
Identification of Genes and Metabolic Pathways Involved in Resin Yield in Masson Pine by Integrative Analysis of Transcriptome, Proteome and Biochemical Characteristics
 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Differences in Photosynthetic Pigment Contents between High and Common Resin Yield
2.2. Differences in Sugar Contents between High and Common Resin Yield
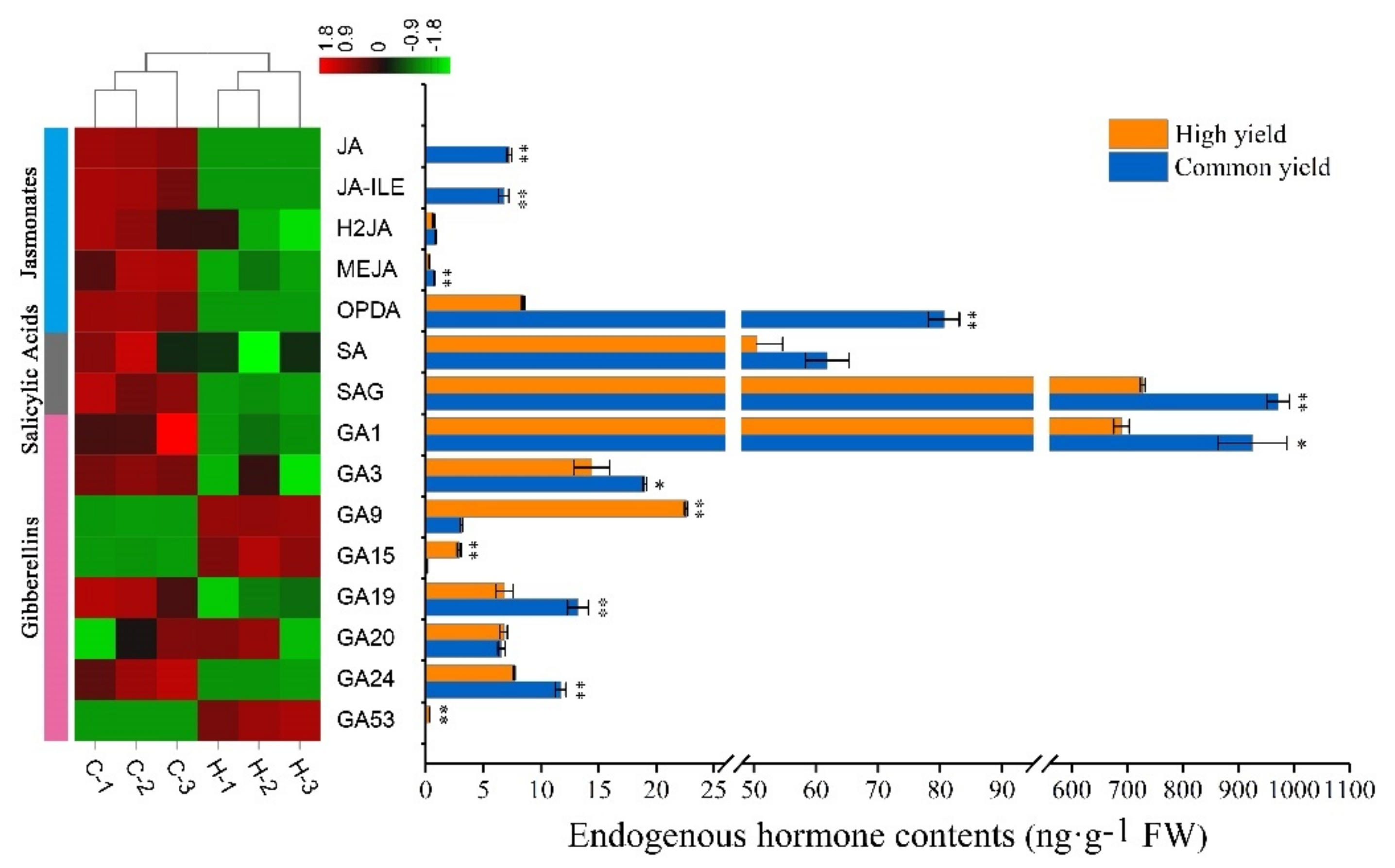
2.3. Differences in Endogenous Hormone Levels between High and Common Resin Yield
2.4. Full-Length Transcriptome Profiling and DEGs Identification
2.5. Proteome Profiling and DEPs Identification
2.6. Correlation of mRNA and Protein Profiles
2.7. Integrated Analysis of Transcriptome and Proteome
2.8. Identification of Key Genes at the Transcriptomic, Proteomic and Biochemical Levels
3. Discussion
3.1. Identification of DEGs and DEPs
3.2. Roles of Terpenoid Biosynthesis Genes in Resin Yield
3.3. Roles of Photosynthesis Genes in Resin Yield
3.4. Roles of Hormone Biosynthesis Genes in Resin Yield
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. Detection of Photosynthetic Pigment Contents
4.3. Detection of Sugar Contents
4.4. Detection of Hormone Contents
4.5. PacBio Iso-Seq Library Construction, Sequencing, and Gene Functional Annotation
4.6. RNA-Seq Library Construction, Sequencing, and Analysis of Differentially Expressed Genes
4.7. Protein Extraction, iTRAQ Labeling, and LC-MS/MS Analysis
4.8. Protein Identification and Functional Annotation
4.9. Quantitative Real-Time PCR Assay
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Franceschi, V.R.; Krokene, P.; Christiansen, E.; Krekling, T. Anatomical and Chemical Defenses of Conifer Bark against Bark Beetles and Other Pests. New Phytol. 2005, 167, 353–376. [Google Scholar] [CrossRef] [PubMed]
- Keeling, C.I.; Bohlmann, J. Genes, Enzymes and Chemicals of Terpenoid Diversity in the Constitutive and Induced Defence of Conifers against Insects and Pathogens. New Phytol. 2006, 170, 657–675. [Google Scholar] [CrossRef] [PubMed]
- Celedon, J.M.; Bohlmann, J. Oleoresin Defenses in Conifers: Chemical Diversity, Terpene Synthases and Limitations of Oleoresin Defense under Climate Change. New Phytol. 2019, 224, 1444–1463. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.A.; Croteau, R.B. Resin-Based Defenses in Conifers. Trends Plant Sci. 1999, 4, 184–190. [Google Scholar] [CrossRef]
- Keeling, C.I.; Bohlmann, J. Diterpene Resin Acids in Conifers. Phytochemistry 2006, 67, 2415–2423. [Google Scholar] [CrossRef]
- Zulak, K.G.; Bohlmann, J. Terpenoid Biosynthesis and Specialized Vascular Cells of Conifer Defense. J. Integr. Plant Biol. 2010, 52, 86–97. [Google Scholar] [CrossRef]
- Neis, F.A.; de Costa, F.; de Almeida, M.R.; Colling, L.C.; de Oliveira Junkes, C.F.; Fett, J.P.; Fett-Neto, A.G. Resin Exudation Profile, Chemical Composition, and Secretory Canal Characterization in Contrasting Yield Phenotypes of Pinus elliottii Engelm. Ind. Crops Prod. 2019, 132, 76–83. [Google Scholar] [CrossRef]
