
Chemoenzymatic Synthesis of Optically Active Ethereal Analog of iso-Moramide—A Novel Potentially Powerful Analgesic †

Abstract
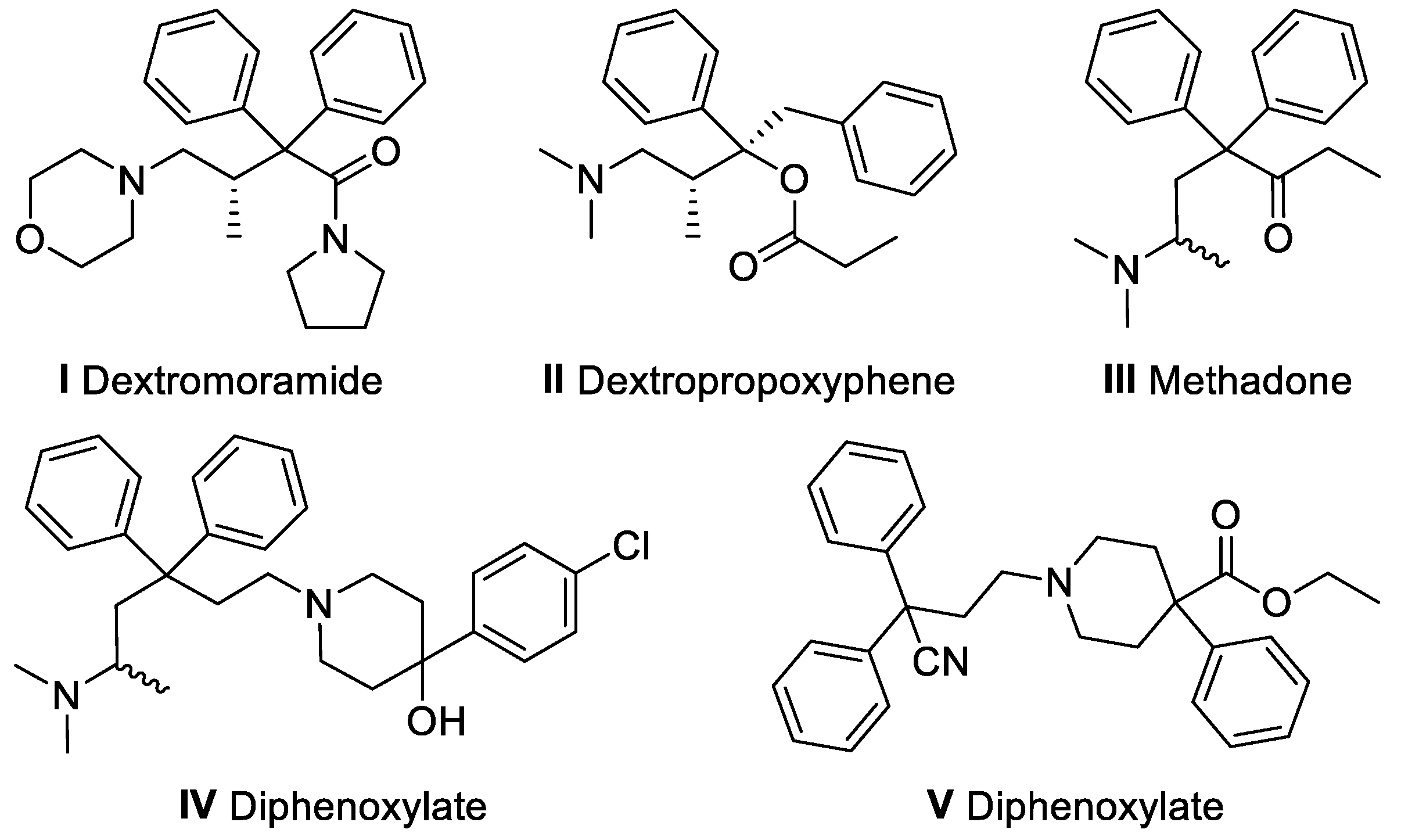
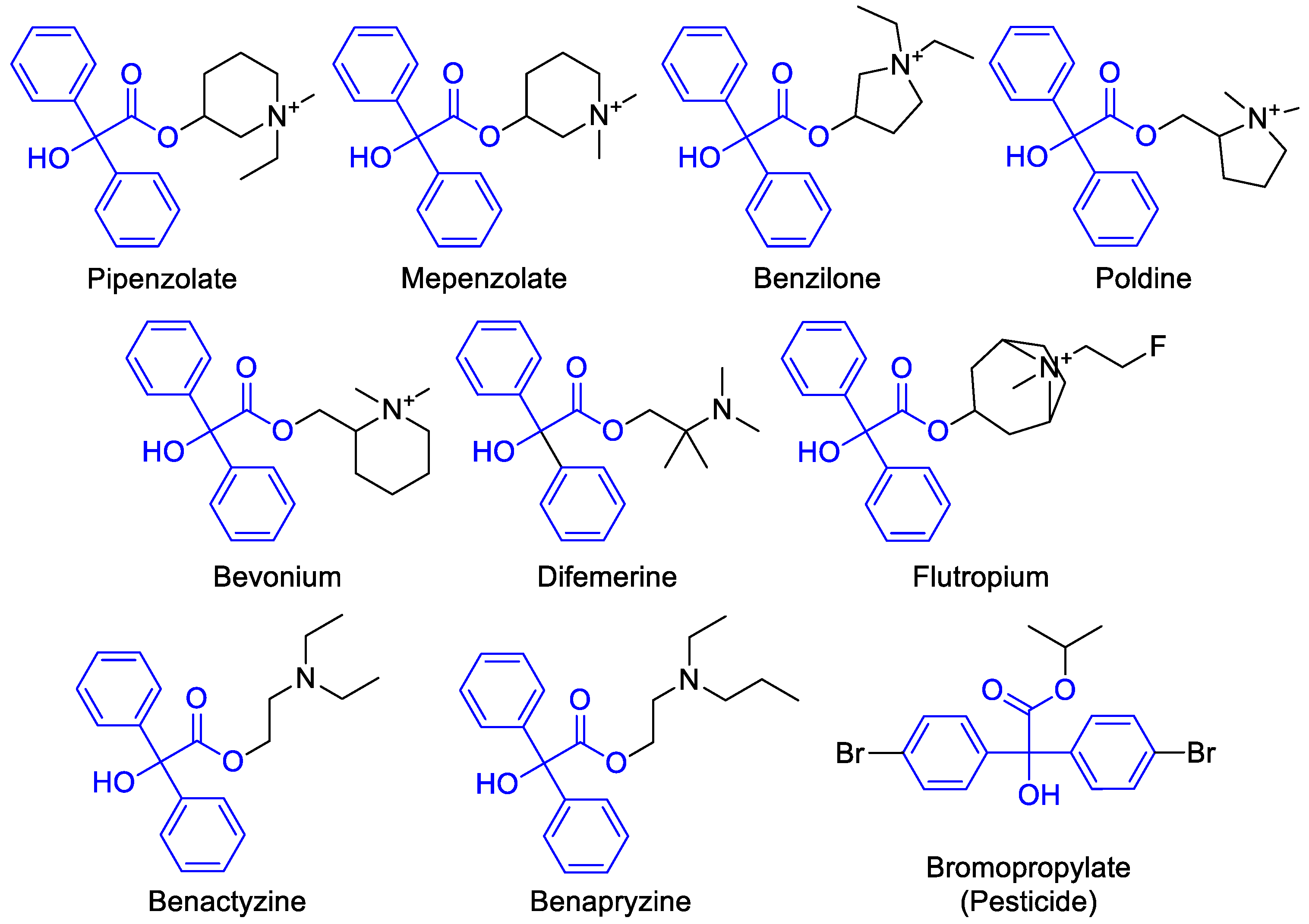
:1. Introduction
2. Results and Discussion
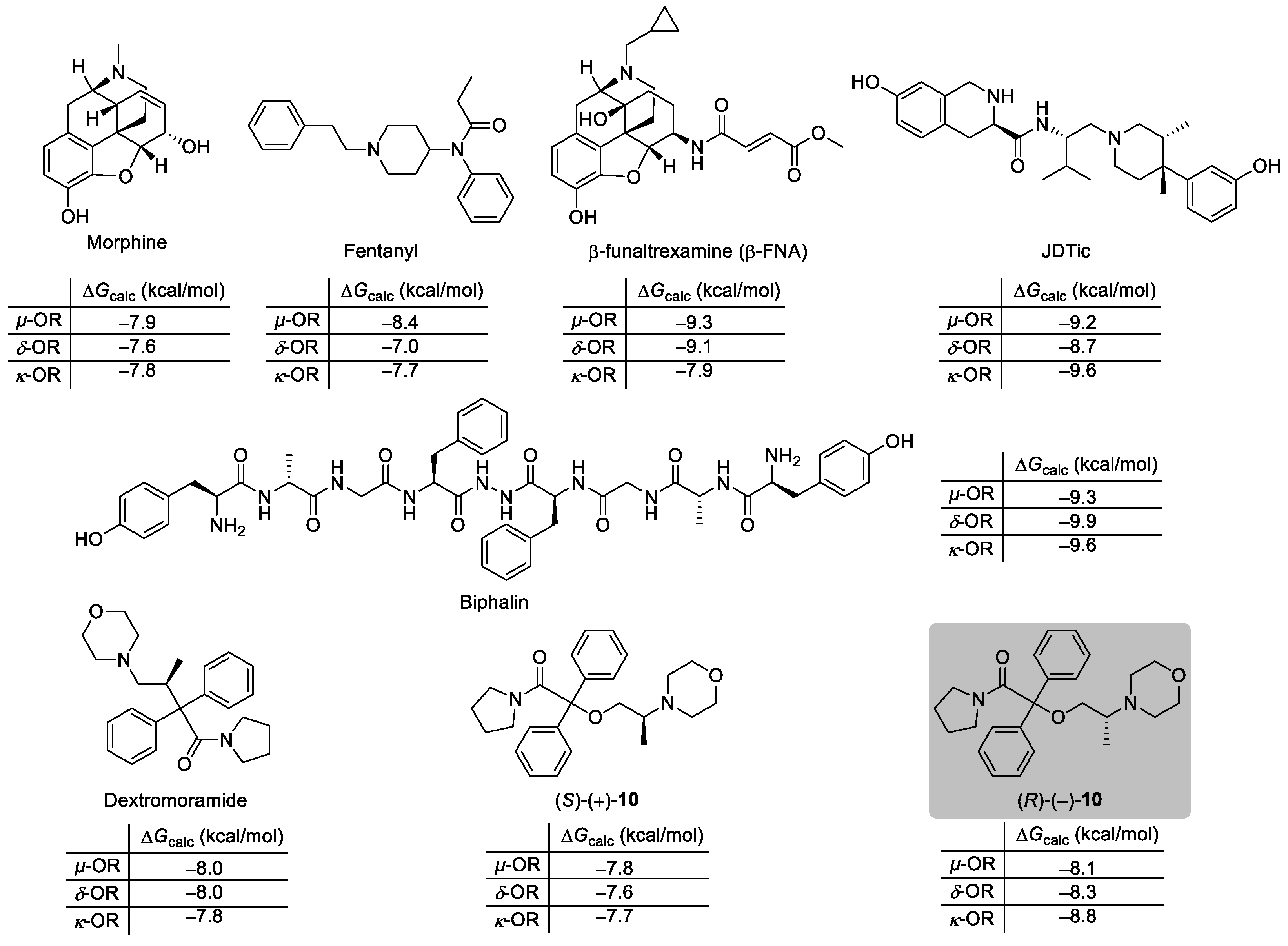
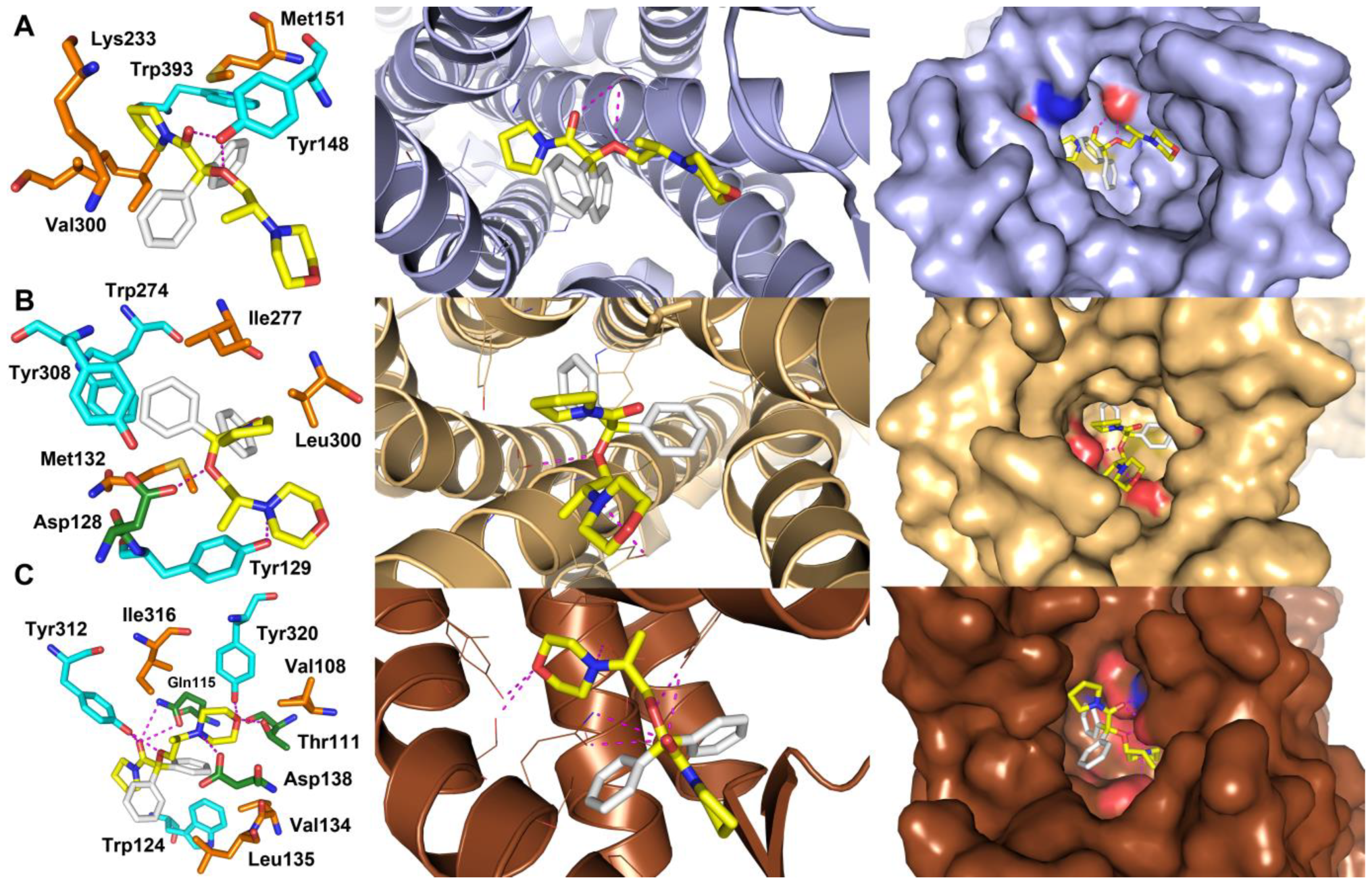
2.1. Molecular Docking Studies
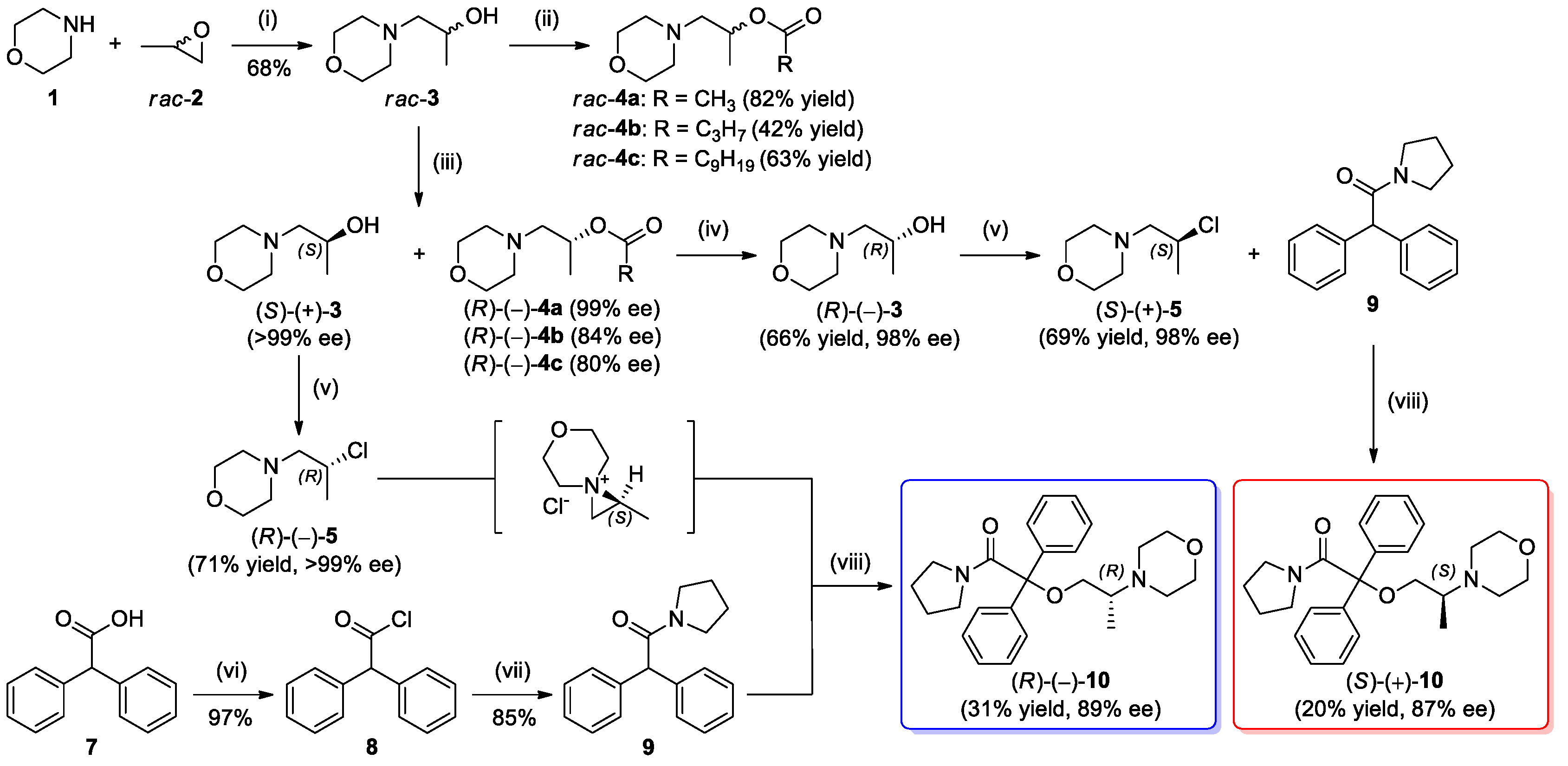
2.2. Synthesis of the Racemic Compounds rac-3 and rac-4a–c
2.3. Lipase-Catalyzed Kinetic Resolution of rac-3 Using Transesterification Methodology

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Lipase Preparation a | t (h) | Conv. (%) b | ees (%) c | eep (%) d | Ee |
|---|---|---|---|---|---|---|
| 1 | Amano PS | 144 | 41 | 62 | 89 | 32 |
| 2 | Amano PS-IM | 120 | 55 | >99 | 81 | 49 |
| 3 | Amano PS-Immobead 150 | 120 | 32 | 46 | >99 | 314 |
| 4 | Novozym 435 | 120 | 67 | >99 | 49 | 14 |
| 5 | Chirazyme L-2, C-2 | 24 | 58 | >99 | 72 | 31 |
| 6 | Chirazyme L-2, C-3 | 24 | 53 | 95 | 84 | 42 |
| 7 | Amano AK | 72 | 48 | 85 | 92 | 65 |
| 8 | Chirazyme L-5 | 72 | 28 | 17 | 44 | 3 |
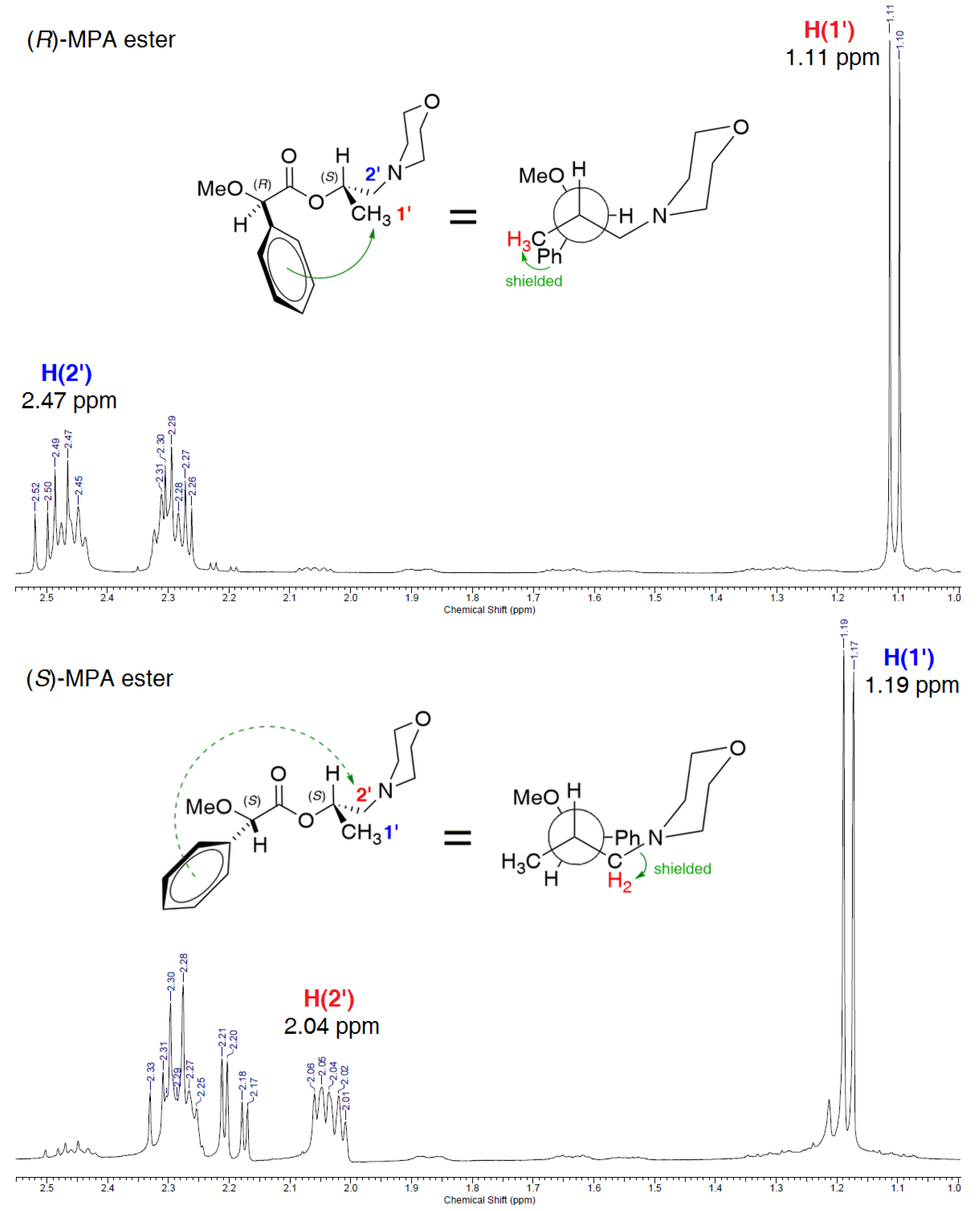
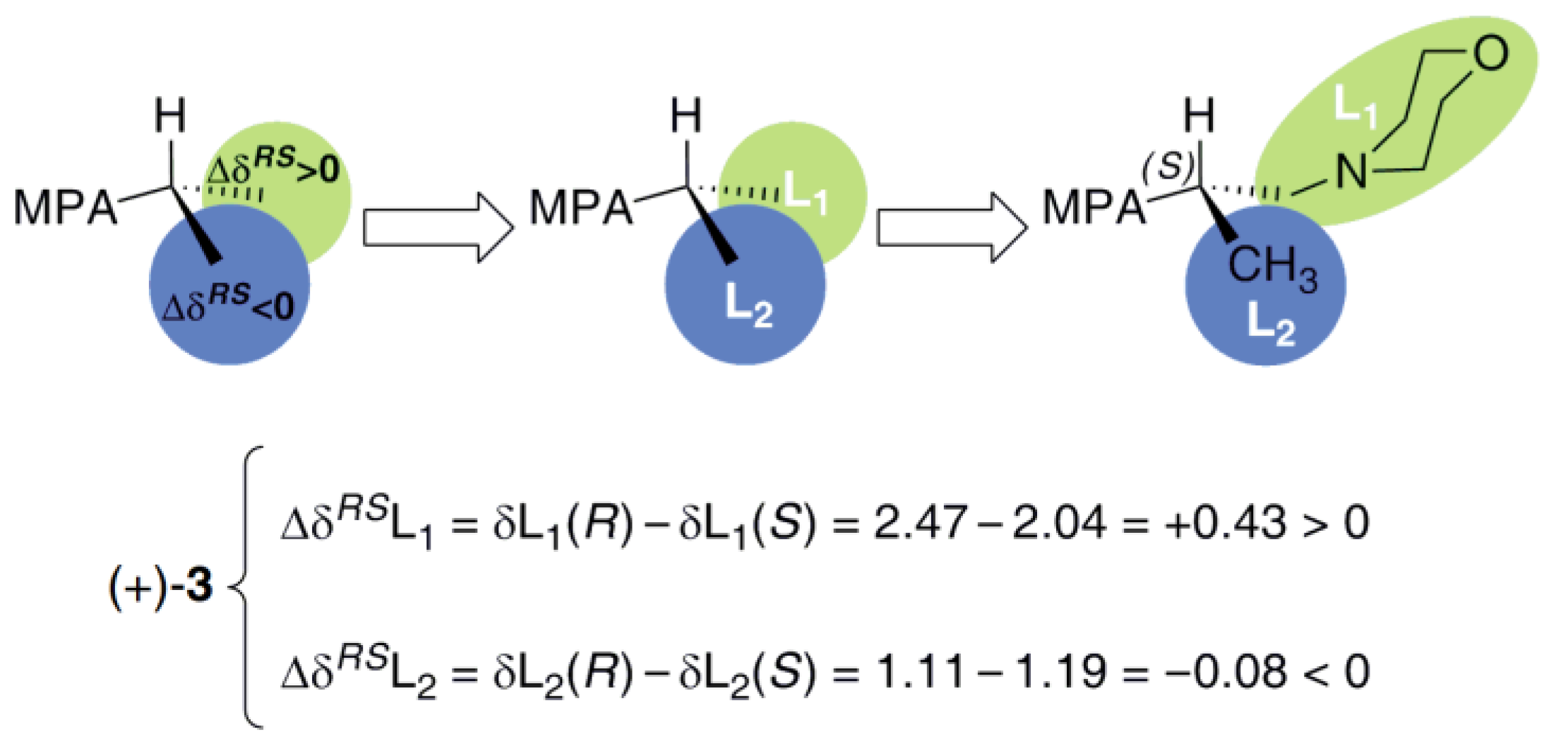
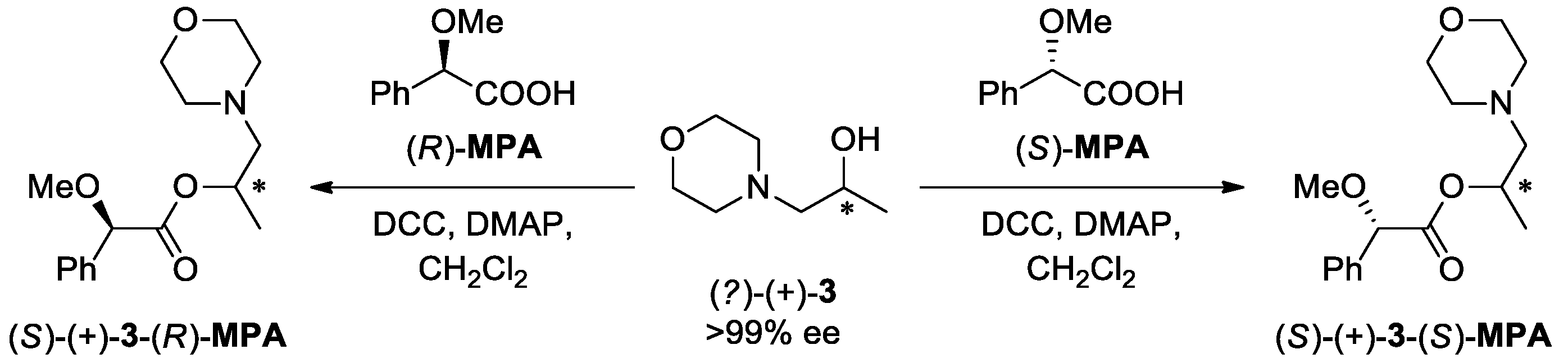
2.4. Determination of the Absolute Configuration of (S)-(+)-3
2.5. Synthesis of Optically Active Ethereal Analog of iso-Moramide (S)-(+)-10 and (R)-(–)-10
3. Materials and Methods
3.1. Molecular Docking
3.2. General Procedure for the Synthesis of 1-(Morpholin-4-yl)propan-2-ol rac-3
3.3. General Procedure for the Synthesis of 1-(Morpholin-4-yl)propan-2-yl acetate rac-4a
3.4. General Procedure for the Synthesis of 1-(Morpholin-4-yl)propan-2-yl butanoate rac-4b
3.5. General Procedure for the Synthesis of 1-(Morpholin-4-yl)propan-2-yl decanoate rac-4c
3.6. General Procedure for Kinetic Resolution of rac-3–Enzyme Screening
3.7. General Procedure for Kinetic Resolution of rac-3–Solvent Screening
3.8. General Procedure for Kinetic Resolution of rac-3–Acyl Donor Screening
3.9. General Procedure for Gram-Scale Kinetic Resolution of rac-3
3.10. General Procedure for Multigram-Scale Kinetic Resolution of rac-3
3.11. General Procedure for Base-Mediated Methanolysis of (R)-(-)-1-(Morpholin-4-yl)propan-2-yl acetate (R)-(–)-4a
3.12. General Procedure for the Determination of the Absolute Configuration of (S)-(+)-1-(Morpholin-4-yl)propan-2-ol (S)-(+)-3 Realized Via Esterification of (S)-(+)-3 with Enantiomers of α-Methoxy-α-phenylacetic Acid (R)-MPA or (S)-MPA
3.13. General Procedure for the Synthesis of 4-[(2R)-(–)-2-Chloropropyl]morpholine (R)-(–)-5 or 4-[(2S)-(+)-2-Chloropropyl]morpholine (S)-(+)-5
