
A Review of Defatting Strategies for Non-Alcoholic Fatty Liver Disease
 , , and
, , and
Abstract
:1. Introduction
2. In Vitro Defatting Techniques
3. Machine Perfusion Defatting Techniques
3.1. Preclinical Studies
3.2. Clinical Trials
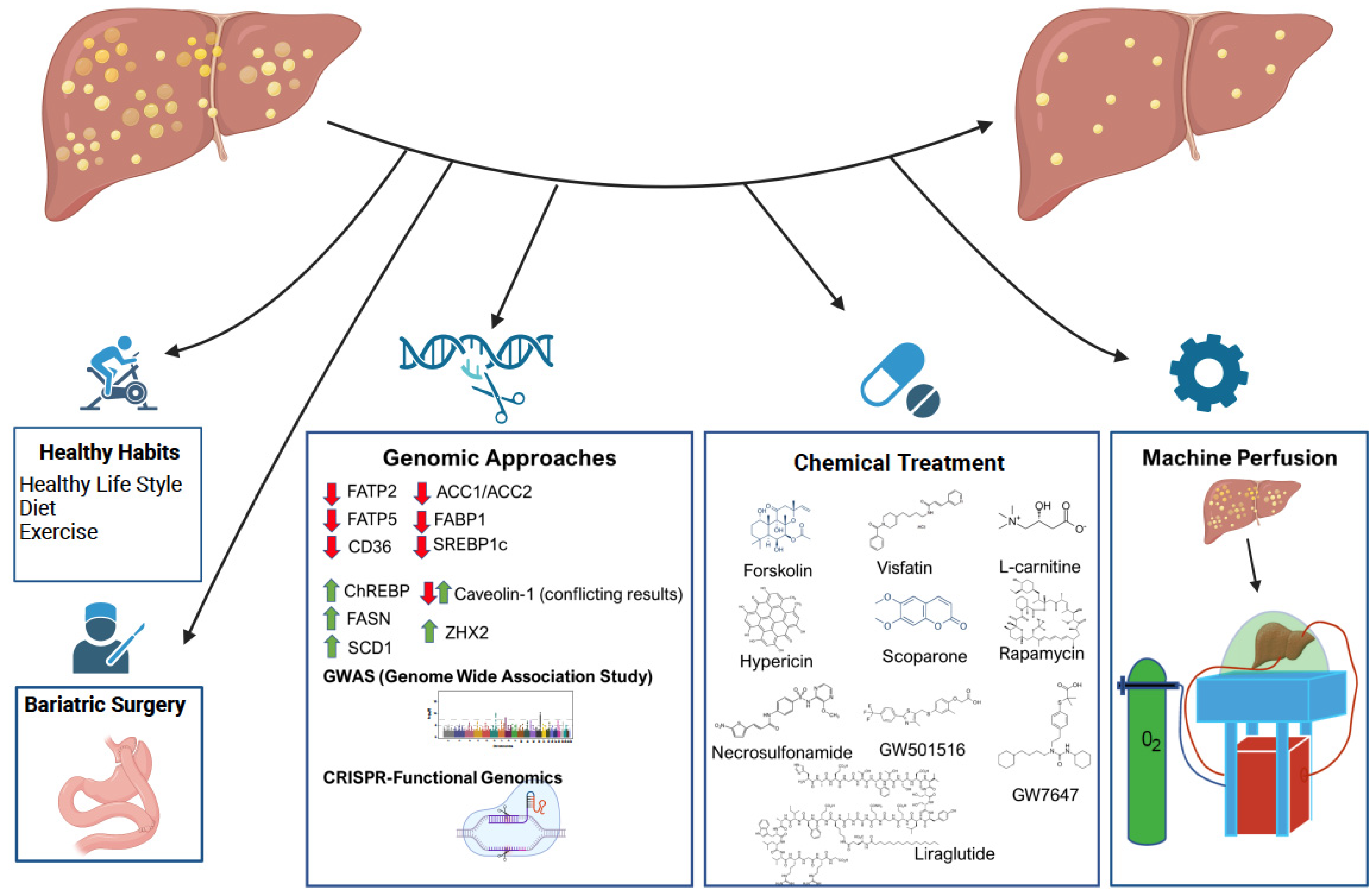
4. Genomic Approaches for Defatting Strategies
5. Additional Interventions for Liver Defatting
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bechmann, L.P.; Hannivoort, R.A.; Gerken, G.; Hotamisligil, G.S.; Trauner, M.; Canbay, A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J. Hepatol. 2012, 56, 952–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, P.; Leray, V.; Diez, M.; Serisier, S.; Le Bloc’H, J.; Siliart, B.; Dumon, H. Liver lipid metabolism. J. Anim. Physiol. Anim. Nutr. 2008, 92, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.V.; Cortez-Pinto, H. Non-alcoholic fatty liver disease: What the clinician needs to know. World J. Gastroenterol. 2014, 20, 12956–12980. [Google Scholar] [CrossRef] [PubMed]
- Maurice, J.; Manousou, P. Non-alcoholic fatty liver disease. Clin. Med. 2018, 18, 245–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, S.; Shimomura, I. Contribution of adipose tissue and de novo lipogenesis to nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1139–1142. [Google Scholar] [CrossRef]
- Goldaracena, N.; Barbas, A.S.; Selzner, M. Normothermic and subnormothermic ex-vivo liver perfusion in liver transplantation. Curr. Opin. Organ Transpl. 2016, 21, 315–321. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Mazilescu, L.I.; Selzner, M.; Selzner, N. Defatting strategies in the current era of liver steatosis. JHEP Rep. 2021, 3, 100265. [Google Scholar] [CrossRef]
- Nativ, N.I.; Yarmush, G.; Chen, A.; Dong, D.; Henry, S.D.; Guarrera, J.V.; Klein, K.M.; Maguire, T.; Schloss, R.; Berthiaume, F.; et al. Rat hepatocyte culture model of macrosteatosis: Effect of macrosteatosis induction and reversal on viability and liver-specific function. J. Hepatol. 2013, 59, 1307–1314. [Google Scholar] [CrossRef]
- Berthiaume, F.; Barbe, L.; Mokuno, Y.; MacDonald, A.D.; Jindal, R.; Yarmush, M.L. Steatosis reversibly increases hepatocyte sensitivity to hypoxia-reoxygenation injury. J. Surg. Res. 2009, 152, 54–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagrath, D.; Xu, H.; Tanimura, Y.; Zuo, R.; Berthiaume, F.; Avila, M.; Yarmush, R.; Yarmush, M.L. Metabolic preconditioning of donor organs: Defatting fatty livers by normothermic perfusion ex vivo. Metab. Eng. 2009, 11, 274–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nativ, N.I.; Yarmush, G.; So, A.; Barminko, J.; Maguire, T.J.; Schloss, R.; Berthiaume, F.; Yarmush, M.L. Elevated sensitivity of macrosteatotic hepatocytes to hypoxia/reoxygenation stress is reversed by a novel defatting protocol. Liver Transpl. 2014, 20, 1000–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarmush, G.; Santos, L.; Yarmush, J.; Koundinyan, S.; Saleem, M.; Nativ, N.I.; Schloss, R.S.; Yarmush, M.L.; Maguire, T.J.; Berthiaume, F. Metabolic Flux Distribution during Defatting of Steatotic Human Hepatoma (HepG2) Cells. Metabolites 2016, 6, 1. [Google Scholar] [CrossRef] [Green Version]
- Boteon, Y.; Wallace, L.; Boteon, A.P.C.S.; Mirza, D.F.; Mergental, H.; Bhogal, R.H.; Afford, S. An effective protocol for pharmacological defatting of primary human hepatocytes which is non-toxic to cholangiocytes or intrahepatic endothelial cells. PLoS ONE 2018, 13, e0201419. [Google Scholar] [CrossRef]