- Da Silva Rodrigues-Corrêa, K.C.; de Lima, J.C.; Fett-Neto, A.G. Pine Oleoresin: Tapping Green Chemicals, Biofuels, Food Protection, and Carbon Sequestration from Multipurpose Trees. Food Energy Secur. 2012, 1, 81–93. [Google Scholar] [CrossRef]
- Bannan, M.W. Vertical Resin Ducts in the Secondary Wood of the Abietineae. New Phytol. 1936, 35, 11–46. [Google Scholar] [CrossRef]
- Wu, H.; Hu, Z.H. Comparative Anatomy of Resin Ducts of the Pinaceae. Trees 1997, 11, 135–143. [Google Scholar] [CrossRef]
- Tadesse, W.; Nanos, N.; Auñon, F.J.; Alía, R.; Gil, L. Evaluation of High Resin Yielders of Pinus pinaster Ait. For. Genet. 2002, 8, 271–278. [Google Scholar]
- Roberds, J.H.; Strom, B.L.; Hain, F.P.; Gwaze, D.P.; McKeand, S.E.; Lott, L.H. Estimates of Genetic Parameters for Oleoresin and Growth Traits in Juvenile Loblolly Pine. Can. J. For. Res. 2003, 33, 2469–2476. [Google Scholar] [CrossRef]
- Westbrook, J.W.; Resende, M.F.R.; Munoz, P.; Walker, A.R.; Wegrzyn, J.L.; Nelson, C.D.; Neale, D.B.; Kirst, M.; Huber, D.A.; Gezan, S.A.; et al. Association Genetics of Oleoresin Flow in Loblolly Pine: Discovering Genes and Predicting Phenotype for Improved Resistance to Bark Beetles and Bioenergy Potential. New Phytol. 2013, 199, 89–100. [Google Scholar] [CrossRef]
- Liu, Q.; Zhou, Z.; Fan, H.; Liu, Y. Genetic Variation and Correlation among Resin Yield, Growth, and Morphologic Traits of Pinus massoniana. Silvae Genet. 2013, 62, 38–44. [Google Scholar] [CrossRef]
- Yang, Z. Genetic Improvement of Key Wood and Resin Properties of Pinus massoniana Lamb; Beijing Forestry University: Beijing, China, 2012; pp. 67–84. (In Chinese) [Google Scholar]
- Wei, Y.; Liu, Q.; Zhou, Z.; Feng, Z. Difference of Photosynthetic Characteristics in Pinus massoniana Clones with Different Resin Yield. J. Northeast. For. Univ. 2015, 43, 36–41. (In Chinese) [Google Scholar]
- Rodríguez-García, A.; López, R.; Martín, J.A.; Pinillos, F.; Gil, L. Resin Yield in Pinus pinaster Is Related to Tree Dendrometry, Stand Density and Tapping-Induced Systemic Changes in Xylem Anatomy. For. Ecol. Manag. 2014, 313, 47–54. [Google Scholar] [CrossRef]
- Yi, M.; Jia, T.; Dong, L.; Zhang, L.; Leng, C.; Liu, S.; Lai, M. Resin Yield in Pinus elliottii Engelm. Is Related to the Resin Flow Rate, Resin Components and Resin Duct Characteristics at Three Locations in Southern China. Ind. Crops Prod. 2021, 160, 113141. [Google Scholar] [CrossRef]
- Karanikas, C.; Walker, V.; Scaltsoyiannes, A.; Comte, G.; Bertrand, C. High vs. Low Yielding Oleoresin Pinus halepensis Mill. Trees GC Terpenoids Profiling as Diagnostic Tool. Ann. For. Sci. 2010, 67, 412. [Google Scholar] [CrossRef]
