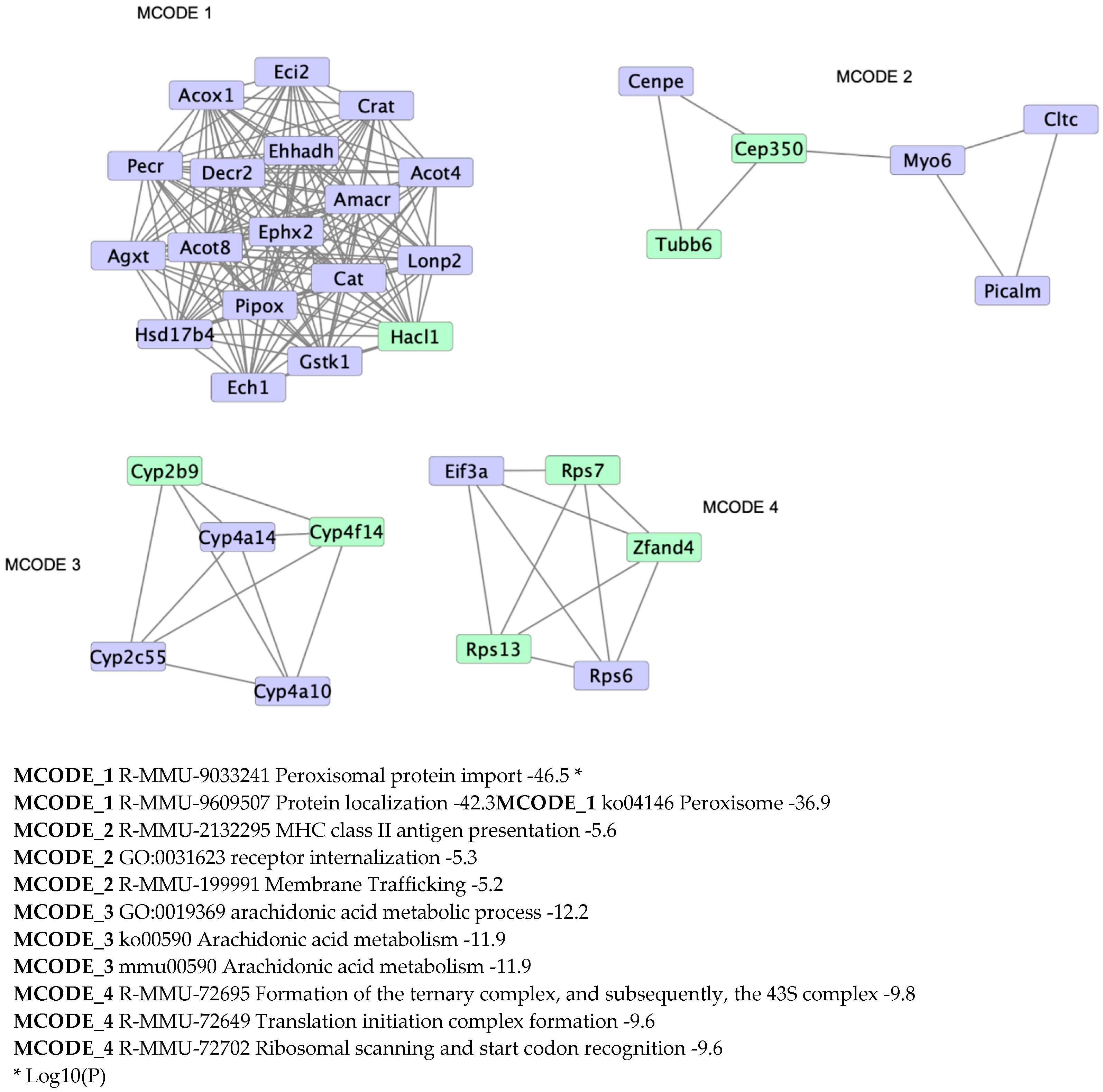
3.3. Hacl1−/− Mouse Liver Proteome
3.4. Hacl1−/− Mouse Kidney Proteome
3.5. Hacl1−/− Mouse Brain Proteome
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data availability statement
Conflicts of Interest
References
- Wanders, R.J.; Komen, J.; Ferdinandusse, S. Phytanic acid metabolism in health and disease. Biochim. Biophys. Acta 2011, 1811, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Miura, Y. The biological significance of omega-oxidation of fatty acids. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2013, 89, 370–382. [Google Scholar] [CrossRef] [Green Version]
- Komen, J.C.; Duran, M.; Wanders, R.J. Omega-hydroxylation of phytanic acid in rat liver microsomes: Implications for Refsum disease. J. Lipid Res. 2004, 45, 1341–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komen, J.C.; Duran, M.; Wanders, R.J. Characterization of phytanic acid omega-hydroxylation in human liver microsomes. Mol. Genet. Metab. 2005, 85, 190–195. [Google Scholar] [CrossRef]
- Wierzbicki, A.S.; Lloyd, M.D.; Schofield, C.J.; Feher, M.D.; Gibberd, F.B. Refsum’s disease: A peroxisomal disorder affecting phytanic acid alpha-oxidation. J. Neurochem. 2002, 80, 727–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, P.J.; Mei, G.; Gibberd, F.B.; Burston, D.; Mayne, P.D.; McClinchy, J.E.; Sidey, M. Diet and Refsum’s disease. The determination of phytanic acid and phytol in certain foods and the application of this knowledge to the choice of suitable convenience foods for patients with Refsum’s disease. J. Hum. Nutr. Diet. 1993, 6, 295–305. [Google Scholar] [CrossRef]
- Dry, J.; Pradalier, A.; Canny, M. Refsum’s disease: 10 years of a diet low in phytanic acid and phytol. Ann. Med. Intern. 1982, 133, 483–487. [Google Scholar]
- Steinberg, D.; Mize, C.E.; Herndon, J.H.; Fales, H.M.; Engel, W.K.; Vroom, F.Q. Phytanic acid in patients with Refsum’s syndrome and response to dietary treatment. Arch. Intern. Med. 1970, 125, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Ferdinandusse, S.; Zomer, A.W.; Komen, J.C.; van den Brink, C.E.; Thanos, M.; Hamers, F.P.; Wanders, R.J.; van der Saag, P.T.; Poll-The, B.T.; Brites, P. Ataxia with loss of Purkinje cells in a mouse model for Refsum disease. Proc. Natl. Acad. Sci. USA 2008, 105, 17712–17717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selkala, E.M.; Nair, R.R.; Schmitz, W.; Kvist, A.P.; Baes, M.; Hiltunen, J.K.; Autio, K.J. Phytol is lethal for Amacr-deficient mice. Biochim. Biophys. Acta 2015, 1851, 1394–1405. [Google Scholar] [CrossRef]
