
Decreased Prosaposin and Progranulin in the Cingulate Cortex Are Associated with Schizophrenia Pathophysiology

Abstract
:1. Introduction
2. Results
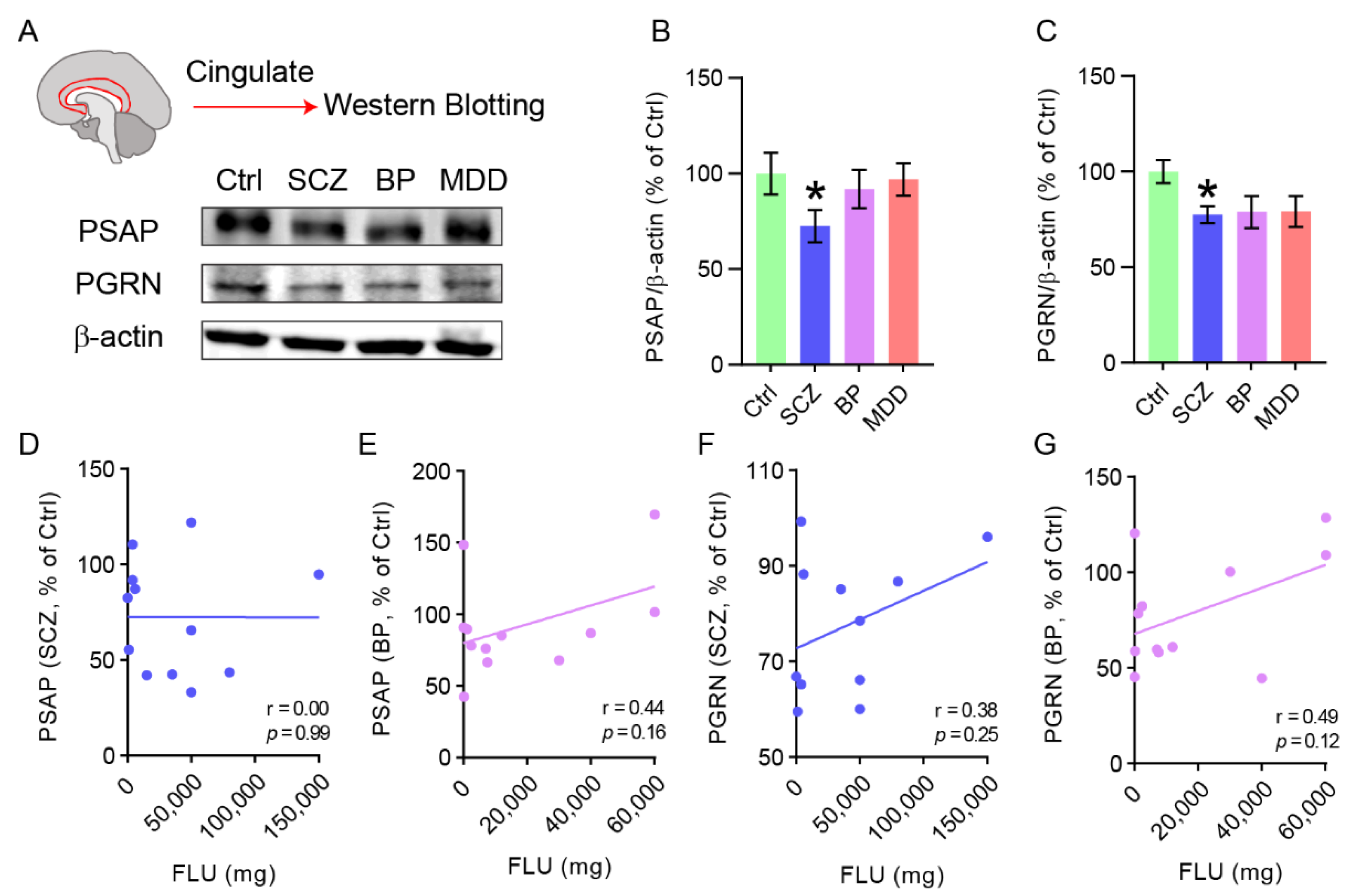
2.1. PSAP and PGRN Are Decreased in the Cingulate Cortex of Schizophrenia Patients
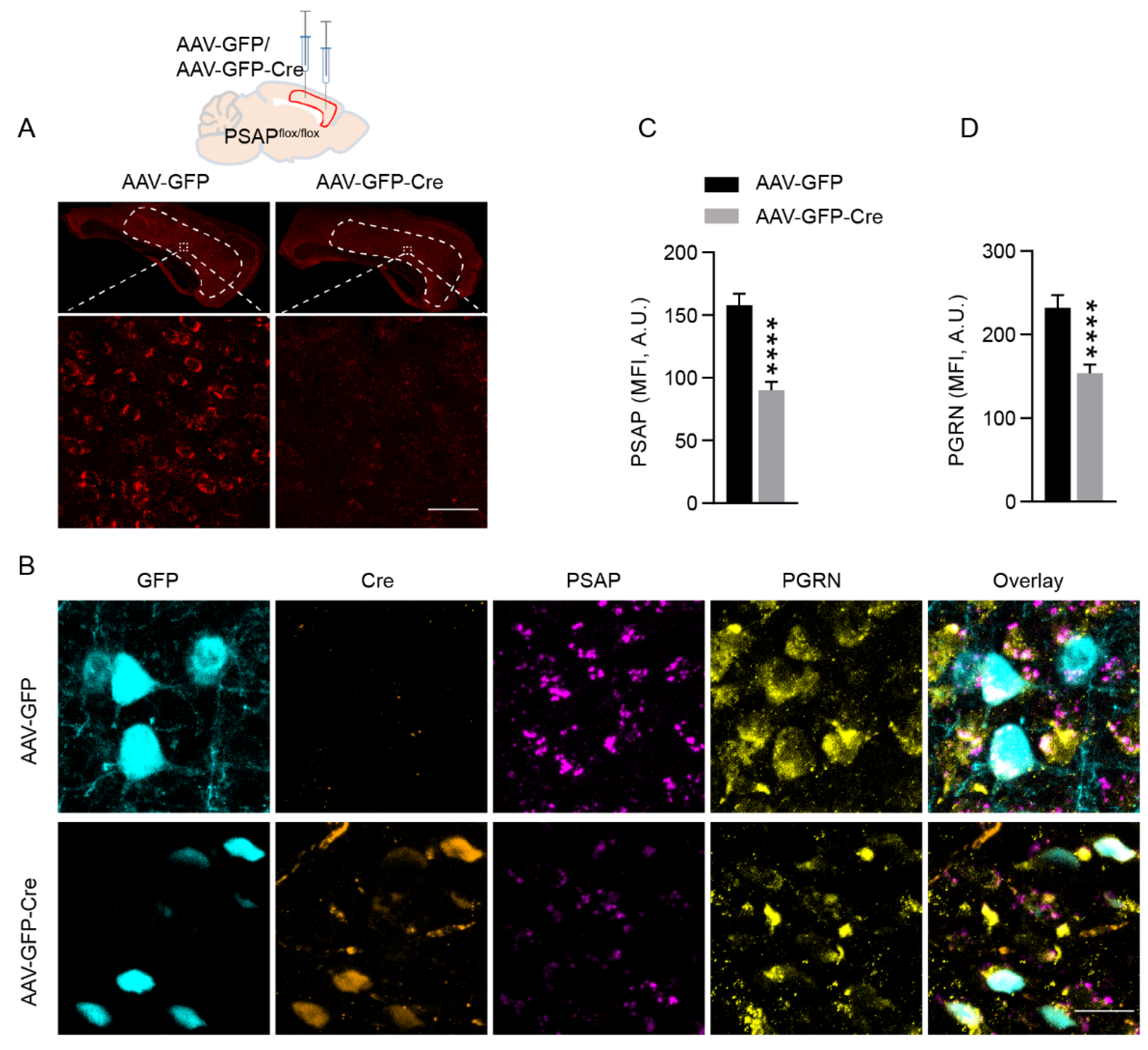
2.2. Deletion of PSAP in the Cingulate Cortex Causes Downregulation of PGRN Levels
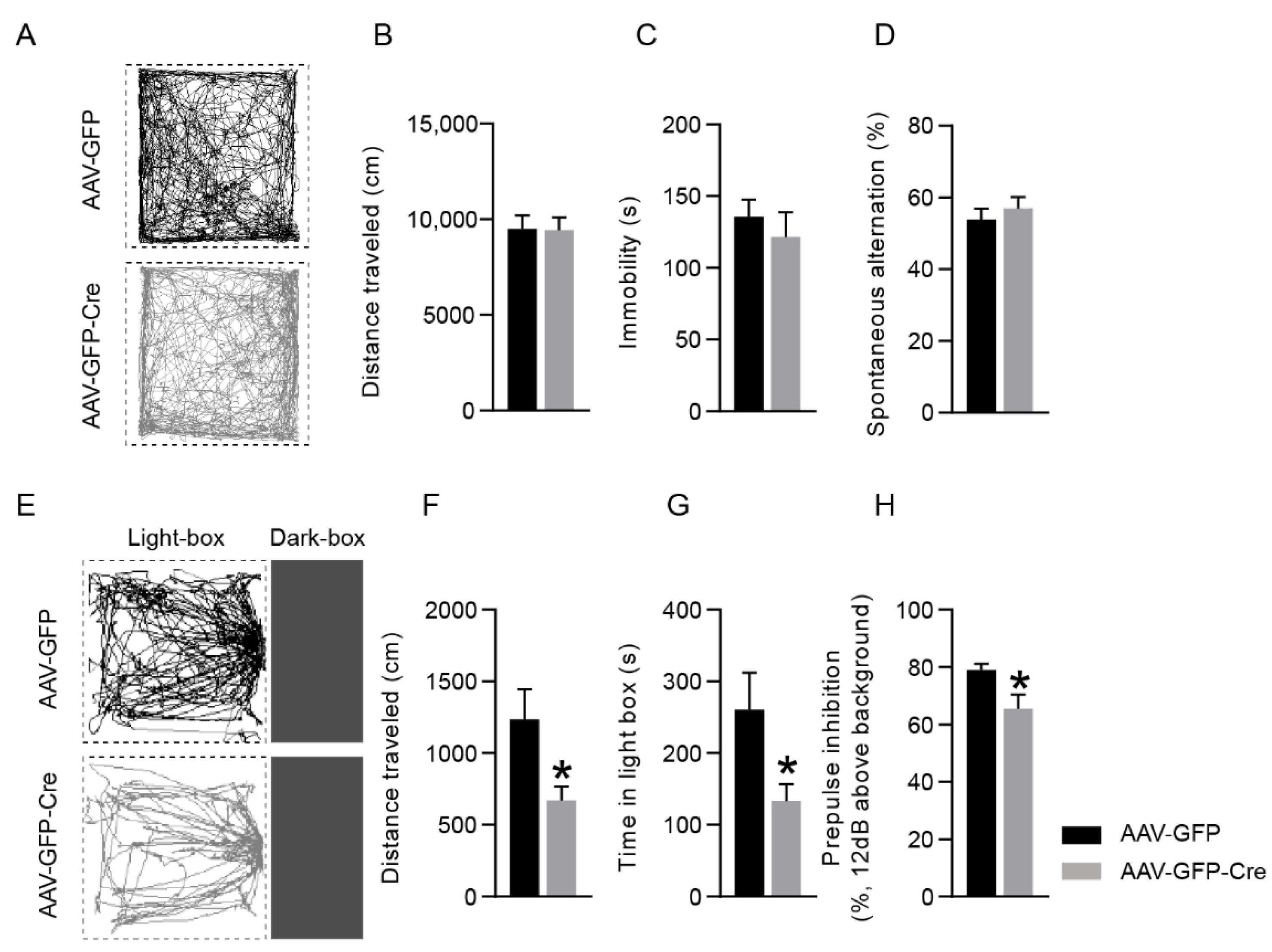
2.3. Mice with PSAP Deficiency in the Cingulate Cortex Display Anxiety-Like Behavior and Sensorimotor Gating Dysfunction
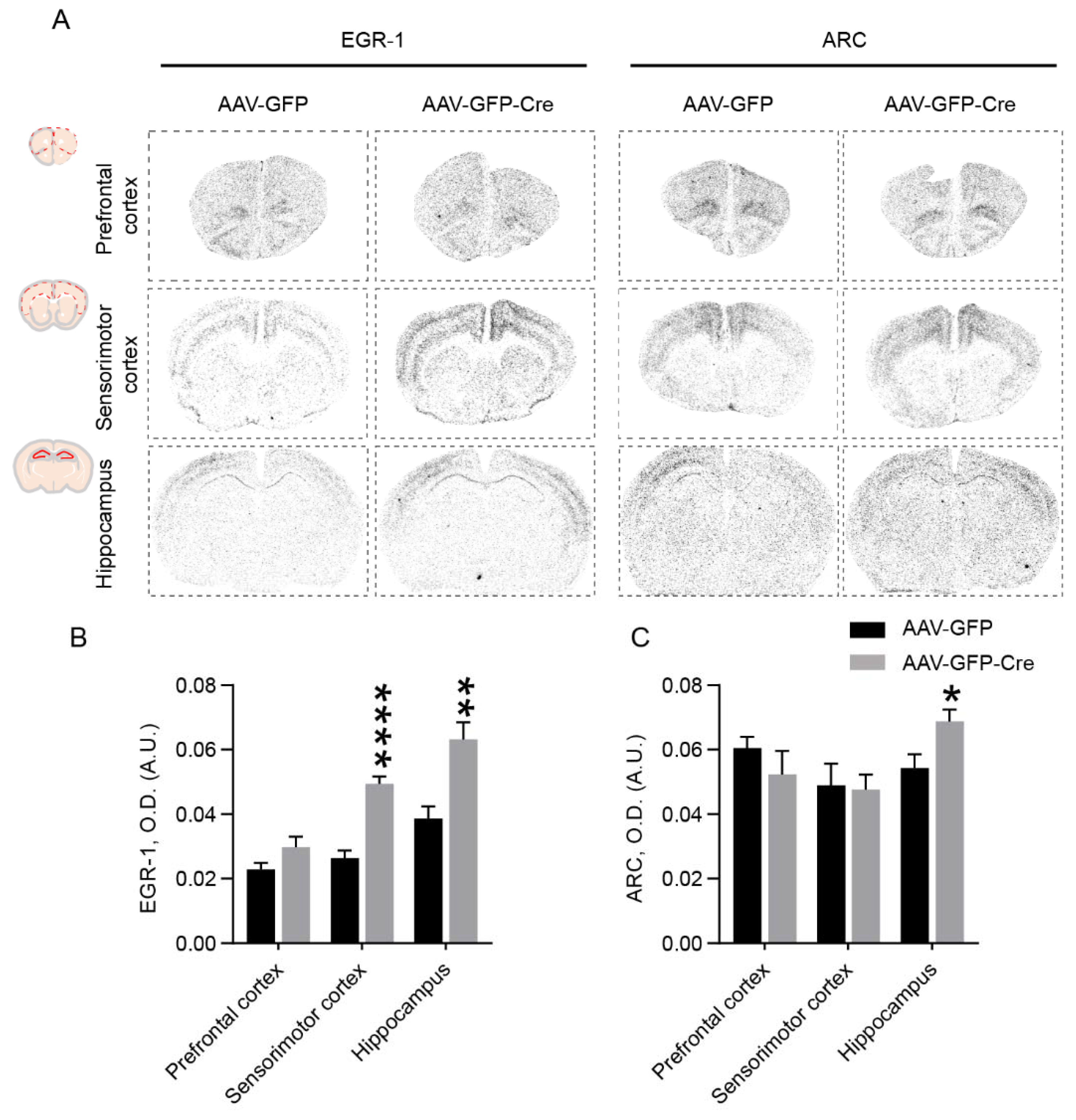
2.4. PSAP Deficiency in the Cingulate Cortex Elevates EGR-1 and ARC Levels in Different Brain Regions
3. Discussion
4. Materials and Methods
4.1. Human Brain Samples
Human Brain Samples
4.2. Western Blotting
4.3. Stereotaxic Surgery on Mice
4.4. Fluorescent In Situ Hybridization (FISH)
4.5. Immunofluorescent Staining
4.6. Radioactive In Situ Hybridization
4.7. Behavioral Tests
4.7.1. Open Field Test (OFT)
4.7.2. Light–Dark Transition Test (LDT)
4.7.3. Prepulse Inhibition (PPI) Test
4.7.4. Y Maze (YM)
4.7.5. Forced Swim Test (FST)
4.7.6. Behavioral Tests
4.8. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jauhar, S.; Johnstone, M.; McKenna, P.J. Schizophrenia. Lancet 2022, 399, 473–486. [Google Scholar] [CrossRef]
- Kahn, R.S.; Sommer, I.E.; Murray, R.M.; Meyer-Lindenberg, A.; Weinberger, D.R.; Cannon, T.D.; O’Donovan, M.; Correll, C.U.; Kane, J.M.; van Os, J.; et al. Schizophrenia. Nature reviews. Dis. Primers 2015, 1, 15067. [Google Scholar] [CrossRef] [PubMed]
- Crossley, N.A.; Mechelli, A.; Fusar-Poli, P.; Broome, M.R.; Matthiasson, P.; Johns, L.C.; Bramon, E.; Valmaggia, L.; Williams, S.C.; McGuire, P.K. Superior temporal lobe dysfunction and frontotemporal dysconnectivity in subjects at risk of psychosis and in first-episode psychosis. Hum. Brain Mapp. 2009, 30, 4129–4137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radua, J.; Borgwardt, S.; Crescini, A.; Mataix-Cols, D.; Meyer-Lindenberg, A.; McGuire, P.K.; Fusar-Poli, P. Multimodal meta-analysis of structural and functional brain changes in first episode psychosis and the effects of antipsychotic medication. Neurosci. Biobehav. Rev. 2012, 36, 2325–2333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owen, M.J.; Sawa, A.; Mortensen, P.B. Schizophrenia. Lancet 2016, 388, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Kishimoto, Y.; Hiraiwa, M.; O’Brien, J.S. Saposins: Structure, function, distribution, and molecular genetics. J. Lipid Res. 1992, 33, 1255–1267. [Google Scholar] [CrossRef]