- Aoudjehane, L.; Gautheron, J.; Le Goff, W.; Goumard, C.; Gilaizeau, J.; Nget, C.S.; Savier, E.; Atif, M.; Lesnik, P.; Morichon, R.; et al. Novel defatting strategies reduce lipid accumulation in primary human culture models of liver steatosis. Dis. Model. Mech. 2020, 13, dmm042663. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-W.; Zhang, H.; Li, M.; Xiong, X.; Chen, X.; Chen, X.; Dong, X.C.; Yin, X.-M. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J. Hepatol. 2013, 58, 993–999. [Google Scholar] [CrossRef] [Green Version]
- Waskowicz, L.R.; Zhou, J.; Landau, D.J.; Brooks, E.D.; Lim, A.; A Yavarow, Z.; Kudo, T.; Zhang, H.; Wu, Y.; Grant, S.; et al. Bezafibrate induces autophagy and improves hepatic lipid metabolism in glycogen storage disease type Ia. Hum. Mol. Genet. 2019, 28, 143–154. [Google Scholar] [CrossRef]
- Zhou, W.; Ye, S. Rapamycin improves insulin resistance and hepatic steatosis in type 2 diabetes rats through activation of autophagy. Cell Biol. Int. 2018, 42, 1282–1291. [Google Scholar] [CrossRef]
- Xu, H.; Du, X.; Liu, G.; Huang, S.; Du, W.; Zou, S.; Tang, D.; Fan, C.; Xie, Y.; Wei, Y.; et al. The pseudokinase MLKL regulates hepatic insulin sensitivity independently of inflammation. Mol. Metab. 2019, 23, 14–23. [Google Scholar] [CrossRef]
- Bessems, M.; Doorschodt, B.M.; Kolkert, J.L.; Vetelainen, R.L.; van Vliet, A.K.; Vreeling, H.; van Marle, J.; van Gulik, T.M. Preservation of steatotic livers: A comparison between cold storage and machine perfusion preservation. Liver Transpl. 2007, 13, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Kron, P.; Schlegel, A.; Mancina, L.; Clavien, P.-A.; Dutkowski, P. Hypothermic oxygenated perfusion (HOPE) for fatty liver grafts in rats and humans. J. Hepatol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, R.W.; Zilvetti, M.; Roy, D.; Hughes, D.; Morovat, A.; Coussios, C.C.; Friend, P.J. Hepatic steatosis and normothermic perfusion-preliminary experiments in a porcine model. Transplantation 2011, 92, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Berendsen, T.; Izamis, M.-L.; Uygun, B.; Yarmush, M.; Uygun, K. Perfusion defatting at subnormothermic temperatures in steatotic rat livers. Transpl. Proc. 2013, 45, 3209–3213. [Google Scholar] [CrossRef] [Green Version]
- Vakili, S.T.T.; Kailar, R.; Rahman, K.; Nezami, B.G.; Mwangi, S.M.; Anania, F.A.; Srinivasan, S. Glial cell line-derived neurotrophic factor-induced mice liver defatting: A novel strategy to enable transplantation of steatotic livers. Liver Transpl. 2016, 22, 459–467. [Google Scholar] [CrossRef] [Green Version]
- Mwangi, S.M.; Peng, S.; Nezami, B.G.; Thorn, N.; Farris, A.B.; Jain, S.; Laroui, H.; Merlin, D.; Anania, F.; Srinivasan, S. Glial cell line-derived neurotrophic factor protects against high-fat diet-induced hepatic steatosis by suppressing hepatic PPAR-γ expression. Am. J. Physiol. Liver Physiol. 2016, 310, G103–G116. [Google Scholar] [CrossRef] [Green Version]
- Raigani, S.; Carroll, C.; Griffith, S.; Pendexter, C.; Rosales, I.; Deirawan, H.; Beydoun, R.; Yarmush, M.; Uygun, K.; Yeh, H. Improvement of steatotic rat liver function with a defatting cocktail during ex situ normothermic machine perfusion is not directly related to liver fat content. PLoS ONE 2020, 15, e0232886. [Google Scholar] [CrossRef]
- Guarrera, J.V.; Henry, S.D.; Samstein, B.; Odeh-Ramadan, R.; Kinkhabwala, M.; Goldstein, M.J.; Ratner, L.E.; Renz, J.F.; Lee, H.T.; Brown, J.R.S.; et al. Hypothermic machine preservation in human liver transplantation: The first clinical series. Am. J. Transpl. 2010, 10, 372–381. [Google Scholar] [CrossRef]