- Henry, L.K.; Thomas, S.T.; Widhalm, J.R.; Lynch, J.H.; Davis, T.C.; Kessler, S.A.; Bohlmann, J.; Noel, J.P.; Dudareva, N. Contribution of Isopentenyl Phosphate to Plant Terpenoid Metabolism. Nat. Plants 2018, 4, 721–729. [Google Scholar] [CrossRef]
- Pazouki, L.; Niinemetst, Ü. Multi-Substrate Terpene Synthases: Their Occurrence and Physiological Significance. Front. Plant Sci. 2016, 7, 1019. [Google Scholar] [CrossRef]
- Ro, D.K.; Arimura, G.I.; Lau, S.Y.W.; Piers, E.; Bohlmann, J. Loblolly Pine Abietadienol/Abietadienal Oxidase PtAO (CYP720B1) Is a Multifunctional, Multisubstrate Cytochrome P450 Monooxygenase. Proc. Natl. Acad. Sci. USA 2005, 102, 8060–8065. [Google Scholar] [CrossRef]
- Sharon, D.; Tilgner, H.; Grubert, F.; Snyder, M. A Single-Molecule Long-Read Survey of the Human Transcriptome. Nat. Biotechnol. 2013, 31, 1009–1014. [Google Scholar] [CrossRef] [Green Version]
- Minoche, A.E.; Dohm, J.C.; Schneider, J.; Holtgräwe, D.; Viehöver, P.; Montfort, M.; Sörensen, R.T.; Weisshaar, B.; Himmelbauer, H. Exploiting Single-Molecule Transcript Sequencing for Eukaryotic Gene Prediction. Genom. Biol. 2015, 16, 184. [Google Scholar] [CrossRef]
- Koren, S.; Schatz, M.C.; Walenz, B.P.; Martin, J.; Howard, J.T.; Ganapathy, G.; Wang, Z.; Rasko, D.A.; McCombie, W.R.; Jarvis, E.D.; et al. Hybrid Error Correction and de novo Assembly of Single-Molecule Sequencing Reads. Nat. Biotechnol. 2012, 30, 693–700. [Google Scholar] [CrossRef]
- Hackl, T.; Hedrich, R.; Schultz, J.; Förster, F. Proovread: Large-Scale High-Accuracy PacBio Correction through Iterative Short Read Consensus. Bioinformatics 2014, 30, 3004–3011. [Google Scholar] [CrossRef]
- Li, Q.; Li, Y.; Song, J.; Xu, H.; Xu, J.; Zhu, Y.; Li, X.; Gao, H.; Dong, L.; Qian, J.; et al. High-Accuracy de novo Assembly and SNP Detection of Chloroplast Genomes Using a SMRT Circular Consensus Sequencing Strategy. New Phytol. 2014, 204, 1041–1049. [Google Scholar] [CrossRef]
- Hao, J.; Guo, H.; Shi, X.; Wang, Y.; Wan, Q.; Song, Y.B.; Zhang, L.; Dong, M.; Shen, C. Comparative Proteomic Analyses of Two Taxus Species (Taxus × media and Taxus mairei) Reveals Variations in the Metabolisms Associated with Paclitaxel and Other Metabolites. Plant Cell Physiol. 2017, 58, 1878–1890. [Google Scholar] [CrossRef]
- Ross, P.L.; Huang, Y.N.; Marchese, J.N.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S.; et al. Multiplexed Protein Quantitation in Saccharomyces Cerevisiae Using Amine–Reactive Isobaric Tagging Reagents. Mol. Cell. Proteom. 2004, 3, 1154–1169. [Google Scholar] [CrossRef]
- Kambiranda, D.; Katam, R.; Basha, S.M.; Siebert, S. iTRAQ–Based Quantitative Proteomics of Developing and Ripening Muscadine Grape Berry. J. Proteom. Res. 2014, 13, 555–569. [Google Scholar] [CrossRef]
- Yahya, G.; Parisi, E.; Flores, A.; Gallego, C.; Aldea, M. A Whi7-Anchored Loop Controls the G1 Cdk-Cyclin Complex at Start. Mol. Cell 2014, 53, 115–126. [Google Scholar] [CrossRef]