- O’Brien, J.S.; Carson, G.S.; Seo, H.C.; Hiraiwa, M.; Kishimoto, Y. Identification of prosaposin as a neurotrophic factor. Proc. Natl. Acad. Sci. USA 1994, 91, 9593–9596. [Google Scholar] [CrossRef] [Green Version]
- Tian, R.; Abarientos, A.; Hong, J.; Hashemi, S.H.; Yan, R.; Drager, N.; Leng, K.; Nalls, M.A.; Singleton, A.B.; Xu, K.; et al. Genome-wide CRISPRi/a screens in human neurons link lysosomal failure to ferroptosis. Nat. Neurosci. 2021, 24, 1020–1034. [Google Scholar] [CrossRef]
- Meyer, R.C.; Giddens, M.M.; Coleman, B.M.; Hall, R.A. The protective role of prosaposin and its receptors in the nervous system. Brain Res. 2014, 1585, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Oji, Y.; Hatano, T.; Ueno, S.I.; Funayama, M.; Ishikawa, K.I.; Okuzumi, A.; Noda, S.; Sato, S.; Satake, W.; Toda, T.; et al. Variants in saposin D domain of prosaposin gene linked to Parkinson’s disease. Brain J. Neurol. 2020, 143, 1190–1205. [Google Scholar] [CrossRef]
- Jungerius, B.J.; Hoogendoorn, M.L.; Bakker, S.C.; Van’t Slot, R.; Bardoel, A.F.; Ophoff, R.A.; Wijmenga, C.; Kahn, R.S.; Sinke, R.J. An association screen of myelin-related genes implicates the chromosome 22q11 PIK4CA gene in schizophrenia. Mol. Psychiatry 2008, 13, 1060–1068. [Google Scholar] [CrossRef] [Green Version]
- Zhuo, C.; Zhao, F.; Tian, H.; Chen, J.; Li, Q.; Yang, L.; Ping, J.; Li, R.; Wang, L.; Xu, Y.; et al. Acid sphingomyelinase/ceramide system in schizophrenia: Implications for therapeutic intervention as a potential novel target. Transl. Psychiatry 2022, 12, 260. [Google Scholar] [CrossRef] [PubMed]
- Tayebi, N.; Lopez, G.; Do, J.; Sidransky, E. Pro-cathepsin D, Prosaposin, and Progranulin: Lysosomal Networks in Parkinsonism. Trends Mol. Med. 2020, 26, 913–923. [Google Scholar] [CrossRef]
- Baker, M.; Mackenzie, I.R.; Pickering-Brown, S.M.; Gass, J.; Rademakers, R.; Lindholm, C.; Snowden, J.; Adamson, J.; Sadovnick, A.D.; Rollinson, S.; et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 2006, 442, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Reho, P.; Koga, S.; Shah, Z.; Chia, R.; Rademakers, R.; Dalgard, C.L.; Boeve, B.F.; Beach, T.G.; Dickson, D.W.; Ross, O.A.; et al. GRN Mutations Are Associated with Lewy Body Dementia. Mov. Disord. Off. J. Mov. Disord. Soc. 2022, 37, 1943–1948. [Google Scholar] [CrossRef]