- Monbaliu, D.; Liu, Q.; Libbrecht, L.; De Vos, R.; Vekemans, K.; Debbaut, C.; Detry, O.; Roskams, T.; Van Pelt, J.; Pirenne, J. Preserving the morphology and evaluating the quality of liver grafts by hypothermic machine perfusion: A proof-of-concept study using discarded human livers. Liver Transpl. 2012, 18, 1495–1507. [Google Scholar] [CrossRef] [Green Version]
- Abudhaise, H.; Davidson, B.R.; Demuylder, P.; Luong, T.V.; Fuller, B. Evolution of dynamic, biochemical, and morphological parameters in hypothermic machine perfusion of human livers: A proof-of-concept study. PLoS ONE 2018, 13, e0203803. [Google Scholar] [CrossRef]
- van Rijn, R.; Schurink, I.J.; de Vries, Y.; Berg, A.P.V.D.; Cerisuelo, M.C.; Murad, S.D.; Erdmann, J.I.; Gilbo, N.; de Haas, R.J.; Heaton, N.; et al. Hypothermic Machine Perfusion in Liver Transplantation—A Randomized Trial. N. Engl. J. Med. 2021, 384, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Czigany, Z.; Pratschke, J.; Froněk, J.; Guba, M.; Schöning, W.; Raptis, D.A.; Andrassy, J.; Kramer, M.; Strnad, P.; Tolba, R.H.; et al. Hypothermic Oxygenated Machine Perfusion Reduces Early Allograft Injury and Improves Post-transplant Outcomes in Extended Criteria Donation Liver Transplantation from Donation After Brain Death: Results from a Multicenter Randomized Controlled Trial (HOPE ECD-DBD). Ann. Surg. 2021, 274, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Ravaioli, M.; De Pace, V.; Angeletti, A.; Comai, G.; Vasuri, F.; Baldassarre, M.; Maroni, L.; Odaldi, F.; Fallani, G.; Caraceni, P.; et al. Hypothermic Oxygenated New Machine Perfusion System in Liver and Kidney Transplantation of Extended Criteria Donors:First Italian Clinical Trial. Sci Rep. 2020, 10, 6063. [Google Scholar] [CrossRef] [Green Version]
- Watson, C.J.E.; Kosmoliaptsis, V.; Pley, C.; Randle, L.; Fear, C.; Crick, K.; Gimson, A.E.; Allison, M.; Upponi, S.; Brais, R.; et al. Observations on the ex situ perfusion of livers for transplantation. Am. J. Transpl. 2018, 18, 2005–2020. [Google Scholar] [CrossRef] [Green Version]
- Mergental, H.; Laing, R.W.; Kirkham, A.J.; Perera, M.T.P.R.; Boteon, Y.L.; Attard, J.; Barton, D.; Curbishley, S.; Wilkhu, M.; Neil, D.A.H.; et al. Transplantation of discarded livers following viability testing with normothermic machine perfusion. Nat. Commun. 2020, 11, 2939. [Google Scholar] [CrossRef] [PubMed]
- Quintini, C.; Del Prete, L.; Simioni, A.; Del Angel, L.; Uso, T.D.; D’Amico, G.; Hashimoto, K.; Aucejo, F.; Fujiki, M.; Eghtesad, B.; et al. Transplantation of declined livers after normothermic perfusion. Surgery 2022, 171, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Nassar, A.; Buccini, L.; Iuppa, G.; Soliman, B.; Pezzati, D.; Hassan, A.; Blum, M.; Baldwin, W.; Bennett, A.; et al. Lipid metabolism and functional assessment of discarded human livers with steatosis undergoing 24 hours of normothermic machine perfusion. Liver Transpl. 2018, 24, 233–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boteon, Y.L.; Attard, J.; Boteon, A.P.C.S.; Wallace, L.; Reynolds, G.; Hubscher, S.; Mirza, D.F.; Mergental, H.; Bhogal, R.H.; Afford, S.C. Manipulation of Lipid Metabolism During Normothermic Machine Perfusion: Effect of Defatting Therapies on Donor Liver Functional Recovery. Liver Transpl. 2019, 25, 1007–1022. [Google Scholar] [CrossRef] [Green Version]
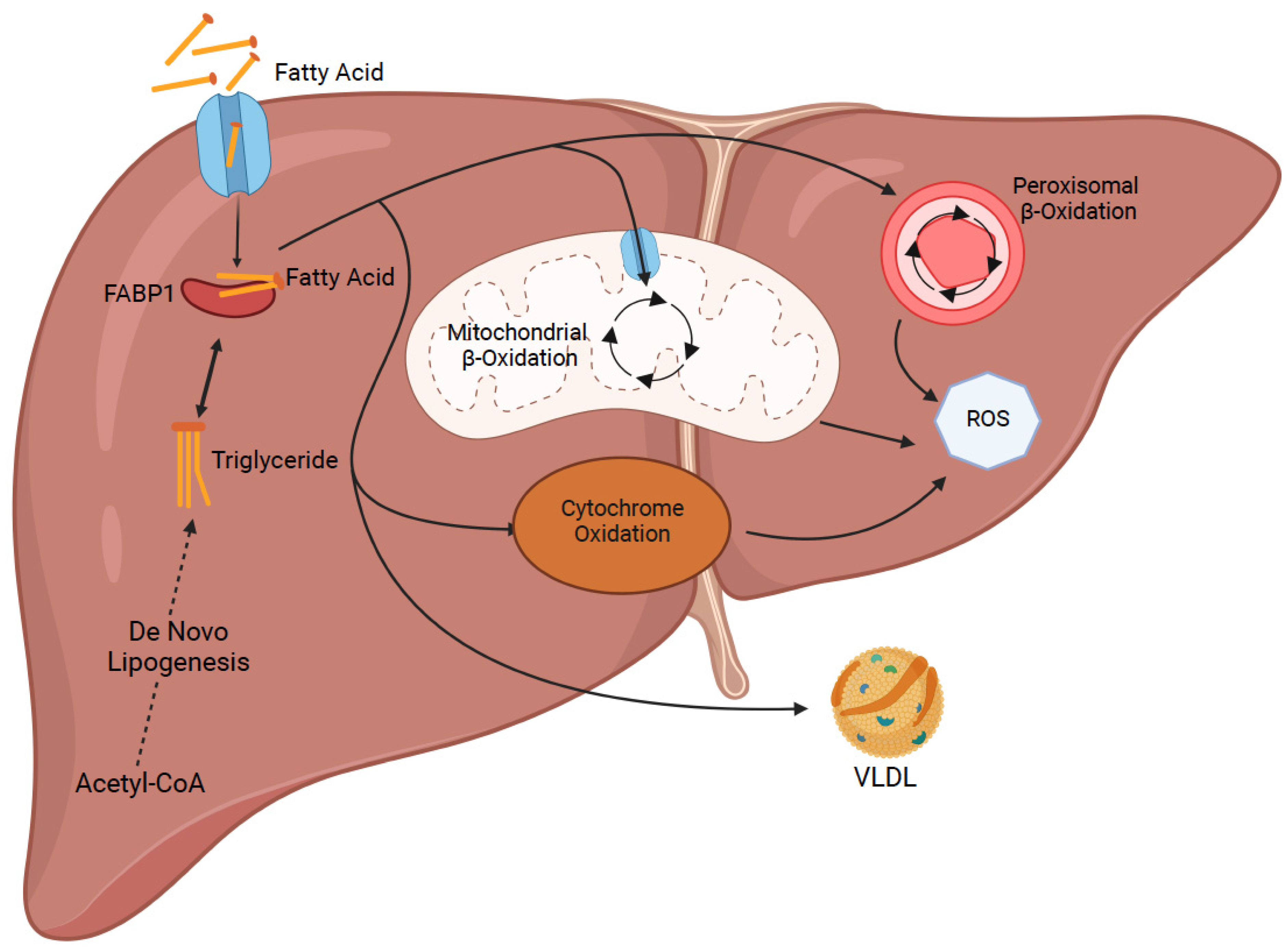
- Mashek, D.G. Hepatic fatty acid trafficking: Multiple forks in the road. Adv. Nutr. 2013, 4, 697–710. [Google Scholar] [CrossRef] [Green Version]
- Koo, S.H. Nonalcoholic fatty liver disease: Molecular mechanisms for the hepatic steatosis. Clin. Mol. Hepatol. 2013, 19, 210–215. [Google Scholar] [CrossRef]