3.14. General Procedure for the Synthesis of (R)- and (S)-4-[2-(Phenylsulfanyl)propyl]morpholine (R)-5–SPh and (S)-5–SPh
3.15. General Procedure for the Synthesis of Diphenylacetic Acid Chloride (8)
3.16. General Procedure for the Synthesis of N-Diphenylacetyl-1-pyrrolidine (9)
3.17. General Procedure for the Synthesis of 2-[(2R)-2-(Morpholin-4-yl)propoxy]-2,2-diphenyl-1-(pyrrolidin-1-yl)ethan-1-one ((R)-(–)-10) and 2-[(2S)-2-(Morpholin-4-yl)propoxy]-2,2-diphenyl-1-(pyrrolidin-1-yl)ethan-1-one ((S)-(+)-10)
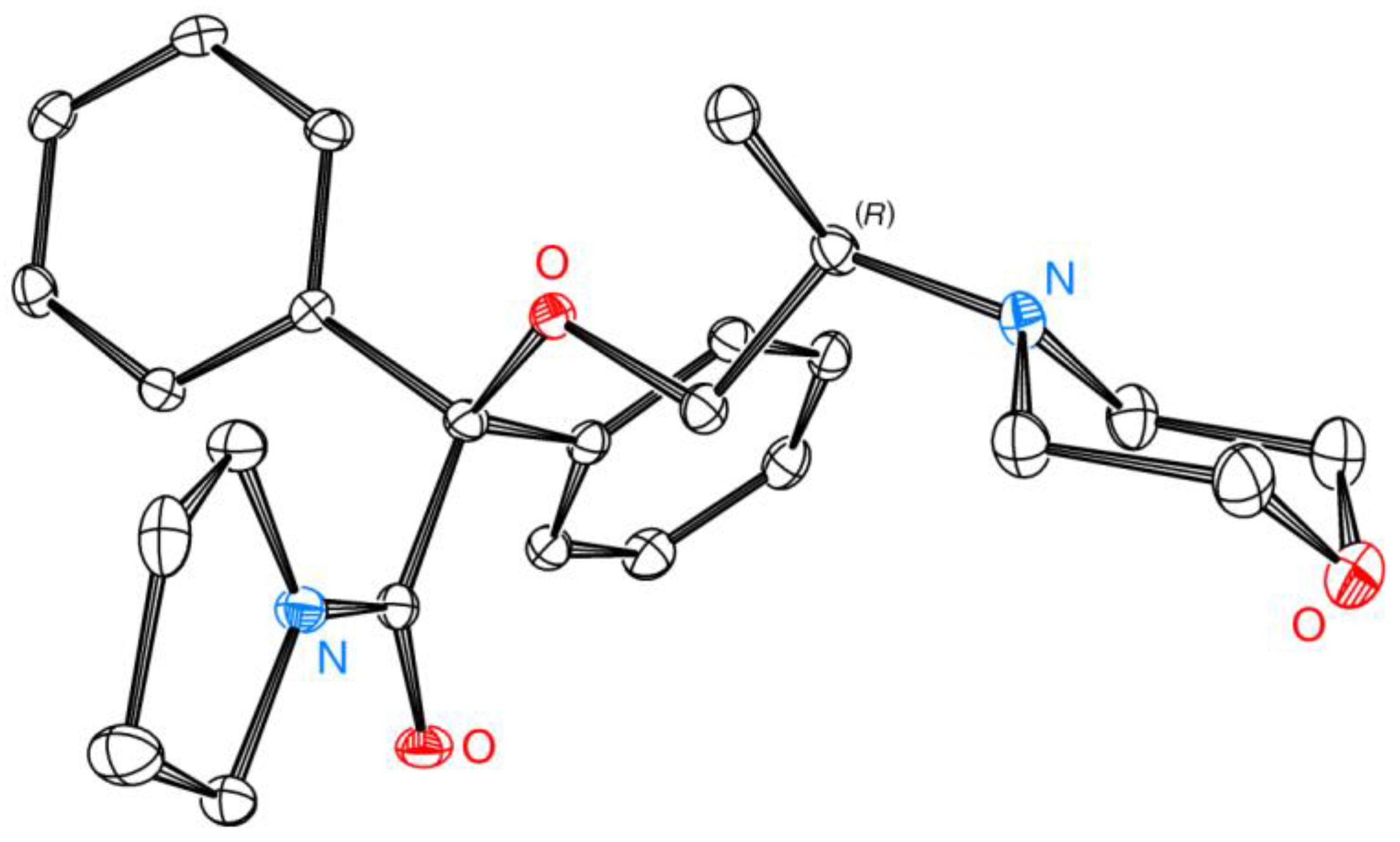
3.18. XRD Analyses
3.18.1. Conditions for Crystal Growth of 2-[(2R)-2-(Morpholin-4-yl)propoxy]-2,2-diphenyl-1-(pyrrolidin-1-yl)ethan-1-one ((R)-(–)-10)
3.18.2. Crystal Structure Determination of (R)-(–)-10
3.18.3. Crystal Data for (R)-(–)-10
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ventafridda, V.; Saita, L.; Ripamonti, C.; Deconno, F. Who Guidelines for the Use of Analgesics in Cancer Pain. Int. J. Tissue React. 1985, 7, 93–96. [Google Scholar] [PubMed]
- Nagase, H. (Ed.) Chemistry of Opioids; Part of Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Holtsman, M. Essentials of Pain Medicine and Regional Anesthesia (Second Edition): Opioid Receptors; Elsevier Inc.: Amsterdam, The Netherlands, 2005; pp. 87–93. [Google Scholar]
- Janssen, P.A.J.; Janseen, J.C. A New Series of Potent Analgesics. J. Am. Chem. Soc. 2002, 78, 3862. [Google Scholar] [CrossRef]
- Janssen, P.A.J.; Jageneau, A.H. A New Series of Potent Analgesics. J. Pharm. Pharmacol. 1957, 9, 381–400. [Google Scholar] [CrossRef]
- Janssen, P.A.J. Synthetic Analgesics Part 1: Diphenylpropylamines; Pergamon Press: New York, NY, USA, 1960; p. 141. [Google Scholar]
- Demoen, P.J. Properties and analysis of dextromoramide and its dosage forms. J. Pharm. Sci. 1961, 50, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Pagani, I.; Barzaghi, N.; Crema, F.; Perucca, E.; Ego, D.; Rovei, V. Pharmacokinetics of dextromoramide in surgical patients. Fund. Clin. Pharmacol. 1989, 3, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Seymour-Shove, R.; Wilson, C.W. Dependence on dextromoramide. British Med. J. 1967, 1, 88–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulyamov, M.; Dalimov, D.N.; Tilyabaev, Z.; Kamaev, F.G.; Abduvakhabov, A.A. Synthesis of some bis(2-morpholino-1-methylethyl) esters of carboxylic acids and study of their interaction with the cholinesterases of warm-blooded animals. Chem. Nat. Compd. 1988, 24, 203–206. [Google Scholar] [CrossRef]
