
A Combination of a Genome-Wide Association Study and a Transcriptome Analysis Reveals circRNAs as New Regulators Involved in the Response to Salt Stress in Maize

 , ,
, ,
Abstract
:1. Introduction
2. Results
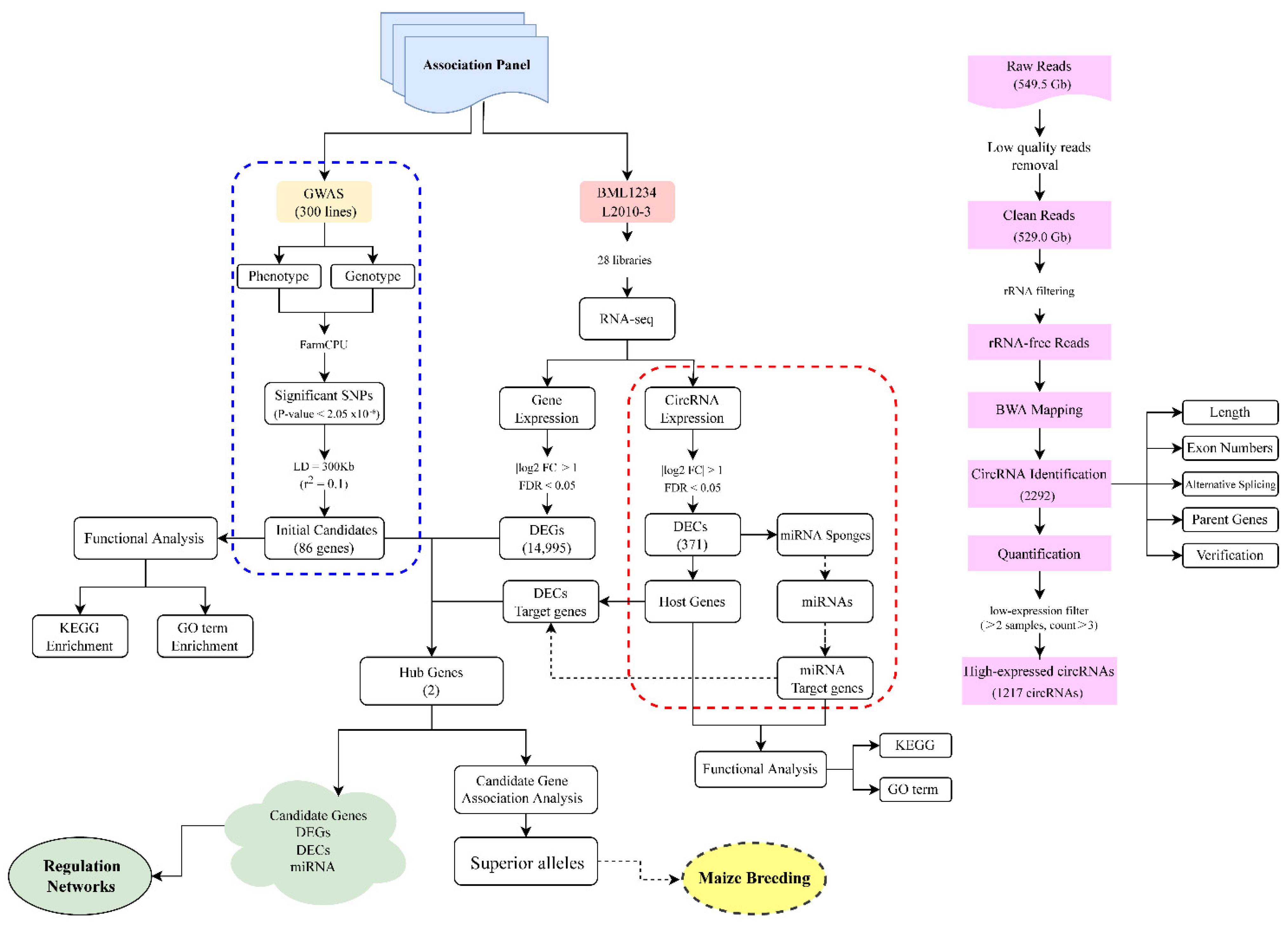
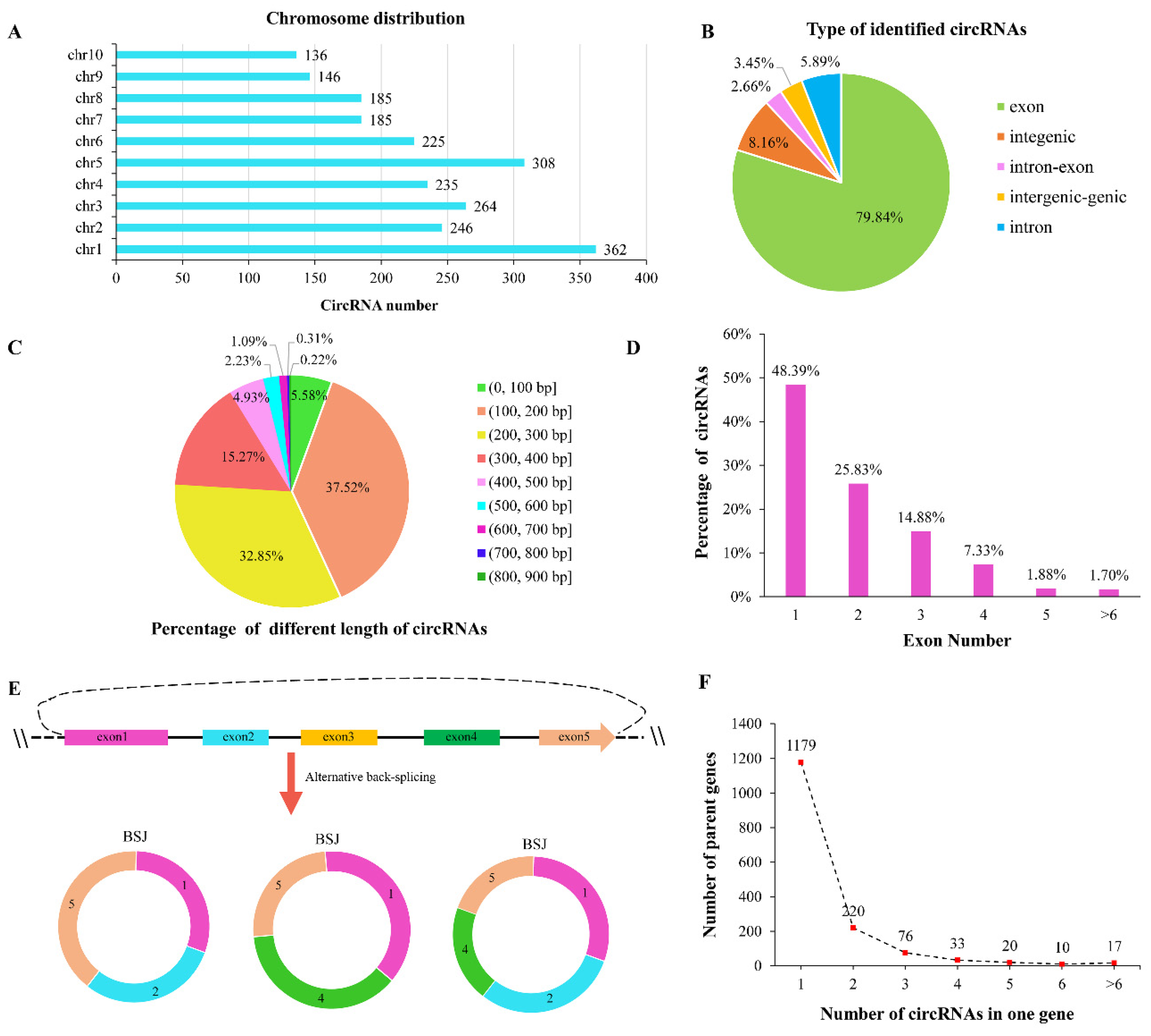