- Georgieva, M.V.; Yahya, G.; Codó, L.; Ortiz, R.; Teixidó, L.; Claros, J.; Jara, R.; Jara, M.; Iborra, A.; Gelpí, J.L.; et al. Inntags: Small Self-Structured Epitopes for Innocuous Protein Tagging. Nat. Methods 2015, 12, 955–958. [Google Scholar] [CrossRef] [PubMed]
- Miyagi, M.; Rao, K.C.S. Proteolytic 18O-Labeling Strategies for Quantitative Proteomics. Mass Spectrom. Rev. 2007, 26, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Tian, Y.; Tan, J.; Jin, D. Identification of Ovarian Tissue Proteins in the Diapause Period of Aspongopus chinensis Dallas by Using LC MS/MS. J. Mt. Agric. Biol. 2021, 40, 1–5. (In Chinese) [Google Scholar]
- Zeng, L.H.; Zhang, Q.; He, B.X.; Lian, H.M.; Cai, Y.L.; Wang, Y.S.; Luo, M. Age Trends in Genetic Parameters for Growth and Resin-Yielding Capacity in Masson Pine. Silvae Genet. 2013, 62, 7–18. [Google Scholar] [CrossRef]
- Liu, Q.; Zhou, Z.; Wei, Y.; Shen, D.; Feng, Z.; Hong, S. Genome-Wide Identification of Differentially Expressed Genes Associated with the High Yielding of Oleoresin in Secondary Xylem of Masson Pine (Pinus massoniana Lamb) by Transcriptomic Analysis. PLoS ONE 2015, 10, e0132624. [Google Scholar] [CrossRef] [PubMed]
- Bai, Q.; He, B.; Cai, Y.; Lian, H.; Zhang, Q. Transcriptomic and Metabolomic Analyses Reveal Several Critical Metabolic Pathways and Candidate Genes Involved in Resin Biosynthesis in Pinus massoniana. Mol. Genet. Genom. 2020, 295, 327–341. [Google Scholar] [CrossRef]
- Mei, L.; Li, Z.; Yan, Y.; Wen, Z.; Wen, X.; Yang, Z.; Feng, Y. Identification and Functional Study of Oleoresin Terpenoid Biosynthesis-Related Genes in Masson Pine (Pinus massoniana L.) Based on Transcriptome Analysis. Tree Genet. Genom. 2020, 16, 53. [Google Scholar] [CrossRef]
- Liu, Q.; Xie, Y.; Liu, B.; Yin, H.; Zhou, Z.; Feng, Z.; Chen, Y. A Transcriptomic Variation Map Provides Insights into the Genetic Basis of Pinus massoniana Lamb. Evolution and the Association with Oleoresin Yield. BMC Plant Biol. 2020, 20, 375. [Google Scholar] [CrossRef]
- Mei, L.; Yan, Y.; Li, Z.; Ran, J.; Shen, L.; Wu, R.; Hou, Q.; Shen, T.; Wen, X.; Yang, Z.; et al. Identification of the Diterpenoid Biosynthesis Genes and Their Expression Status in Relation to Oleoresin Yield of Masson Pine. Ind. Crops Prod. 2021, 170, 113827. [Google Scholar] [CrossRef]
- Zhang, C.; Wei, Y.; Xiao, D.; Gao, L.; Lyu, S.; Hou, X.; Bouuema, G. Transcriptomic and Proteomic Analyses Provide New Insights into the Regulation Mechanism of Low-Temperature-Induced Leafy Head Formation in Chinese Cabbage. J. Proteom. 2016, 144, 1–10. [Google Scholar] [CrossRef]
- Ding, Z.; Fu, L.; Tie, W.; Yan, Y.; Wu, C.; Hu, W.; Zhang, J. Extensive Post-Transcriptional Regulation Revealed by Transcriptomic and Proteomic Integrative Analysis in Cassava under Drought. J. Agric. Food Chem. 2019, 67, 3521–3534. [Google Scholar] [CrossRef]