- Gafurov, M.B.; Dalimov, D.N.; Kamaev, F.G.; Vaizburg, G.M.; Abduvakhabov, A.A. Synthesis, structure, and anticholinesterase activities of methylphosphonothionates of N-β-hydroxyethylmorpholine and N-β-hydroxypropylmorpholine. Chem. Nat. Compd. 1989, 25, 465–468. [Google Scholar] [CrossRef]
- Gielen, H.; Min-Jian Li, V.; Rosentreter, U.; Schlemmer, K.H.; Allerheiligen, S.; Telan, L.; Bärfacker, L.; Keldenich, J.; Fitzgerald, M.F.; Nash, K.; et al. Dihydropyridine Derivatives for Use as Human Neutrophil Elastase Inhibitors. WO Patent 020412, 11 March 2004. [Google Scholar]
- Amishiro, N.; Fukuda, Y.; Kinpara, K.; Mie, M.; Tagaya, H.; Takahashi, T. Kynurenine Production Inhibitor. U.S. Patent 237584A1, 29 September 2011. [Google Scholar]
- Courtney, S.M.; Prime, M.; Mitchell, W.; Brown, C.J.; De Aguiar Pena, P.C.; Johnson, P.; Dominguez, C.; Toledo-Sherman, L.M.; Munoz, I. Kynurenine-3-monooxygenase Inhibitors, Pharmaceutical Compositions, and Methods of Use Thereof. WO Patent 033085A1, 7 March 2013. [Google Scholar]
- Booker, S.; D’angelo, N.; D’amico, D.C.; Kim, T.-S.; Liu, L.; Meagher, K.; Norman, M.H.; Panter, K.; Schenkel, L.B.; Smith, A.L.; et al. PI3 Kinase Modulators and Methods of Use. WO Patent 017822A2, 5 May 2009. [Google Scholar]
- Stieber, F.; Schadt, O.; Dorsch, D.; Blaukat, A. Pyridazinone Derivatives. U.S. Patent 0257181 A1, 15 October 2009. [Google Scholar]
- Hutchison, A.; Yuan, J.; Lee, K.; Maynard, G.; Chenard, B.L.; Liu, N.; Guo, Q.; Guo, Z.; Hrnciar, P. 3-Substituted-6-aryl Pyridines as Ligands of C5a Receptors. WO Patent 043925 A2, 27 May 2004. [Google Scholar]
- Vasudevan, A.; Wodka, D.; Verzal, M.K.; Souers, A.J.; Gao, J.; Brodjian, S.; Fry, D.; Dayton, B.; Marsh, K.C.; Hernandez, L.E.; et al. Synthesis and evaluation of 2-amino-8-alkoxy quinolines as MCHr1 antagonists. Part 2. Bioorg. Med. Chem. Lett. 2004, 14, 4879–4882. [Google Scholar] [CrossRef] [PubMed]
- Andrews, M.D.; Blagg, J.; Brennan, P.E.; Fish, P.V.; Roberts, L.R.; Storer, R.I.; Whitlock, G.A. Pyrimido [4, 5-D] Azepine Derivatives as 5-HT2C Agonists. WO Patent 117169 A1, 2 October 2008. [Google Scholar]
- Harbottle, G.W.; Feeder, N.; Gibson, K.R.; Glossop, M.; Maw, G.N.; Million, W.A.; Morel, F.F.; Osborne, S.; Poinsard, C. Microwave-assisted synthesis of mGluR1 ligands: Carbon, nitrogen and oxygen linked derivatives of pyrido[3,4-d]pyrimidin-4-ylamines. Tetrahedron Lett. 2007, 48, 4293–4296. [Google Scholar] [CrossRef]
- Grauert, M.; Bakker, R.; Breitfelder, S.; Buettner, F.; Eickelmann, P.; Fox, T.; Grundl, M.; Lehmann-Lintz, T.; Rist, W. Pyridazinones as GPR119 Agonists, W. WO Patent 138427 A2, 10 November 2011. [Google Scholar]