- Falcon, A.; Doege, H.; Fluitt, A.; Tsang, B.; Watson, N.; Kay, M.A.; Stahl, A. FATP2 is a hepatic fatty acid transporter and peroxisomal very long-chain acyl-CoA synthetase. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E384–E393. [Google Scholar] [CrossRef] [Green Version]
- Doege, H.; Baillie, R.A.; Ortegon, A.M.; Tsang, B.; Wu, Q.; Punreddy, S.; Hirsch, D.; Watson, N.; Gimeno, R.E.; Stahl, A. Targeted deletion of FATP5 reveals multiple functions in liver metabolism: Alterations in hepatic lipid homeostasis. Gastroenterology 2006, 130, 1245–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doege, H.; Grimm, D.; Falcon, A.; Tsang, B.; Storm, T.A.; Xu, H.; Ortegon, A.M.; Kazantzis, M.; Kay, M.A.; Stahl, A. Silencing of hepatic fatty acid transporter protein 5 in vivo reverses diet-induced non-alcoholic fatty liver disease and improves hyperglycemia. J. Biol. Chem. 2008, 283, 22186–22192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverstein, R.L.; Febbraio, M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci. Signal. 2009, 2, re3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, C.G.; Tran, J.L.; Erion, D.M.; Vera, N.B.; Febbraio, M.; Weiss, E.J. Hepatocyte-Specific Disruption of CD36 Attenuates Fatty Liver and Improves Insulin Sensitivity in HFD-Fed Mice. Endocrinology 2016, 157, 570–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, Y.; Liu, S.; Chen, H.-T.; Yu, C.-H.; Teng, X.-D.; Yao, H.-T.; Xu, G.-Q. Upregulation of caveolin-1 and SR-B1 in mice with non-alcoholic fatty liver disease. Hepatobiliary Pancreat. Dis. Int. 2013, 12, 630–636. [Google Scholar] [CrossRef]
- Li, M.; Chen, D.; Huang, H.; Wang, J.; Wan, X.; Xu, C.; Li, C.; Ma, H.; Yu, C.; Li, Y. Caveolin1 protects against diet induced hepatic lipid accumulation in mice. PLoS ONE 2017, 12, e0178748. [Google Scholar] [CrossRef] [Green Version]
- Xue, W.; Wang, J.; Jiang, W.; Shi, C.; Wang, X.; Huang, Y.; Hu, C. Caveolin-1 alleviates lipid accumulation in NAFLD associated with promoting autophagy by inhibiting the Akt/mTOR pathway. Eur. J. Pharmacol. 2020, 871, 172910. [Google Scholar] [CrossRef]
- Wang, G.; Bonkovsky, H.L.; de Lemos, A.; Burczynski, F.J. Recent insights into the biological functions of liver fatty acid binding protein 1. J. Lipid Res. 2015, 56, 2238–2247. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, N.; Kato, M.; Tanaka, M.; Miyazaki, M.; Takao, S.; Kohjima, M.; Kotoh, K.; Enjoji, M.; Nakamuta, M.; Takayanagi, R. Effects of insulin resistance and hepatic lipid accumulation on hepatic mRNA expression levels of apoB, MTP and L-FABP in non-alcoholic fatty liver disease. Exp. Ther. Med. 2011, 2, 1077–1081. [Google Scholar] [CrossRef]
- Martin, G.G.; Atshaves, B.P.; Huang, H.; McIntosh, A.L.; Williams, B.J.; Pai, P.-J.; Russell, D.H.; Kier, A.B.; Schroeder, F. Hepatic phenotype of liver fatty acid binding protein gene-ablated mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G1053–G1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukai, T.; Egawa, M.; Takeuchi, T.; Yamashita, H.; Kusudo, T. Silencing of FABP1 ameliorates hepatic steatosis, inflammation, and oxidative stress in mice with nonalcoholic fatty liver disease. FEBS Open Biol. 2017, 7, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Sanders, F.W.; Griffin, J.L. De novo lipogenesis in the liver in health and disease: More than just a shunting yard for glucose. Biol. Rev. Camb. Philos. Soc. 2016, 91, 452–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohjima, M.; Enjoji, M.; Higuchi, N.; Kato, M.; Kotoh, K.; Yoshimoto, T.; Fujino, T.; Yada, M.; Yada, R.; Harada, N.; et al. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int. J. Mol. Med. 2007, 20, 351–358. [Google Scholar] [CrossRef] [Green Version]