- Yahya, G.; Menges, P.; Ngandiri, D.A.; Schulz, D.; Wallek, A.; Kulak, N.; Mann, M.; Cramer, P.; Savage, V.; Raeschle, M.; et al. Scaling of Cellular Proteome with Ploidy. bioRxiv 2021. [Google Scholar] [CrossRef]
- Tupý, J. Stimulatory Effects of 2,4-Dichlorophenoxyacetic Acid and of 1-Naph-Thylacetic Acid on Sucrose Level, Invertase Activity and Sucrose Utilization in the Latex of Hevea brasiliensis. Planta 1969, 88, 144–153. [Google Scholar] [CrossRef]
- Zheng, Y.; Hong, R. The Rhythmic Changes in the Resin-Producing Capacity, Growth Increment, Sugar Content (in Needles) and Starch Content (in Needles) of Pinus massoniana. Fujian For. Sci. Technol. 1995, 22, 12–16. (In Chinese) [Google Scholar]
- Vranová, E.; Coman, D.; Gruissem, W. Network Analysis of the MVA and MEP Pathways for Isoprenoid Synthesis. Annu. Rev. Plant Biol. 2013, 64, 665–700. [Google Scholar] [CrossRef]
- Bohlmann, J.; Meyer-Gauen, G.; Croteau, R. Plant Terpenoid Synthases: Molecular Biology and Phylogenetic Analysis. Proc. Natl. Acad. Sci. USA 1998, 95, 4126–4133. [Google Scholar] [CrossRef]
- Davis, E.M.; Croteau, R. Cyclization Enzymes in the Biosynthesis of Monoterpenes, Sesquiterpenes, and Diterpenes. Top. Curr. Chem. 2000, 209, 53–95. [Google Scholar] [CrossRef]
- Hamberger, B.; Ohnishi, T.; Hamberger, B.; Séguin, A.; Bohlmann, J. Evolution of Diterpene Metabolism: Sitka Spruce CYP720B4 Catalyzes Multiple Oxidations in Resin Acid Biosynthesis of Conifer Defense against Insects. Plant Physiol. 2011, 157, 1677–1695. [Google Scholar] [CrossRef]
- Warren, R.L.; Keeling, C.I.; Yuen, M.M.S.; Raymond, A.; Taylor, G.A.; Vandervalk, B.P.; Mohamadi, H.; Paulino, D.; Chiu, R.; Jackman, S.D.; et al. Improved White Spruce (Picea glauca) Genome Assemblies and Annotation of Large Gene Families of Conifer Terpenoid and Phenolic Defense Metabolism. Plant J. 2015, 83, 189–212. [Google Scholar] [CrossRef]
- Geisler, K.; Jensen, N.B.; Yuen, M.M.S.; Madilao, L.; Bohlmann, J. Modularity of Conifer Diterpene Resin Acid Biosynthesis: P450 Enzymes of Different CYP720B Clades Use Alternative Substrates and Converge on the Same Products. Plant Physiol. 2016, 171, 152–164. [Google Scholar] [CrossRef]
- Liu, B.; Xie, Y.; Yin, H.; Zhou, Z.; Liu, Q. Identification and Defensive Characterization of PmCYP720B11v2 from Pinus massoniana. Int. J. Mol. Sci. 2022, 23, 6640. [Google Scholar] [CrossRef]
- Alicandri, E.; Covino, S.; Sebastiani, B.; Paolacci, A.R.; Badiani, M.; Manti, F.; Bonsignore, C.P.; Sorgonà, A.; Ciaffi, M. Diterpene Resin Acids and Olefins in Calabrian Pine (Pinus nigra subsp. laricio (Poiret) Maire) Oleoresin: GC-MS Profiling of Major Diterpenoids in Different Plant Organs, Molecular Identification and Expression Analysis of Diterpene Synthase Genes. Plants 2021, 10, 2391. [Google Scholar] [CrossRef]
- Zhang, Y.; Ding, X.; Luan, Q.; Jiang, J.; Diao, S. Identification and Tissue-Specific Expression Analysis of CYP720B Subfamily Genes in Slash Pine and Loblolly Pine. Forests 2022, 13, 283. [Google Scholar] [CrossRef]