- Sparks, M.A.; Freidinger, R.M.; Perlow, D.S.; Williams, P.D. Tocolytic Oxytocin Receptor Antagonists. U.S. Patent 5,726,172, 10 November 1998. [Google Scholar]
- Ismaiel, A.M.; Gad, L.M.; Ghareib, S.A.; Bamanie, F.H.; Moustafa, M.A. Synthesis of piperazino and morpholino derivatives of aryloxypropane with potential analgesic and possible antimigraine activities. Med. Chem. Res. 2010, 20, 381–387. [Google Scholar] [CrossRef]
- Beshore, D.C.; Dudkin, V.; Garbaccio, R.M.; Johnson, A.W.; Kuduk, S.D.; Skudlarek, J.W.; Wang, C.; Fraley, M.E. Ether Benzotriazole Derivatives. U.S. Patent 0135977 A1, 31 May 2012. [Google Scholar]
- Bai, H.; Bailey, S.; Bhumralkar, D.R.; Bi, F.; Guo, F.; He, M.; Humphries, P.S.; Ling, A.L.; Lou, J.; Nukui, S.; et al. Fused Phenyl amido Heterocyclic Compounds for the Prevention and Treatment of Glucokinase-Mediated Diseases. WO Patent 122482 A1, 1 November 2007. [Google Scholar]
- Bossert, F.; Meyer, H.; Vater, W. 1,4-Dihydropyridine Carboxylic Acid Esters Useful as Coronary Vessel Dilators and Anti-Hypertensives. U.S. Patent 3,974,275, 10 August 1976. [Google Scholar]
- Cirillo, P.F.; Hickey, E.R.; Regan, J.R. Aromatic Heterocyclic Compounds as Anti-Inflammatory Agents. U.S. Patent 6,297,381 B1, 2 October 2001. [Google Scholar]
- Salakhutdinov, B.A.; Dalimov, D.N.; Aripov, T.F.; Tukfatullina, I.I.; Ziyatdinova, R.K.; Dzhuraev, A.Z.; Kamaev, F.G.; Izotova, L.Y.; Ibragimov, B.T.; Mavridis, I.; et al. Synthesis, Structure, and Membrane Activity of New Glycyrrhetic Acid Derivatives. Chem. Nat. Compd. 2002, 38, 249–256. [Google Scholar] [CrossRef]
- Argoff, C.E.; Viscusi, E.R. The use of opioid analgesics for chronic pain: Minimizing the risk for harm. Am. J. Gastroenterol. Suppl. 2014, 2, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Kieffer, B.L.; Evans, C.J. Opioid receptors: From binding sites to visible molecules in vivo. Neuropharmacology 2009, 56 (Suppl. S1), 205–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasternak, G.W. Opioids and their receptors: Are we there yet? Neuropharmacology 2014, 76 Pt B, 198–203. [Google Scholar] [CrossRef] [Green Version]
- Raehal, K.M.; Schmid, C.L.; Groer, C.E.; Bohn, L.M. Functional selectivity at the mu-opioid receptor: Implications for understanding opioid analgesia and tolerance. Pharmacol. Rev. 2011, 63, 1001–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, J.T.; Ingram, S.L.; Henderson, G.; Chavkin, C.; von Zastrow, M.; Schulz, S.; Koch, T.; Evans, C.J.; Christie, M.J. Regulation of mu-opioid receptors: Desensitization, phosphorylation, internalization, and tolerance. Pharmacol. Rev. 2013, 65, 223–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costigan, M.; Woolf, C.J. Pain: Molecular mechanisms. J. Pain 2000, 1, 35–44. [Google Scholar] [CrossRef]