- Mezzar, S.; De Schryver, E.; Asselberghs, S.; Meyhi, E.; Morvay, P.L.; Baes, M.; Van Veldhoven, P.P. Phytol-induced pathology in 2-hydroxyacyl-CoA lyase (HACL1) deficient mice. Evidence for a second non-HACL1-related lyase. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 972–990. [Google Scholar] [CrossRef] [PubMed]
- Skarnes, W.C.; Rosen, B.; West, A.P.; Koutsourakis, M.; Bushell, W.; Iyer, V.; Mujica, A.O.; Thomas, M.; Harrow, J.; Cox, T.; et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature 2011, 474, 337–342. [Google Scholar] [CrossRef] [Green Version]
- de Angelis, M.H.; Nicholson, G.; Selloum, M.; White, J.K.; Morgan, H.; Ramirez-Solis, R.; Sorg, T.; Wells, S.; Fuchs, H.; Fray, M.; et al. Analysis of mammalian gene function through broad-based phenotypic screens across a consortium of mouse clinics. Nat. Genet. 2015, 47, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Bliss, E.; Heywood, W.E.; Benatti, M.; Sebire, N.J.; Mills, K. An optimised method for the proteomic profiling of full thickness human skin. Biol. Proced. Online 2016, 18, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Goedhart, J.; Luijsterburg, M.S. VolcaNoseR is a web app for creating, exploring, labeling and sharing volcano plots. Sci. Rep. 2020, 10, 20560. [Google Scholar] [CrossRef]
- ten Brink, H.J.; Schor, D.S.; Kok, R.M.; Poll-The, B.T.; Wanders, R.J.; Jakobs, C. Phytanic acid alpha-oxidation: Accumulation of 2-hydroxyphytanic acid and absence of 2-oxophytanic acid in plasma from patients with peroxisomal disorders. J. Lipid Res. 1992, 33, 1449–1457. [Google Scholar] [CrossRef]
- Ellinghaus, P.; Wolfrum, C.; Assmann, G.; Spener, F.; Seedorf, U. Phytanic acid activates the peroxisome proliferator-activated receptor alpha (PPARalpha) in sterol carrier protein 2-/ sterol carrier protein x-deficient mice. J. Biol. Chem. 1999, 274, 2766–2772. [Google Scholar] [CrossRef] [Green Version]
- Wierzbicki, A.S.; Mayne, P.D.; Lloyd, M.D.; Burston, D.; Mei, G.; Sidey, M.C.; Feher, M.D.; Gibberd, F.B. Metabolism of phytanic acid and 3-methyl-adipic acid excretion in patients with adult Refsum disease. J. Lipid Res. 2003, 44, 1481–1488. [Google Scholar] [CrossRef] [Green Version]
- Bell, D.R.; Plant, N.J.; Rider, C.G.; Na, L.; Brown, S.; Ateitalla, I.; Acharya, S.K.; Davies, M.H.; Elias, E.; Jenkins, N.A.; et al. Species-specific induction of cytochrome P-450 4A RNAs: PCR cloning of partial guinea-pig, human and mouse CYP4A cDNAs. Biochem. J. 1993, 294, 173–180. [Google Scholar] [CrossRef] [Green Version]
- Ferdinandusse, S.; Jimenez-Sanchez, G.; Koster, J.; Denis, S.; Van Roermund, C.W.; Silva-Zolezzi, I.; Moser, A.B.; Visser, W.F.; Gulluoglu, M.; Durmaz, O.; et al. A novel bile acid biosynthesis defect due to a deficiency of peroxisomal ABCD3. Hum. Mol. Genet. 2015, 24, 361–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Roermund, C.W.; Ijlst, L.; Wagemans, T.; Wanders, R.J.; Waterham, H.R. A role for the human peroxisomal half-transporter ABCD3 in the oxidation of dicarboxylic acids. Biochim. Biophys. Acta 2014, 1841, 563–568. [Google Scholar] [CrossRef]