- Feng, Y.; Yan, P.; Yang, Z.; Huang, Y.; Meng, L. Study on Evaluation Parameters of Rosin Production Capacity of Pinus massoniana. Guangxi For. Sci. 2021, 50, 440–446. (In Chinese) [Google Scholar]
- Kovács, L.; Damkjaer, J.; Kereïche, S.; Ilioaia, C.; Ruban, A.V.; Boekema, E.J.; Jansson, S.; Horton, P. Lack of the Light-Harvesting Complex CP24 Affects the Structure and Function of the Grana Membranes of Higher Plant Chloroplasts. Plant Cell 2006, 18, 3106–3120. [Google Scholar] [CrossRef] [Green Version]
- Chu, P.; Yan, G.X.; Yang, Q.; Zhai, L.N.; Zhang, C.; Zhang, F.Q.; Guan, R.Z. iTRAQ-Based Quantitative Proteomics Analysis of Brassica napus Leaves Reveals Pathways Associated with Chlorophyll Deficiency. J. Proteom. 2015, 113, 244–259. [Google Scholar] [CrossRef]
- Shi, L.; Hall, M.; Funk, C.; Schröder, W.P. Photosystem II, a Growing Complex: Updates on Newly Discovered Components and Low Molecular Mass Proteins. Biochim. Biophys. Acta J. 2012, 1817, 13–25. [Google Scholar] [CrossRef]
- Dong, L.; Tu, W.; Liu, K.; Sun, R.; Liu, C.; Wang, K.; Yang, C. The PsbS Protein Plays Important Roles in Photosystem II Supercomplex Remodeling under Elevated Light Conditions. J. Plant Physiol. 2015, 172, 33–41. [Google Scholar] [CrossRef]
- Hahn, A.; Vonck, J.; Mills, D.J.; Meier, T.; Kühlbrandt, W. Structure, Mechanism, and Regulation of the Chloroplast ATP Synthase. Science 2018, 360, eaat4318. [Google Scholar] [CrossRef]
- Palacio, S.; Maestro, M.; Montserrat-Martí, G. Relationship between Shoot-Rooting and Root-Sprouting Abilities and the Carbohydrate and Nitrogen Reserves of Mediterranean Dwarf Shrubs. Ann. Bot. 2007, 100, 865–874. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Bai, M.Y.; Oh, E.; Zhu, J.Y. Brassinosteroid Signaling Network and Regulation of Photomorphogenesis. Annu. Rev. Genet. 2012, 46, 701–724. [Google Scholar] [CrossRef] [PubMed]
- Shalygo, N.; Czarnecki, O.; Peter, E.; Grimm, B. Expression of Chlorophyll Synthase Is Also Involved in Feedback-Control of Chlorophyll Biosynthesis. Plant Mol. Biol. 2009, 71, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Cai, B.; Liang, Y. A Study on the Relationships between Chlorophyll Content and Resin Productivity of Masson Pine. J. Fujian Coll. For. 1998, 18, 58–61. (In Chinese) [Google Scholar]
- Tanaka, A.; Ito, H.; Tanaka, R.; Tanaka, N.; Yoshida, K.; Okada, K. Chlorophyll a Oxygenase (CAO) Is Involved in Chlorophyll b Formation from Chlorophyll a. Proc. Natl. Acad. Sci. USA 1998, 95, 12719–12723. [Google Scholar] [CrossRef]
- Valladares, F.; Niinemets, Ü. Shade Tolerance, a Key Plant Feature of Complex Nature and Consequences. Annu. Rev. Ecol. Evol. Syst. 2008, 39, 237–257. [Google Scholar] [CrossRef]