- Hutchinson, M.R.; Shavit, Y.; Grace, P.M.; Rice, K.C.; Maier, S.F.; Watkins, L.R. Exploring the neuroimmunopharmacology of opioids: An integrative review of mechanisms of central immune signaling and their implications for opioid analgesia. Pharmacol. Rev. 2011, 63, 772–810. [Google Scholar] [CrossRef] [Green Version]
- Vanderah, T.W. Delta and kappa opioid receptors as suitable drug targets for pain. Clin. J. Pain 2010, 26 (Suppl. S10), S10–S15. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, G.W.; Pan, Y.X. Mu opioids and their receptors: Evolution of a concept. Pharmacol. Rev. 2013, 65, 1257–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, P.Y.; Hoon, M.A. Molecular genetics of kappa opioids in pain and itch sensations. In The Kappa Opioid Receptor; Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2022; Volume 271, pp. 255–274. [Google Scholar] [CrossRef]
- Inan, S.; Cowan, A. Antipruritic effects of kappa opioid receptor agonists: Evidence from rodents to humans. In The Kappa Opioid Receptor; Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2022; Volume 271, pp. 275–292. [Google Scholar]
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granier, S.; Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Weis, W.I.; Kobilka, B.K. Structure of the delta-opioid receptor bound to naltrindole. Nature 2012, 485, 400–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Wacker, D.; Mileni, M.; Katritch, V.; Han, G.W.; Vardy, E.; Liu, W.; Thompson, A.A.; Huang, X.P.; Carroll, F.I.; et al. Structure of the human kappa-opioid receptor in complex with jdtic. Nature 2012, 485, 327–332. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. Autodock vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comp. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Azizi, N.; Saidi, M.R. Highly chemoselective addition of amines to epoxides in water. Org. Lett. 2005, 7, 3649–3651. [Google Scholar] [CrossRef]
- Chen, C.S.; Fujimoto, Y.; Girdaukas, G.; Sih, C.J. Quantitative analyses of biochemical kinetic resolutions of enantiomers. J. Am. Chem. Soc. 2002, 104, 7294–7299. [Google Scholar] [CrossRef]
- Subileau, M.; Jan, A.H.; Drone, J.; Rutyna, C.; Perrier, V.; Dubreucq, E. What makes a lipase a valuable acyltransferase in water abundant medium? Catal. Sci. Technol. 2017, 7, 2566–2578. [Google Scholar] [CrossRef]