- Shimano, H.; Horton, J.D.; Shimomura, I.; Hammer, R.E.; Brown, M.S.; Goldstein, J.L. Isoform 1c of sterol regulatory element binding protein is less active than isoform 1a in livers of transgenic mice and in cultured cells. J. Clin. Investig. 1997, 99, 846–854. [Google Scholar] [CrossRef] [Green Version]
- Liang, G.; Yang, J.; Horton, J.D.; Hammer, R.E.; Goldstein, J.L.; Brown, M.S. Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. J. Biol. Chem. 2002, 277, 9520–9528. [Google Scholar] [CrossRef] [Green Version]
- Iizuka, K.; Bruick, R.K.; Liang, G.; Horton, J.D.; Uyeda, K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc. Natl. Acad. Sci. USA 2004, 101, 7281–7286. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Tong, X.; Vandommelen, K.; Gupta, N.; Stamper, K.; Brady, G.F.; Meng, Z.; Lin, J.; Rui, L.; Omary, B.; et al. Lipogenic transcription factor ChREBP mediates fructose-induced metabolic adaptations to prevent hepatotoxicity. J. Clin. Investig. 2017, 127, 2855–2867. [Google Scholar] [CrossRef] [Green Version]
- Ducheix, S.; Vegliante, M.C.; Villani, G.; Napoli, N.; Sabbà, C.; Moschetta, A. Is hepatic lipogenesis fundamental for NAFLD/NASH? A focus on the nuclear receptor coactivator PGC-1β. Cell Mol. Life Sci. 2016, 73, 3809–3822. [Google Scholar] [CrossRef]
- Benhamed, F.; Denechaud, P.-D.; Lemoine, M.; Robichon, C.; Moldes, M.; Bertrand-Michel, J.; Ratziu, V.; Serfaty, L.; Housset, C.; Capeau, J.; et al. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J. Clin. Investig. 2012, 122, 2176–2194. [Google Scholar] [CrossRef]
- Mao, J.; DeMayo, F.J.; Li, H.; Abu-Elheiga, L.; Gu, Z.; Shaikenov, T.E.; Kordari, P.; Chirala, S.S.; Heird, W.C.; Wakil, S.J. Liver-specific deletion of acetyl-CoA carboxylase 1 reduces hepatic triglyceride accumulation without affecting glucose homeostasis. Proc. Natl. Acad. Sci. USA 2006, 103, 8552–8557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savage, D.B.; Choi, C.S.; Samuel, V.T.; Liu, Z.-X.; Zhang, D.; Wang, A.; Zhang, X.-M.; Cline, G.W.; Yu, X.X.; Geisler, J.G.; et al. Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2. J. Clin. Investig. 2006, 116, 817–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, M.; Yashiro, H.; Ogino, H.; Aoyama, K.; Nambu, T.; Nakamura, S.; Nishida, M.; Wang, X.; Erion, D.M.; Kaneko, M. Acetyl-CoA carboxylase 1 and 2 inhibition ameliorates steatosis and hepatic fibrosis in a MC4R knockout murine model of nonalcoholic steatohepatitis. PLoS ONE 2020, 15, e0228212. [Google Scholar] [CrossRef] [PubMed]
- Tamura, Y.O.; Sugama, J.; Iwasaki, S.; Sasaki, M.; Yasuno, H.; Aoyama, K.; Watanabe, M.; Erion, D.M.; Yashiro, H. Selective Acetyl-CoA Carboxylase 1 Inhibitor Improves Hepatic Steatosis and Hepatic Fibrosis in a Preclinical Nonalcoholic Steatohepatitis Model. J. Pharmacol. Exp. Ther. 2021, 379, 280–289. [Google Scholar] [CrossRef]
- Li, L.; Pilo, G.M.; Li, X.; Cigliano, A.; Latte, G.; Che, L.; Joseph, C.; Mela, M.; Wang, C.; Jiang, L.; et al. Inactivation of fatty acid synthase impairs hepatocarcinogenesis driven by AKT in mice and humans. J. Hepatol. 2016, 64, 333–341. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.Z.; Berk, M.; McIntyre, T.M.; Feldstein, A.E. Hepatic lipid partitioning and liver damage in nonalcoholic fatty liver disease: Role of stearoyl-CoA desaturase. J. Biol. Chem. 2009, 284, 5637–5644. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Gao, L.; Jiang, C.; Chen, J.; Qin, Z.; Zhong, F.; Yan, Y.; Tong, R.; Zhou, M.; Yuan, A.; et al. The transcription factor zinc fingers and homeoboxes 2 alleviates NASH by transcriptional activation of phosphatase and tensin homolog. Hepatology 2022, 75, 939–954. [Google Scholar] [CrossRef]