- Rhinn, H.; Tatton, N.; McCaughey, S.; Kurnellas, M.; Rosenthal, A. Progranulin as a therapeutic target in neurodegenerative diseases. Trends Pharmacol. Sci. 2022, 43, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Jian, J.; Hettinghouse, A.; Zhao, X.; Setchell, K.D.R.; Sun, Y.; Liu, C.J. Progranulin associates with hexosaminidase A and ameliorates GM2 ganglioside accumulation and lysosomal storage in Tay-Sachs disease. J. Mol. Med. 2018, 96, 1359–1373. [Google Scholar] [CrossRef] [PubMed]
- Arrant, A.E.; Roth, J.R.; Boyle, N.R.; Kashyap, S.N.; Hoffmann, M.Q.; Murchison, C.F.; Ramos, E.M.; Nana, A.L.; Spina, S.; Grinberg, L.T.; et al. Impaired beta-glucocerebrosidase activity and processing in frontotemporal dementia due to progranulin mutations. Acta Neuropathol. Commun. 2019, 7, 218. [Google Scholar] [CrossRef] [Green Version]
- Valdez, C.; Ysselstein, D.; Young, T.J.; Zheng, J.; Krainc, D. Progranulin mutations result in impaired processing of prosaposin and reduced glucocerebrosidase activity. Hum. Mol. Genet. 2019, 29, 716–726. [Google Scholar] [CrossRef]
- Velakoulis, D.; Walterfang, M.; Mocellin, R.; Pantelis, C.; McLean, C. Frontotemporal dementia presenting as schizophrenia-like psychosis in young people: Clinicopathological series and review of cases. Br. J. Psychiatry 2009, 194, 298–305. [Google Scholar] [CrossRef]
- Geser, F.; Robinson, J.L.; Malunda, J.A.; Xie, S.X.; Clark, C.M.; Kwong, L.K.; Moberg, P.J.; Moore, E.M.; Van Deerlin, V.M.; Lee, V.M.; et al. Pathological 43-kDa transactivation response DNA-binding protein in older adults with and without severe mental illness. Arch. Neurol. 2010, 67, 1238–1250. [Google Scholar] [CrossRef] [PubMed]
- Momeni, P.; DeTucci, K.; Straub, R.E.; Weinberger, D.R.; Davies, P.; Grafman, J.; Hardy, J.; Huey, E.D. Progranulin (GRN) in two siblings of a Latino family and in other patients with schizophrenia. Neurocase 2010, 16, 273–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galimberti, D.; Dell’Osso, B.; Fenoglio, C.; Villa, C.; Cortini, F.; Serpente, M.; Kittel-Schneider, S.; Weigl, J.; Neuner, M.; Volkert, J.; et al. Progranulin gene variability and plasma levels in bipolar disorder and schizophrenia. PLoS ONE 2012, 7, e32164. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, R.S.; Berk, M.; Brietzke, E.; Goldstein, B.I.; López-Jaramillo, C.; Kessing, L.V.; Malhi, G.S.; Nierenberg, A.A.; Rosenblat, J.D.; Majeed, A.; et al. Bipolar disorders. Lancet 2020, 396, 1841–1856. [Google Scholar] [CrossRef]