- Niyogi, K.K.; Truong, T.B. Evolution of Flexible Non-Photochemical Quenching Mechanisms that Regulate Light Harvesting in Oxygenic Photosynthesis. Curr. Opin. Plant Biol. 2013, 16, 307–314. [Google Scholar] [CrossRef]
- Dall’Osto, L.; Cazzaniga, S.; Havaux, M.; Bassi, R. Enhanced Photoprotection by Protein-Bound vs Free Xanthophyll Pools: A Comparative Analysis of Chlorophyll b and Xanthophyll Biosynthesis Mutants. Mol. Plant 2010, 3, 576–593. [Google Scholar] [CrossRef]
- Rüdiger, W.; Benz, J.; Guthoff, C. Detection and Partial Characterization of Activity of Chlorophyll Synthetase in Etioplast Membranes. Eur. J. Biochem. 1980, 109, 193–200. [Google Scholar] [CrossRef]
- Wu, Z.; Zhang, X.; He, B.; Diao, L.; Sheng, S.; Wang, J.; Guo, X.; Su, N.; Wang, L.; Jiang, L.; et al. A Chlorophyll-Deficient Rice Mutant with Impaired Chlorophyllide Esterification in Chlorophyll Biosynthesis. Plant Physiol. 2007, 145, 29–40. [Google Scholar] [CrossRef]
- Iriti, M.; Faoro, F. Chemical Diversity and Defence Metabolism: How Plants Cope with Pathogens and Ozone Pollution. Int. J. Mol. Sci. 2009, 10, 3371–3399. [Google Scholar] [CrossRef]
- Biondi, S.; Antognoni, F.; Perellino, N.C.; Sacchetti, G.; Minghetti, A.; Poli, F. Medium Composition and Methyl Jasmonate Influence the Amount and Spectrum of Secondary Metabolites in Callus Cultures. Plant Biosyst. 2004, 138, 117–124. [Google Scholar] [CrossRef]
- Zhai, Q.; Zhang, X.; Wu, F.; Feng, H.; Deng, L.; Xu, L.; Zhang, M.; Wang, Q.; Li, C. Transcriptional Mechanism of Jasmonate Receptor COI1-Mediated Delay of Flowering Time in Arabidopsis. Plant Cell 2015, 27, 2814–2828. [Google Scholar] [CrossRef]
- Takaoka, Y.; Iwahashi, M.; Chini, A.; Saito, H.; Ishimaru, Y.; Egoshi, S.; Kato, N.; Tanaka, M.; Bashir, K.; Seki, M.; et al. A Rationally Designed JAZ Subtype-Selective Agonist of Jasmonate Perception. Nat. Commun. 2018, 9, 3654. [Google Scholar] [CrossRef]
- Chen, J.H.; Tian, Y.R.; Li, A.J.; Xia, X.L.; Yin, W.L. Jasmonic Acid Signaling and Its Research Progress in Woody Plants. Sci. Sin. Vitae 2020, 50, 215–226. (In Chinese) [Google Scholar] [CrossRef]
- Chini, A.; Fonseca, S.; Fernández, G.; Adie, B.; Chico, J.M.; Lorenzo, O.; García-Casado, G.; López-Vidriero, I.; Lozano, F.M.; Ponce, M.R.; et al. The JAZ Family of Repressors Is the Missing Link in Jasmonate Signalling. Nature 2007, 448, 666–671. [Google Scholar] [CrossRef]
- Katsir, L.; Schilmiller, A.L.; Staswick, P.E.; He, S.Y.; Howe, G.A. COI1 Is a Critical Component of a Receptor for Jasmonate and the Bacterial Virulence Factor Coronatine. Proc. Natl. Acad. Sci. USA 2008, 105, 7100–7105. [Google Scholar] [CrossRef] [Green Version]
- Banyai, W.; Mii, M.; Supaibulwatana, K. Enhancement of Artemisinin Content and Biomass in Artemisia annua by Exogenous GA3 Treatment. Plant Growth Regul. 2011, 63, 45–54. [Google Scholar] [CrossRef]