- Hilgendorf, K.I.; Johnson, C.T.; Han, K.; Rabiee, A.; Demeter, J.; Cheng, R.; Zhu, Y.; Jiang, Z.; Svensson, K.J.; Bassik, M.C.; et al. A CRISPR-based genome-wide screen for adipogenesis reveals new insights into mitotic expansion and lipogenesis. bioRxiv 2020. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Darlay, R.; Cockell, S.; Meroni, M.; Govaere, O.; Tiniakos, D.; Burt, A.D.; Bedossa, P.; Palmer, J.; Liu, Y.-L.; et al. Genome-wide association study of non-alcoholic fatty liver and steatohepatitis in a histologically characterised cohort☆. J. Hepatol. 2020, 73, 505–515. [Google Scholar] [CrossRef]
- Namjou, B.; Lingren, T.; Huang, Y.; Parameswaran, S.; Cobb1, B.L.; Stanaway, I.B.; Connolly, J.J.; Mentch, F.D.; Benoit, B.; Niu, X.; et al. eMERGE Network, Xanthakos SA, Harley JB. GWAS and enrichment analyses of non-alcoholic fatty liver disease identify new trait-associated genes and pathways across eMERGE Network. BMC Med. 2019, 17, 135. [Google Scholar] [CrossRef]
- Mathurin, P.; Gonzalez, F.; Kerdraon, O.; Leteurtre, E.; Arnalsteen, L.; Hollebecque, A.; Louvet, A.; Dharancy, S.; Cocq, P.; Jany, P.; et al. The evolution of severe steatosis after bariatric surgery is related to insulin resistance. Gastroenterology 2006, 130, 1617–1624. [Google Scholar] [CrossRef] [PubMed]
- Lassailly, G.; Caiazzo, R.; Buob, D.; Pigeyre, M.; Verkindt, H.; Labreuche, J.; Raverdy, V.; Leteurtre, E.; Dharancy, S.; Louvet, A.; et al. Bariatric surgery reduces features of nonalcoholic steatohepatitis in morbidly obese patients. Gastroenterology 2015, 149, 379–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanyal, A.J.; Brunt, E.M.; Kleiner, D.E.; Kowdley, K.V.; Chalasani, N.; Lavine, J.E.; Ratziu, V.; McCullough, A. Endpoints and clinical trial design for nonalcoholic steatohepatitis. Hepatology 2011, 54, 344–353. [Google Scholar] [CrossRef] [Green Version]
- Lassailly, G.; Caiazzo, R.; Ntandja-Wandji, L.-C.; Gnemmi, V.; Baud, G.; Verkindt, H.; Ningarhari, M.; Louvet, A.; Leteurtre, E.; Raverdy, V.; et al. Bariatric Surgery Provides Long-term Resolution of Nonalcoholic Steatohepatitis and Regression of Fibrosis. Gastroenterology 2020, 159, 1290–1301. [Google Scholar] [CrossRef] [PubMed]
- Gastaldelli, A.; Cusi, K. From NASH to diabetes and from diabetes to NASH: Mechanisms and treatment options. JHEP Rep. 2019, 1, 312–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, M.J. BASL and the Dame Sheila Sherlock Award 2016. Glucagon-like peptide-1 analogues in nonalcoholic steatohepatitis: From bench to bedside. Clin. Liver Dis. 2017, 10, 32–35. [Google Scholar] [CrossRef] [Green Version]
- Sanyal, A.J. Past, present and future perspectives in nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 377–386. [Google Scholar] [CrossRef]
- Drucker, D.J. The biology of incretin hormones. Cell Metab. 2006, 3, 153–165. [Google Scholar] [CrossRef] [Green Version]
- Cusi, K. Incretin-Based Therapies for the Management of Nonalcoholic Fatty Liver Disease in Patients with Type 2 Diabetes. Hepatology 2019, 69, 2318–2322. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; Abouda, G.; A Aldersley, M.; et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef]
- Ozempic (Semaglutide) Prescribing Information. January 2020. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/209637s003lbl.pdf (accessed on 24 August 2022).