- Terleph, T.A.; Tremere, L.A. The Use of Immediate Early Genes as Mapping Tools for Neuronal Activation: Concepts and Methods. In Immediate Early Genes in Sensory Processing, Cognitive Performance and Neurological Disorders; Pinaud, R., Tremere, L.A., Eds.; Springer: Boston, MA, USA, 2006; pp. 1–10. [Google Scholar]
- Fromer, M.; Pocklington, A.J.; Kavanagh, D.H.; Williams, H.J.; Dwyer, S.; Gormley, P.; Georgieva, L.; Rees, E.; Palta, P.; Ruderfer, D.M.; et al. De novo mutations in schizophrenia implicate synaptic networks. Nature 2014, 506, 179–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cattane, N.; Minelli, A.; Milanesi, E.; Maj, C.; Bignotti, S.; Bortolomasi, M.; Bocchio Chiavetto, L.; Gennarelli, M. Altered gene expression in schizophrenia: Findings from transcriptional signatures in fibroblasts and blood. PLoS ONE 2015, 10, e0116686. [Google Scholar] [CrossRef]
- Kwon, W.; Kim, H.S.; Jeong, J.; Sung, Y.; Choi, M.; Park, S.; Lee, J.; Jang, S.; Kim, S.H.; Lee, S.; et al. Tet1 overexpression leads to anxiety-like behavior and enhanced fear memories via the activation of calcium-dependent cascade through Egr1 expression in mice. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2018, 32, 390–403. [Google Scholar]
- Iwakura, Y.; Kawahara-Miki, R.; Kida, S.; Sotoyama, H.; Gabdulkhaev, R.; Takahashi, H.; Kunii, Y.; Hino, M.; Nagaoka, A.; Izumi, R.; et al. Elevation of EGR1/zif268, a Neural Activity Marker, in the Auditory Cortex of Patients with Schizophrenia and its Animal Model. Neurochem. Res. 2022, 47, 2715–2722. [Google Scholar] [CrossRef]
- Smeland, O.B.; Shadrin, A.; Bahrami, S.; Broce, I.; Tesli, M.; Frei, O.; Wirgenes, K.V.; O’Connell, K.S.; Krull, F.; Bettella, F.; et al. Genome-wide Association Analysis of Parkinson’s Disease and Schizophrenia Reveals Shared Genetic Architecture and Identifies Novel Risk Loci. Biol. Psychiatry 2021, 89, 227–235. [Google Scholar] [CrossRef]
- Kuusimaki, T.; Al-Abdulrasul, H.; Kurki, S.; Hietala, J.; Hartikainen, S.; Koponen, M.; Tolppanen, A.M.; Kaasinen, V. Increased Risk of Parkinson’s Disease in Patients With Schizophrenia Spectrum Disorders. Mov. Disord. Off. J. Mov. Disord. Soc. 2021, 36, 1353–1361. [Google Scholar] [CrossRef]