- Kitamoto, Y.; Kuruma, Y.; Suzuki, K.; Hattori, T. Effect of solvent polarity on enantioselectivity in Candida antarctica lipase B catalyzed kinetic resolution of primary and secondary alcohols. J. Org. Chem. 2015, 80, 521–527. [Google Scholar] [CrossRef]
- Carrea, G.; Riva, S. Properties and Synthetic Applications of Enzymes in Organic Solvents. Angew. Chem. Int. Ed. 2000, 39, 2226–2254. [Google Scholar] [CrossRef]
- Ema, T.; Maeno, S.; Takaya, Y.; Sakai, T.; Utaka, M. Significant effect of acyl groups on enantioselectivity in lipase-catalyzed transesterifications. Tetrahedron Asymm. 1996, 7, 625–628. [Google Scholar] [CrossRef]
- Flores, M.V.; Sewalt, J.J.W.; Janssen, A.E.M.; van der Padt, A. The nature of fatty acid modifies the equilibrium position in the esterification catalyzed by lipase. Biotechnol. Bioeng. 2000, 67, 364–371. [Google Scholar] [CrossRef]
- Ottosson, J.; Hult, K. Influence of acyl chain length on the enantioselectivity of Candida antarctica lipase B and its thermodynamic components in kinetic resolution of sec-alcohols. J. Mol. Catal. B Enzym. 2001, 11, 1025–1028. [Google Scholar] [CrossRef]
- Melais, N.; Aribi-Zouioueche, L.; Riant, O. The effect of the migrating group structure on enantioselectivity in lipase-catalyzed kinetic resolution of 1-phenylethanol. Comptes Rend. Chim. 2016, 19, 971–977. [Google Scholar] [CrossRef]
- Noro, J.; Castro, T.G.; Cavaco-Paulo, A.; Silva, C. Substrate hydrophobicity and enzyme modifiers play a major role in the activity of lipase from Thermomyces lanuginosus. Catal. Sci. Technol. 2020, 10, 5913–5924. [Google Scholar] [CrossRef]
- Baumann, M.; Leslie, A.; Moody, T.S.; Smyth, M.; Wharry, S. Tandem continuous flow curtius rearrangement and subsequent enzyme-mediated impurity tagging. Organic Proc. Res. Dev. 2020, 25, 452–456. [Google Scholar] [CrossRef]
- Resa, J.M.; González, C.; Ortiz de Landaluce, S.; Goenaga, J.M. Density, refractive index, speed of sound, and vapor−liquid equilibria for binary mixtures of methanol + vinyl propionate and vinyl acetate + vinyl propionate. Vapor pressures of vinyl propionate. J. Chem. Eng. Data 2005, 50, 319–324. [Google Scholar] [CrossRef]
- Grayson, D.; Lewis, F.; Eichler, M. Synthesis of β-Amino Alcohols via the Reduction of Lactamides Derived from Ethyl (2S)-Lactate with Borane-Methyl Sulfide. Synlett 2009, 2009, 1923–1928. [Google Scholar] [CrossRef]
- Seco, J.M.; Quiñoá, E.; Riguera, R. A practical guide for the assignment of the absolute configuration of alcohols, amines and carboxylic acids by NMR. Tetrahedron Asymm. 2001, 12, 2915–2925. [Google Scholar] [CrossRef]