- Kushner, R.F.; Calanna, S.; Davies, M.; Dicker, D.; Garvey, W.T.; Goldman, B.; Lingvay, I.; Thomsen, M.; Wadden, T.A.; Wharton, S.; et al. Semaglutide 2.4 mg for the Treatment of Obesity: Key Elements of the STEP Trials 1 to 5. Obesity 2020, 28, 1050–1061. [Google Scholar] [CrossRef] [PubMed]
- Capehorn, M.; Catarig, A.-M.; Furberg, J.; Janez, A.; Price, H.; Tadayon, S.; Vergès, B.; Marre, M. Efficacy and safety of once-weekly semaglutide 1.0mg vs once-daily liraglutide 1.2mg as add-on to 1-3 oral antidiabetic drugs in subjects with type 2 diabetes (SUSTAIN 10). Diabetes Metab. 2020, 46, 100–109. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, P.M.; Birkenfeld, A.L.; McGowan, B.; Mosenzon, O.; Pedersen, S.D.; Wharton, S.; Carson, C.G.; Jepsen, C.H.; Kabisch, M.; Wilding, J.P.H. Efficacy and safety of semaglutide compared with liraglutide and placebo for weight loss in patients with obesity: A randomised, double-blind, placebo and active controlled, dose-ranging, phase 2 trial. Lancet 2018, 392, 637–649. [Google Scholar] [CrossRef]
- Nauck, M.A.; Meier, J.J. Management of endocrine Disease: Are all GLP-1 agonists equal in the treatment of type 2 diabetes? Eur. J. Endocrinol. 2019, 181, R211–R234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newsome, P.; Francque, S.; Harrison, S.; Ratziu, V.; Van Gaal, L.; Calanna, S.; Hansen, M.; Linder, M.; Sanyal, A. Effect of semaglutide on liver enzymes and markers of inflammation in subjects with type 2 diabetes and/or obesity. Aliment. Pharmacol. Ther. 2019, 50, 193–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newsome, P.N.; Buchholtz, K.; Cusi, K.; Linder, M.; Okanoue, T.; Ratziu, V.; Sanyal, A.J.; Sejling, A.-S.; Harrison, S.A. A Placebo-Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2021, 384, 1113–1124. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| First Author | Year | In Vitro Model | Defatting Strategy | Effects of Agents |
|---|---|---|---|---|
| Mao et al. [10] | 2013 | Rat hepatocytes | Forskolin, PPARα and δ agonist, scoparone, hypericin, visfatin, amino acids | Faster steatosis reduction; recovery of urea secretion and bile canalicular formation |
| Nativ et al. [13] | 2014 | Rat hepatocytes | Forskolin, PPARα and δ agonist, scoparone, hypericin, visfatin, amino acids ± L-carnitine ± 90% O2 | Higher reduction in TGs, increase in β-oxidation and ATP levels with L-carnitine and hyperoxia |
| Yarmush et al. [14] | 2016 | Human hepatoma cells | Forskolin, PPARα and δ agonist, scoparone, hypericin, visfatin, amino acids ± 90% O2 | Decreased TGs, increased β-oxidation, TCA cycle and urea cycle, especially with hyperoxia |
| Boteon et al. [15] | 2018 | PHH, HIEC, human cholangiocytes | Forskolin, PPARα and δ agonist, scoparone, hypericin, visfatin, L-carnitine | PHH-decrease in lipids and TGs; increased viability of PHH and cholangiocytes; no cytotoxic effects on HIEC |
| Aoudjehane et al. [16] | 2020 | PHH, PHH from human fatty liver, human PCLS | Forskolin, L-carnitine, PPAR α and δ agonist, rapamycin, necrosulfonamide | Decrease in lipids and TGs and endoplasmic reticulum stress and production of reactive oxygen species |
| First Author | Year | Temperature of Perfusion | Additional Agents | Effects of Perfusion |
|---|---|---|---|---|
| Bessems et al. [21] | 2007 | Hypothermic | None | Less cell damage; increased bile production, ammonia clearance, urea production, O2 consumption, and ATP levels |
| Kron et al. [22] | 2017 | HOPE | None | HOPE: less oxidative stress, nuclear injury, macrophage activation and fibrosis; no decrease in steatosis |
| HNPE | HNPE: loss of protective effects seen with HOPE therapy | |||
| Jamieson et al. [23] | 2011 | Normothermic | None | 13% reduction in steatosis |
| Nagrath et al. [12] | 2009 | Normothermic | PPARα and δ ligands, hypericin, scoparone, forskolin and visfatin | 65% reduction in TG content |
| Liu et al. [24] | 2013 | Subnormothermic | PPARα and δ ligands, hypericin, scoparone, forskolin and visfatin | No significant reduction in steatosis |
| Vakili et al. [25] | 2016 | Normothermic | GDNF or PPARα and δ ligands, hypericin, scoparone, forskolin and visfatin | GDNF: equally effective as defatting agents at lowering TGs, and caused less liver damage (rise in LDH activity) |
| Raigani et al. [27] | 2020 | Normothermic | PPARα and δ ligands, hypericin, scoparone, forskolin, visfatin, L-carnitine and amino acids | Decreased perfusate lactate, better bile quality, and decreased inflammatory markers; increased β-oxidation markers; no significant reduction in steatosis |