- Kim, K.; Kim, S.; Myung, W.; Shim, I.; Lee, H.; Kim, B.; Cho, S.K.; Yoon, J.; Kim, D.K.; Won, H.H. Shared Genetic Background between Parkinson’s Disease and Schizophrenia: A Two-Sample Mendelian Randomization Study. Brain Sci. 2021, 11, 1042. [Google Scholar] [CrossRef] [PubMed]
- Schoder, D.; Hannequin, D.; Martinaud, O.; Opolczynski, G.; Guyant-Maréchal, L.; Le Ber, I.; Campion, D. Morbid risk for schizophrenia in first-degree relatives of people with frontotemporal dementia. Br. J. Psychiatry 2010, 197, 28–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibuya, Y.; Kumar, K.K.; Mader, M.M.; Yoo, Y.; Ayala, L.A.; Zhou, M.; Mohr, M.A.; Neumayer, G.; Kumar, I.; Yamamoto, R.; et al. Treatment of a genetic brain disease by CNS-wide microglia replacement. Sci. Transl. Med. 2022, 14, eabl9945. [Google Scholar] [CrossRef] [PubMed]
- Townley, R.A.; Boeve, B.F.; Benarroch, E.E. Progranulin: Functions and neurologic correlations. Neurology 2018, 90, 118–125. [Google Scholar] [CrossRef]
- Filiou, M.D.; Zhang, Y.; Teplytska, L.; Reckow, S.; Gormanns, P.; Maccarrone, G.; Frank, E.; Kessler, M.S.; Hambsch, B.; Nussbaumer, M.; et al. Proteomics and metabolomics analysis of a trait anxiety mouse model reveals divergent mitochondrial pathways. Biol. Psychiatry 2011, 70, 1074–1082. [Google Scholar] [CrossRef]
- Filiou, M.D.; Asara, J.M.; Nussbaumer, M.; Teplytska, L.; Landgraf, R.; Turck, C.W. Behavioral extremes of trait anxiety in mice are characterized by distinct metabolic profiles. J. Psychiatr. Res. 2014, 58, 115–122. [Google Scholar] [CrossRef]
- Klumpp, H.; Ho, S.S.; Taylor, S.F.; Phan, K.L.; Abelson, J.L.; Liberzon, I. Trait anxiety modulates anterior cingulate activation to threat interference. Depress. Anxiety 2011, 28, 194–201. [Google Scholar] [CrossRef] [Green Version]
- Blair, K.S.; Geraci, M.; Smith, B.W.; Hollon, N.; DeVido, J.; Otero, M.; Blair, J.R.; Pine, D.S. Reduced dorsal anterior cingulate cortical activity during emotional regulation and top-down attentional control in generalized social phobia, generalized anxiety disorder, and comorbid generalized social phobia/generalized anxiety disorder. Biol. Psychiatry 2012, 72, 476–482. [Google Scholar] [CrossRef] [Green Version]