- Miura, H.; Mizuguchi, H.; Amano-Iwashita, M.; Maeda-Kogure, R.; Negishi, A.; Sakai, A.; Toyama, T.; Kawai, H.; Mitsumoto, A.; Kudo, N. Clofibric acid increases molecular species of phosphatidylethanolamine containing arachidonic acid for biogenesis of peroxisomal membranes in peroxisome proliferation in the liver. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158963. [Google Scholar] [CrossRef]
- Rakhshandehroo, M.; Knoch, B.; Muller, M.; Kersten, S. Peroxisome proliferator-activated receptor alpha target genes. Cell. Mol. Life Sci. CMLS 2004, 61, 393–416. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.; Min, L.; Yang, J.; Dai, M.; Song, D.; Hua, H.; Xu, G.; Gonzalez, F.J.; Liu, A. Peroxisome Proliferator-Activated Receptor alpha Activation Suppresses Cytochrome P450 Induction Potential in Mice Treated with Gemfibrozil. Basic Clin. Pharmacol. Toxicol. 2017, 121, 169–174. [Google Scholar] [CrossRef]
- Hariparsad, N.; Chu, X.; Yabut, J.; Labhart, P.; Hartley, D.P.; Dai, X.; Evers, R. Identification of pregnane-X receptor target genes and coactivator and corepressor binding to promoter elements in human hepatocytes. Nucleic Acids Res. 2009, 37, 1160–1173. [Google Scholar] [CrossRef]
- Chang, T.K.; Bandiera, S.M.; Chen, J. Constitutive androstane receptor and pregnane X receptor gene expression in human liver: Interindividual variability and correlation with CYP2B6 mRNA levels. Drug. Metab. Dispos. 2003, 31, 7–10. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, J.P.; Mota, L.C.; Baldwin, W.S. Activation of CAR and PXR by Dietary, Environmental and Occupational Chemicals Alters Drug Metabolism, Intermediary Metabolism, and Cell Proliferation. Curr. Pharm. Person. Med. 2009, 7, 81–105. [Google Scholar] [CrossRef] [Green Version]
- Mi, J.; Kirchner, E.; Cristobal, S. Quantitative proteomic comparison of mouse peroxisomes from liver and kidney. Proteomics 2007, 7, 1916–1928. [Google Scholar] [CrossRef]
- Jones, J.M.; Morrell, J.C.; Gould, S.J. Identification and characterization of HAOX1, HAOX2, and HAOX3, three human peroxisomal 2-hydroxy acid oxidases. J. Biol. Chem. 2000, 275, 12590–12597. [Google Scholar] [CrossRef] [Green Version]
- Draye, J.P.; Van Hoof, F.; de Hoffmann, E.; Vamecq, J. Peroxisomal oxidation of L-2-hydroxyphytanic acid in rat kidney cortex. Eur. J. Biochem. 1987, 167, 573–578. [Google Scholar] [CrossRef]