| First Author | Year | Temperature of Perfusion | Additional Agents | Effects of Perfusion |
|---|---|---|---|---|
| Guarrera et al. [28] | 2010 | Hypothermic | None | May improve graft function |
| Monbaliu et al. [29] | 2012 | Hypothermic | None | Discarded livers had higher levels of injury markers in the perfusate |
| Abudhaise et al. [30] | 2018 | |||
| van Rijn et al. [31] | 2021 | HOPE | None | Less occurrence of biliary strictures, post reperfusion syndrome, and allograft dysfunction |
| Czigany et al. [32] | 2021 | HOPE | None | Lower levels of liver injury enzymes, graft dysfunction, 90-day complications, and hospital stay |
| Ravaioli et al. [33] | 2020 | |||
| Watson et al. [34] | 2018 | Normothermic | None | Discarded livers were suitable for transplantation after perfusion |
| Mergental et al. [35] | 2020 | |||
| Quintini et al. [36] | 2022 | |||
| Liu et al. [37] | 2018 | Normothermic | None | Increased perfusate TG levels during treatment; no significant decrease in steatosis histologically |
| Boteon et al. [38] | 2019 | Normothermic | PPARα and δ ligands, hypericin, scoparone, forskolin and visfatin | Reduction in TGs and macrosteatosis, increased β-oxidation, higher ATP levels, and enhanced viability |
| Gene | First Author | Year | Type of Modification | Effect of Genomic Modification |
|---|---|---|---|---|
| FATP2 | Falcon et al. [41] | 2010 | Knockdown | Decreased lipid uptake and lower liver TGs |
| FATP5 | Doege et al. [42] | 2006 | Knockout | Decreased lipid uptake, lower TGs, and reverses steatosis |
| Doege et al. [43] | 2008 | |||
| CD36 | Wilson et al. [45] | 2016 | Knockout | Decreased lipid uptake, lower TGs, improved insulin sensitivity and reduced inflammatory markers |
| Caveolin-1 | Li et al. [47] | 2017 | Knockdown | Increased steatosis, plasma cholesterol, and liver injury enzymes |
| Li et al. [47] | 2017 | Overexpression | Decreased lipid accumulation | |
| FABP1 | Martin et al. [51] | 2009 | Knockout | Lower TGs and decreased lipid disposal pathways |
| Mukai et al. [52] | 2017 | Knockout | Decreased expression of inflammatory markers | |
| SREBP1c | Shimano et al. [55] | 1997 | Overexpression | Higher TG levels |
| Liang et al. [56] | 2002 | Knockout | Decreased ACC and FASN mRNA levels (needed for DNL) | |
| ChREBP | Lizuka et al. [57] | 2004 | Knockout | Lower TGs, but higher insulin resistance, delayed glucose clearance and simple sugar intolerance |
| Zhang et al. [58] | 2017 | Knockout | Protection against steatosis but enhanced hepatic damage | |
| Benhamed et al. [60] | 2012 | Overexpression | Produced steatosis but maintained insulin sensitivity and glucose tolerance | |
| ACC1/ACC2 | Mao et al. [61] | 2006 | Knockout | Less production of malonyl-CoA, less TG accumulation; increased synthesis of lipogenic enzymes |
| Savage et al. [62] | 2006 | Knockdown | Reversed steatosis, reduced malonyl-CoA, improved insulin sensitivity, and increased β-oxidation | |
| Matsumoto et al. [63] | 2020 | Small molecule inhibitors (ACC1/2) | Lower TGs, reduced fibrosis and lowered liver injury markers; higher plasma TGs | |
| Tamura et al. [64] | 2021 | Small molecule inhibitors (ACC1) | Reduction in steatosis and fibrosis; no change in plasma TGs | |
| FASN | Li et al. [65] | 2016 | Knockout | Hypoglycemia, liver steatosis, and decreased β-oxidation; decline in cell proliferation and rise in apoptosis |
| SCD1 | Li et al. [66] | 2009 | Knockout | Increased fibrosis and cellular apoptosis |
| ZHX2 | Zhao et al. [67] | 2022 | Overexpression | Stopped progression of steatosis and reduced liver inflammation |
| Knockout | Increased lipid accumulation, increased fibrosis and enhanced liver inflammation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Young, E.N.; Dogan, M.; Watkins, C.; Bajwa, A.; Eason, J.D.; Kuscu, C.; Kuscu, C. A Review of Defatting Strategies for Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 11805. https://doi.org/10.3390/ijms231911805
Young EN, Dogan M, Watkins C, Bajwa A, Eason JD, Kuscu C, Kuscu C. A Review of Defatting Strategies for Non-Alcoholic Fatty Liver Disease. International Journal of Molecular Sciences. 2022; 23(19):11805. https://doi.org/10.3390/ijms231911805
Chicago/Turabian StyleYoung, Erin Nicole, Murat Dogan, Christine Watkins, Amandeep Bajwa, James D. Eason, Canan Kuscu, and Cem Kuscu. 2022. "A Review of Defatting Strategies for Non-Alcoholic Fatty Liver Disease" International Journal of Molecular Sciences 23, no. 19: 11805. https://doi.org/10.3390/ijms231911805