- Yee, B.K. Cytotoxic lesion of the medial prefrontal cortex abolishes the partial reinforcement extinction effect, attenuates prepulse inhibition of the acoustic startle reflex and induces transient hyperlocomotion, while sparing spontaneous object recognition memory in the rat. Neuroscience 2000, 95, 675–689. [Google Scholar]
- Tapias-Espinosa, C.; Rio-Alamos, C.; Sanchez-Gonzalez, A.; Oliveras, I.; Sampedro-Viana, D.; Castillo-Ruiz, M.D.M.; Canete, T.; Tobena, A.; Fernandez-Teruel, A. Schizophrenia-like reduced sensorimotor gating in intact inbred and outbred rats is associated with decreased medial prefrontal cortex activity and volume. Neuropsychopharmacology 2019, 44, 1975–1984. [Google Scholar] [CrossRef]
- Kim, Y.; Noh, Y.W.; Kim, K.; Kim, E. Hyperactive ACC-MDT Pathway Suppresses Prepulse Inhibition in Mice. Schizophr. Bull. 2021, 47, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Rolls, E.T. The cingulate cortex and limbic systems for emotion, action, and memory. Brain Struct. Funct. 2019, 224, 3001–3018. [Google Scholar] [CrossRef] [PubMed]
- Logan, T.; Simon, M.J.; Rana, A.; Cherf, G.M.; Srivastava, A.; Davis, S.S.; Low, R.L.Y.; Chiu, C.L.; Fang, M.; Huang, F.; et al. Rescue of a lysosomal storage disorder caused by Grn loss of function with a brain penetrant progranulin biologic. Cell 2021, 184, 4651–4668.e25. [Google Scholar] [CrossRef] [PubMed]
- Narayan, S.; Head, S.R.; Gilmartin, T.J.; Dean, B.; Thomas, E.A. Evidence for disruption of sphingolipid metabolism in schizophrenia. J. Neurosci. Res. 2009, 87, 278–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, A.; Wilczek, K.; Blennow, K.; Maras, A.; Jatzko, A.; Petroianu, G.; Braus, D.F.; Gattaz, W.F. Altered thalamic membrane phospholipids in schizophrenia: A postmortem study. Biol. Psychiatry 2004, 56, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, E.; Prabakaran, S.; Whitfield, P.; Major, H.; Leweke, F.M.; Koethe, D.; McKenna, P.; Bahn, S. High throughput lipidomic profiling of schizophrenia and bipolar disorder brain tissue reveals alterations of free fatty acids, phosphatidylcholines, and ceramides. J. Proteome Res. 2008, 7, 4266–4277. [Google Scholar] [CrossRef]
- Esaki, K.; Balan, S.; Iwayama, Y.; Shimamoto-Mitsuyama, C.; Hirabayashi, Y.; Dean, B.; Yoshikawa, T. Evidence for Altered Metabolism of Sphingosine-1-Phosphate in the Corpus Callosum of Patients with Schizophrenia. Schizophr. Bull 2020, 46, 1172–1181. [Google Scholar] [CrossRef]