- Jia, Y.; Qi, C.; Zhang, Z.; Hashimoto, T.; Rao, M.S.; Huyghe, S.; Suzuki, Y.; Van Veldhoven, P.P.; Baes, M.; Reddy, J.K. Overexpression of peroxisome proliferator-activated receptor-alpha (PPARalpha)-regulated genes in liver in the absence of peroxisome proliferation in mice deficient in both L- and D-forms of enoyl-CoA hydratase/dehydrogenase enzymes of peroxisomal beta-oxidation system. J. Biol. Chem. 2003, 278, 47232–47239. [Google Scholar] [CrossRef] [Green Version]
- Kersten, S. Integrated physiology and systems biology of PPARalpha. Mol. Metab. 2014, 3, 354–371. [Google Scholar] [CrossRef]
- Huyghe, S.; Casteels, M.; Janssen, A.; Meulders, L.; Mannaerts, G.P.; Declercq, P.E.; van Veldhoven, P.P.; Baes, M. Prenatal and postnatal development of peroxisomal lipid-metabolizing pathways in the mouse. Biochem. J. 2001, 353, 673–680. [Google Scholar] [CrossRef]
- Yamaori, S.; Jiang, R.; Maeda, C.; Ogawa, R.; Okazaki, H.; Aramaki, H.; Watanabe, K. Expression levels of 39 Cyp mRNAs in the mouse brain and neuroblastoma cell lines, C-1300N18 and NB2a-strong expression of Cyp1b1. Fundam. Toxicol. Sci. 2017, 4, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Sandhir, R.; Khan, M.; Singh, I. Identification of the pathway of alpha-oxidation of cerebronic acid in peroxisomes. Lipids 2000, 35, 1127–1133. [Google Scholar] [CrossRef]
- Wanders, R.J.A.; Waterham, H.R.; Leroy, B.P. Refsum Disease. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Kariminejad, A.; Nafissi, S.; Nilipoor, Y.; Tavasoli, A.; Van Veldhoven, P.P.; Bonnard, C.; Ng, Y.T.; Majoie, C.B.; Reversade, B.; Hennekam, R.C. Intellectual disability, muscle weakness and characteristic face in three siblings: A newly described recessive syndrome mapping to 3p24.3-p25.3. Am. J. Med. Genet. A 2015, 167, 2508–2515. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KEGG Pathway | Count | p-Value | Gene |
|---|---|---|---|
| Peroxisome | 22 | 1.60 × 10−25 | Pecr, Gstk1, Acot8, Pex19, Abcd3, Acsl1, Pex11a, Ephx2, Eci2, Ech1, Hsd17b4, Pipox, Acaa1b, Acaa1a, Amacr, Acox1, Ehhadh, Cat, Decr2, Agxt, Crat, Nudt12 |
| Fatty acid degradation | 9 | 1.30 × 10−8 | Aldh3a2, Acsl1, Acox1, Ehhadh, Cyp4a10, Eci2, Acaa1b, Cyp4a14, Acaa1a |
| Biosynthesis of unsaturated fatty acids | 6 | 4.30 × 10−6 | Pecr, Acox1, Acaa1b, Scd1, Acaa1a, Acot4 |
| Fatty acid metabolism | 7 | 7.00 × 10−6 | Pecr, Acsl1, Acox1, Ehhadh, Acaa1b, Scd1, Acaa1a |
| PPAR signaling pathway | 8 | 8.60 × 10−6 | Acsl1, Acox1, Ehhadh, Cyp4a10, Acaa1b, Cyp4a14, Scd1, Acaa1a |
| Metabolic pathways | 25 | 3.40 × 10−4 | Hsd17b4, H2-Ke6, Acaa1b, Acaa1a, Plb1, Cyp2c55, Rdh16, Dlat, Acot8, Acsl1, Ephx2, Cyp4a10, Pmm2, Pipox, Cyp4a14, Kmo, Aldh3a2, Amacr, Acox1, Ehhadh, Agxt, Gart, Mgll, Acot4, Uox |
| alpha-Linolenic acid metabolism | 4 | 1.60 × 10−3 | Acox1, Acaa1b, Acaa1a, Plb1 |