- Latypov, S.K.; Seco, J.M.; Quinoa, E.; Riguera, R. Conformational Structure and Dynamics of Arylmethoxyacetates: DNMR Spectroscopy and Aromatic Shielding Effect. J. Org. Chem. 2002, 60, 504–515. [Google Scholar] [CrossRef]
- Chuang, G.J.; Wang, W.; Lee, E.; Ritter, T. A dinuclear palladium catalyst for alpha-hydroxylation of carbonyls with O2. J. Am. Chem. Soc. 2011, 133, 1760–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.F.; Jiao, N. Highly efficient C-H hydroxylation of carbonyl compounds with oxygen under mild conditions. Angew. Chem. 2014, 53, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Russell, G.A.; Janzen, E.G.; Becker, H.-D.; Smentowski, F.J. Autoxidation and Condensation Reactions of Carbanions in Dimethyl Sulfoxide Solution. J. Am. Chem. Soc. 2002, 84, 2652–2653. [Google Scholar] [CrossRef]
- Russell, G.A. Detection of paramagnetic intermediates in the oxidation of carbanions. Pure App. Chem. 1967, 15, 185–206. [Google Scholar] [CrossRef]
- Farrugia, L.J. Wingxandortep for windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Williams, D.B.; Lawton, M. Drying of organic solvents: Quantitative evaluation of the efficiency of several desiccants. J. Org. Chem. 2010, 75, 8351–8354. [Google Scholar] [CrossRef]
- Bojarska, J.; New, R.; Borowiecki, P.; Remko, M.; Breza, M.; Madura, I.D.; Fruzinski, A.; Pietrzak, A.; Wolf, W.M. The first insight into the supramolecular system of d,l-alpha-difluoromethylornithine: A new antiviral perspective. Front. Chem. 2021, 9, 679776. [Google Scholar] [CrossRef] [PubMed]
- Borowiecki, P.; Zdun, B.; Dranka, M. Chemoenzymatic enantioselective and stereo-convergent syntheses of lisofylline enantiomers via lipase-catalyzed kinetic resolution and optical inversion approach. Mol. Catal. 2021, 504, 111451. [Google Scholar] [CrossRef]
- Buehler, C.A.; Smith, H.A.; Kryger, A.C.; Wells, R.L.; Thames, S.F. Physiologically active compounds. V. Aminothiol esters of substituted acetic, chloroacetic, benzilic, and related acids. J. Med. Chem. 1963, 6, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Ide, W.S.; Lorz, E.; Phillips, A.P.; Russell, P.B.; Baltzly, R.; Blumfeld, R. Unsymmetrically substituted piperazines. Xii.1 benzhydrylpiperazines and related compounds with spasmolytic and anti-fibrillatory action. J. Org. Chem. 2002, 24, 459–463. [Google Scholar] [CrossRef]
- CRYSALISPRO Software System; Rigaku: Oxford, UK, 2022.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. Olex2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Shelxt—integrated space-group and crystal-structure determination. Acta Cryst. Sec. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with shelxl. Acta Cryst. Sec. C Struct. Chem. 2015, 71, 3–8. [Google Scholar]