- Checa, A.; Malmqvist, A.; Flyckt, L.; Schwieler, L.; Samuelsson, M.; Skogh, E.; Cervenka, S.; Dahl, M.L.; Piehl, F.; Karolinska Schizophrenia Project, C.; et al. Cerebrospinal fluid levels of sphingolipids associate with disease severity in first episode psychosis patients. Schizophr. Res. 2018, 199, 438–441. [Google Scholar] [CrossRef]
- Smesny, S.; Schmelzer, C.E.; Hinder, A.; Kohler, A.; Schneider, C.; Rudzok, M.; Schmidt, U.; Milleit, B.; Milleit, C.; Nenadic, I.; et al. Skin ceramide alterations in first-episode schizophrenia indicate abnormal sphingolipid metabolism. Schizophr. Bull. 2013, 39, 933–941. [Google Scholar] [CrossRef] [Green Version]
- Alquezar, C.; de la Encarnacion, A.; Moreno, F.; Lopez de Munain, A.; Martin-Requero, A. Progranulin deficiency induces overactivation of WNT5A expression via TNF-alpha/NF-kappaB pathway in peripheral cells from frontotemporal dementia-linked granulin mutation carriers. J. Psychiatry Neurosci. 2016, 41, 225–239. [Google Scholar] [CrossRef] [Green Version]
- Krabbe, G.; Minami, S.S.; Etchegaray, J.I.; Taneja, P.; Djukic, B.; Davalos, D.; Le, D.; Lo, I.; Zhan, L.; Reichert, M.C.; et al. Microglial NFkappaB-TNFalpha hyperactivation induces obsessive-compulsive behavior in mouse models of progranulin-deficient frontotemporal dementia. Proc. Natl. Acad. Sci. USA 2017, 114, 5029–5034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ketharanathan, T.; Pereira, A.; Reets, U.; Walker, D.; Sundram, S. Brain changes in NF-κB1 and epidermal growth factor system markers at peri-pubescence in the spiny mouse following maternal immune activation. Psychiatry Res. 2021, 295, 113564. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Tian, L.; Baranova, A.; Cao, H.; Yao, Y.; Rao, S.; Zhang, F. Convergent lines of evidence supporting involvement of NFKB1 in schizophrenia. Psychiatry Res. 2022, 312, 114588. [Google Scholar] [CrossRef] [PubMed]
- Mataga, N.; Fujishima, S.; Condie, B.G.; Hensch, T.K. Experience-dependent plasticity of mouse visual cortex in the absence of the neuronal activity-dependent marker egr1/zif268. J. Neurosci. Off. J. Soc. Neurosci. 2001, 21, 9724–9732. [Google Scholar] [CrossRef] [Green Version]
- Kelly, M.P.; Deadwyler, S.A. Experience-dependent regulation of the immediate-early gene arc differs across brain regions. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 6443–6451. [Google Scholar] [CrossRef] [Green Version]