- Lv, Z.; Guo, Z.; Zhang, L.; Zhang, F.; Jiang, W.; Shen, Q.; Fu, X.; Yan, T.; Shi, P.; Hao, X.; et al. Interaction of bZIP Transcription Factor TGA6 with Salicylic Acid Signaling Modulates Artemisinin Biosynthesis in Artemisia annua. J. Exp. Bot. 2019, 70, 3969–3979. [Google Scholar] [CrossRef]
- Hedden, P.; Thomas, S.G. Gibberellin Biosynthesis and Its Regulation. Biochem. J. 2012, 444, 11–25. [Google Scholar] [CrossRef]
- Dere, Ş.; Güneş, T.; Sivaci, R. Spectrophotometric Determination of Chlorophyll-A, B and Total Carotenoid Contents of Some Algae Species Using Different Solvents. Turk. J. Bot. 1998, 22, 13–17. [Google Scholar]
- Wang, H.; Wu, F.; Li, M.; Zhu, X.; Shi, C.; Shao, C.; Ding, G. Structure and Chlorophyll Fluorescence of Heteroblastic Foliage Affect First-Year Growth in Pinus massoniana Lamb. Seedlings. Plant Physiol. Biochem. 2022, 170, 206–217. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate Transcript Quantification from RNA-Seq Data with or without a Reference Genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential Expression Analysis for Sequence Count Data. Genom. Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PacBio | ||
|---|---|---|
| Before Correct | After Correct | |
| Subreads base (G) | 23.92 | |
| Subreads number | 15,216,963 | |
| Average subreads length | 1572 | |
| N50 (subreads) | 1893 | |
| CCS_number | 520,718 | |
| Full length | 446,770 | |
| FLNC | 416,262 | |
| Average FLNC read length | 1719 | |
| Consensus reads | 38,784 | |
| Total_nucleotides | 72,040,170 | 72,013,662 |
| Total_number | 38,784 | 38,784 |
| Mean_length | 1858 | 1857 |
| Min_length | 75 | 75 |
| Max_length | 9057 | 9060 |
| N50 (consensus) | 2143 | 2142 |
| N90 (consensus) | 1139 | 1138 |
| Number of transcripts | 38,784 | |
| Number of Genes | 17,266 | |
| Number of genes annotated | 98.30% | |
| Number of genes unannotated | 1.70% | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Shen, L.; Hou, Q.; Zhou, Z.; Mei, L.; Zhao, H.; Wen, X. Identification of Genes and Metabolic Pathways Involved in Resin Yield in Masson Pine by Integrative Analysis of Transcriptome, Proteome and Biochemical Characteristics. Int. J. Mol. Sci. 2022, 23, 11420. https://doi.org/10.3390/ijms231911420
Li Z, Shen L, Hou Q, Zhou Z, Mei L, Zhao H, Wen X. Identification of Genes and Metabolic Pathways Involved in Resin Yield in Masson Pine by Integrative Analysis of Transcriptome, Proteome and Biochemical Characteristics. International Journal of Molecular Sciences. 2022; 23(19):11420. https://doi.org/10.3390/ijms231911420
Chicago/Turabian StyleLi, Zhengchun, Luonan Shen, Qiandong Hou, Zijing Zhou, Lina Mei, Hong Zhao, and Xiaopeng Wen. 2022. "Identification of Genes and Metabolic Pathways Involved in Resin Yield in Masson Pine by Integrative Analysis of Transcriptome, Proteome and Biochemical Characteristics" International Journal of Molecular Sciences 23, no. 19: 11420. https://doi.org/10.3390/ijms231911420