| Valine, leucine and isoleucine degradation | 5 | 1.70 × 10−3 | Aldh3a2, Ehhadh, Acaa1b, Acaa1a, Aacs |
| Arachidonic acid metabolism | 5 | 9.50 × 10−3 | Cyp2c55, Ephx2, Cyp4a10, Cyp4a14, Plb1 |
| Tryptophan metabolism | 4 | 9.50 × 10−3 | Aldh3a2, Ehhadh, Cat, Kmo |
| Primary bile acid biosynthesis | 3 | 9.50 × 10−3 | Acot8, Amacr, hsd17b4 |
| Lysine degradation | 4 | 1.30 × 10−2 | Aldh3a2, Setd2, Ehhadh, Pipox |
| Retinol metabolism | 4 | 5.00 × 10−2 | Cyp2c55, Cyp4a10, Rdh16, Cyp4a14 |
| Pyruvate metabolism | 3 | 5.10 × 10−2 | Aldh3a2, Acot12, Dlat |
| Carbon metabolism | 4 | 9.50 × 10−2 | Ehhadh, Cat, Dlat, Agxt |
| p-Value | Genes | |
|---|---|---|
| GO Biological Process | ||
| Fatty acid metabolic process | 2.60 × 10−3 | Acsl3, Crot, Hsd17b4 |
| Lipid metabolic process | 2.10 × 10−2 | Acsl3, Crot, Hsd17b4 |
| Fatty acid beta-oxidation | 2.20 × 10−2 | Crot, Hsd17b4 |
| GO Cellular Compartment | ||
| Peroxisome | 6.00 × 10−7 | Acsl3, Crot, Hsd17b4, Scp2, Zadh2 |
| Intracellular membrane-bounded organelle | 7.30 × 10−3 | Acsl3, Crot, Hsd17b4, Scp2 |
| Mitochondrion | 1.20 × 10−2 | Acsl3, Crot, Hsd17b4, Scp2, Zadh2 |
| Peroxisomal membrane | 2.90 × 10−2 | Acsl3, Hsd17b4 |
| GO Molecular Function | ||
| Receptor binding | 1.40 × 10−2 | Crot, Hsd17b4, Scp2 |
| Actin filament binding | 5.90 × 10−2 | Myo1b, Svil |
| Transferase activity, transferring acyl groups | 7.40 × 10−2 | Crot, Scp2 |
| KEGG Pathway | ||
| Peroxisome | 1.20 × 10−6 | Acsl3, Crot, Hsd17b4, Scp2 |
| Primary bile acid biosynthesis | 6.20 × 10−3 | Hsd17b4, Scp2 |
| PPAR signaling pathway | 3.10 × 10−2 | Acsl3, Scp2 |
| Metabolic pathways | 7.30 × 10−2 | Acsl3, Hsd17b4, Scp2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalil, Y.; Carrino, S.; Lin, F.; Ferlin, A.; Lad, H.V.; Mazzacuva, F.; Falcone, S.; Rivers, N.; Banks, G.; Concas, D.; et al. Tissue Proteome of 2-Hydroxyacyl-CoA Lyase Deficient Mice Reveals Peroxisome Proliferation and Activation of ω-Oxidation. Int. J. Mol. Sci. 2022, 23, 987. https://doi.org/10.3390/ijms23020987
Khalil Y, Carrino S, Lin F, Ferlin A, Lad HV, Mazzacuva F, Falcone S, Rivers N, Banks G, Concas D, et al. Tissue Proteome of 2-Hydroxyacyl-CoA Lyase Deficient Mice Reveals Peroxisome Proliferation and Activation of ω-Oxidation. International Journal of Molecular Sciences. 2022; 23(2):987. https://doi.org/10.3390/ijms23020987
Chicago/Turabian StyleKhalil, Youssef, Sara Carrino, Fujun Lin, Anna Ferlin, Heena V. Lad, Francesca Mazzacuva, Sara Falcone, Natalie Rivers, Gareth Banks, Danilo Concas, and et al. 2022. "Tissue Proteome of 2-Hydroxyacyl-CoA Lyase Deficient Mice Reveals Peroxisome Proliferation and Activation of ω-Oxidation" International Journal of Molecular Sciences 23, no. 2: 987. https://doi.org/10.3390/ijms23020987