- Han, J.H.; Kushner, S.A.; Yiu, A.P.; Cole, C.J.; Matynia, A.; Brown, R.A.; Neve, R.L.; Guzowski, J.F.; Silva, A.J.; Josselyn, S.A. Neuronal competition and selection during memory formation. Science 2007, 316, 457–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Q.; McHugh, R.; Morgan, E.; Sutherland, R.J.; McDonald, R.J. Behaviour-driven Arc expression is greater in dorsal than ventral CA1 regardless of task or sex differences. Behav. Brain Res. 2022, 423, 113790. [Google Scholar] [CrossRef] [PubMed]
- García-Díaz, C.; Sánchez-Catalán, M.J.; Castro-Salazar, E.; García-Avilés, A.; Albert-Gascó, H.; Sánchez-Sarasúa de la Bárcena, S.; Sánchez-Pérez, A.M.; Gundlach, A.L.; Olucha-Bordonau, F.E. Nucleus incertus ablation disrupted conspecific recognition and modified immediate early gene expression patterns in ‘social brain’ circuits of rats. Behav. Brain Res. 2019, 356, 332–347. [Google Scholar] [CrossRef]
- Torrey, E.F.; Webster, M.; Knable, M.; Johnston, N.; Yolken, R.H. The stanley foundation brain collection and neuropathology consortium. Schizophr. Res. 2000, 44, 151–155. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Healthy Control | Schizophrenia | Bipolar Disorder | Major Depressive Disorder | |
|---|---|---|---|---|
| Age (y) | 49.4 ± 3.2 | 43.6 ± 4.2 | 40.0 ± 3.4 | 46.3 ± 2.4 |
| Gender (M/F) | 7/5 | 7/5 | 8/4 | 8/4 |
| Age of Onset (y) | N/A | 23.5 ± 2.6 | 20.9 ± 2.5 | 32.0 ± 3.3 |
| Psychosis (%) | 0 | 100 | 75 | 0 |
| FLU (mg) | 0 | 37083 ± 12738 | 18367 ± 6691 | 0 |
| PMI (H) | 23.6 ± 2.7 | 31.4 ± 4.0 | 32.1 ± 4.3 | 27.3 ± 3.4 |
| pH | 6.33 ± 0.06 | 6.19 ± 0.08 | 6.20 ± 0.07 | 6.20 ± 0.06 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Y.; Zhang, X.; Flais, I.; Svenningsson, P. Decreased Prosaposin and Progranulin in the Cingulate Cortex Are Associated with Schizophrenia Pathophysiology. Int. J. Mol. Sci. 2022, 23, 12056. https://doi.org/10.3390/ijms231912056
He Y, Zhang X, Flais I, Svenningsson P. Decreased Prosaposin and Progranulin in the Cingulate Cortex Are Associated with Schizophrenia Pathophysiology. International Journal of Molecular Sciences. 2022; 23(19):12056. https://doi.org/10.3390/ijms231912056
Chicago/Turabian StyleHe, Yachao, Xiaoqun Zhang, Ivana Flais, and Per Svenningsson. 2022. "Decreased Prosaposin and Progranulin in the Cingulate Cortex Are Associated with Schizophrenia Pathophysiology" International Journal of Molecular Sciences 23, no. 19: 12056. https://doi.org/10.3390/ijms231912056