- Flack, H.D. On enantiomorph-polarity estimation. Acta Cryst. Sec. A Found. Cryst. 1983, 39, 876–881. [Google Scholar] [CrossRef]
- Parsons, S.; Flack, H. Precise absolute-structure determination in light-atom crystals. Acta Cryst. Sec. A Found. Cryst. 2004, 60, s61. [Google Scholar] [CrossRef] [Green Version]
- Spek, A.L. Structure validation in chemical crystallography. Acta Cryst. Sec. D Biol. Cryst. 2009, 65, 148–155. [Google Scholar] [CrossRef] [Green Version]
- Hooft, R.W.; Straver, L.H.; Spek, A.L. Determination of absolute structure using bayesian statistics on bijvoet differences. J. Appl. Cryst. 2008, 41, 96–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazlauskas, R.J.; Weissfloch, A.N.E.; Rappaport, A.T.; Cuccia, L.A. A rule to predict which enantiomer of a secondary alcohol reacts faster in reactions catalyzed by cholesterol esterase, lipase from pseudomonas cepacia, and lipase from candida rugosa. J. Org. Chem. 2002, 56, 2656–2665. [Google Scholar] [CrossRef]











| Entry | Solvent a (logP) b | t (h) | Conv. (%) c | ees (%) d | eep (%) e | Ef |
|---|---|---|---|---|---|---|
| 1 | 1,4-Dioxane (–0.31) | 96 | 56 | 76 | 60 | 9 |
| 2 | CH3CN (0.17) | 96 | 57 | 93 | 70 | 19 |
| 3 | Acetone (0.20) | 96 | 53 | 87 | 77 | 21 |
| 4 | THF (0.40) | 72 | 54 | 97 | 83 | 45 |
| 5 | Et2O (0.76) | 72 | 57 | 98 | 74 | 30 |
| 6 | MTBE (0.96) | 72 | 57 | 99 | 75 | 35 |
| 7 | tert-Amyl alcohol (1.09) | 96 | 55 | 97 | 79 | 35 |
| 8 | n-Hexane (3.00) | 72 | 55 | 95 | 78 | 29 |

| Entry | Acyl Donor a | Conv. (%) b | ees (%) c | eep (%) d | Ee |
|---|---|---|---|---|---|
| 1 | Vinyl acetate | 55 | >99 | 81 | 49 |
| 2 | Vinyl butanoate | 54 | >99 | 84 | 59 |
| 3 | Vinyl decanoate | 55 | >99 | 80 | 46 |

| Entry | Scale (g) | Lipase Preparation | t (h) | Conv. (%) a | ees (%) b/ Yield c (%) | eep (%) d/ Yield c (%) | Ee |
|---|---|---|---|---|---|---|---|
| 1 | 1.0 f | Chirazyme L-2, C-2 | 16 | 57 | >99/34 | 75/40 | 35 |
| 2 | 1.0 g | Amano PS-Immobead 150 | 144 | 33 | 49/62 | 99/28 | 324 |
| 3 | 10.0 h | Chirazyme L-2, C-2 | 27 | 56 | >99/36 | 79/38 | 44 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borowiecki, P.
Chemoenzymatic Synthesis of Optically Active Ethereal Analog of iso-Moramide—A Novel Potentially Powerful Analgesic
Borowiecki P.
Chemoenzymatic Synthesis of Optically Active Ethereal Analog of iso-Moramide—A Novel Potentially Powerful Analgesic
Borowiecki, Paweł.
2022. "Chemoenzymatic Synthesis of Optically Active Ethereal Analog of iso-Moramide—A Novel Potentially Powerful Analgesic