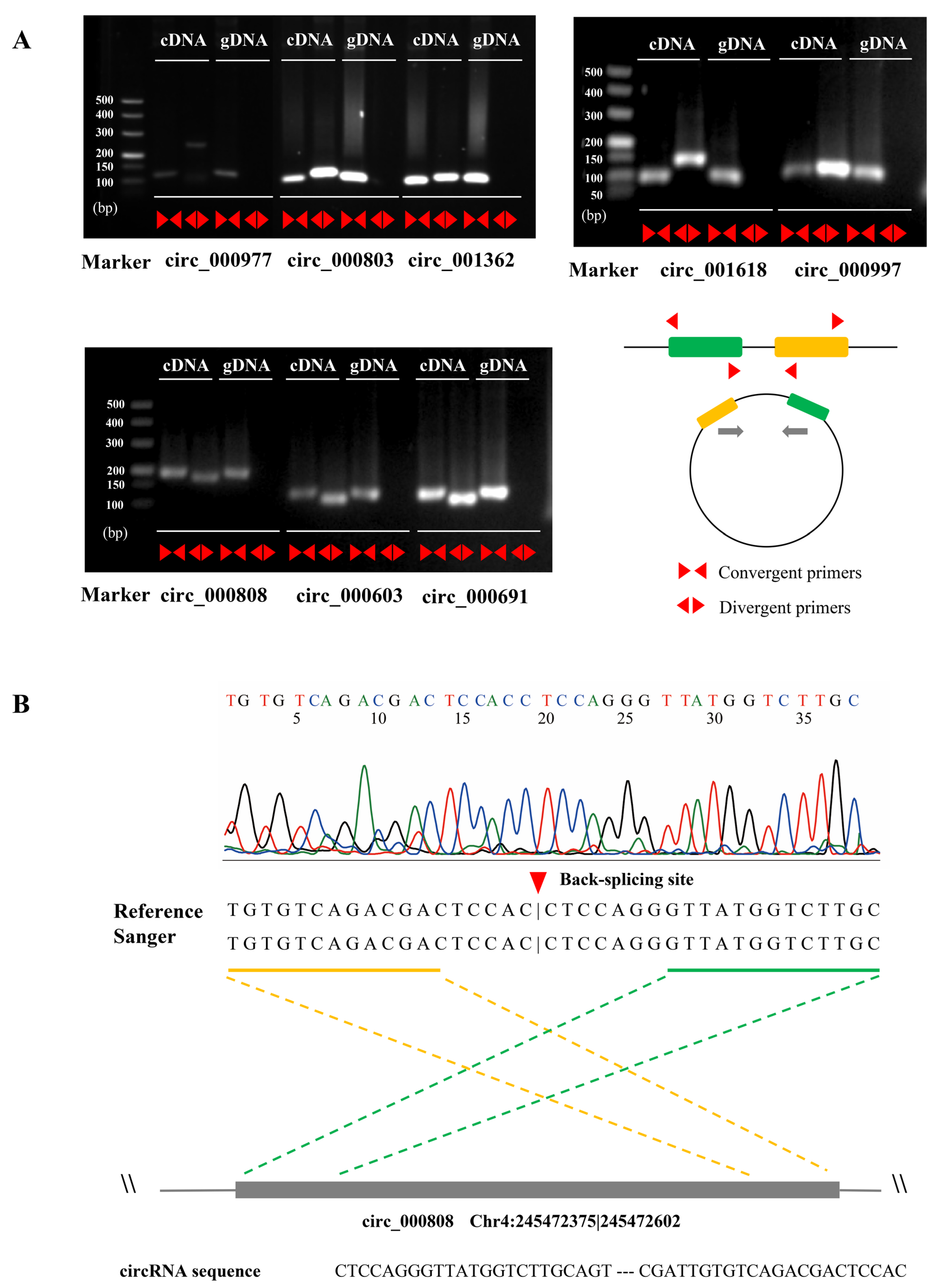
2.1. Identification, Characterization, and Validation of circRNAs in Maize Seedlings
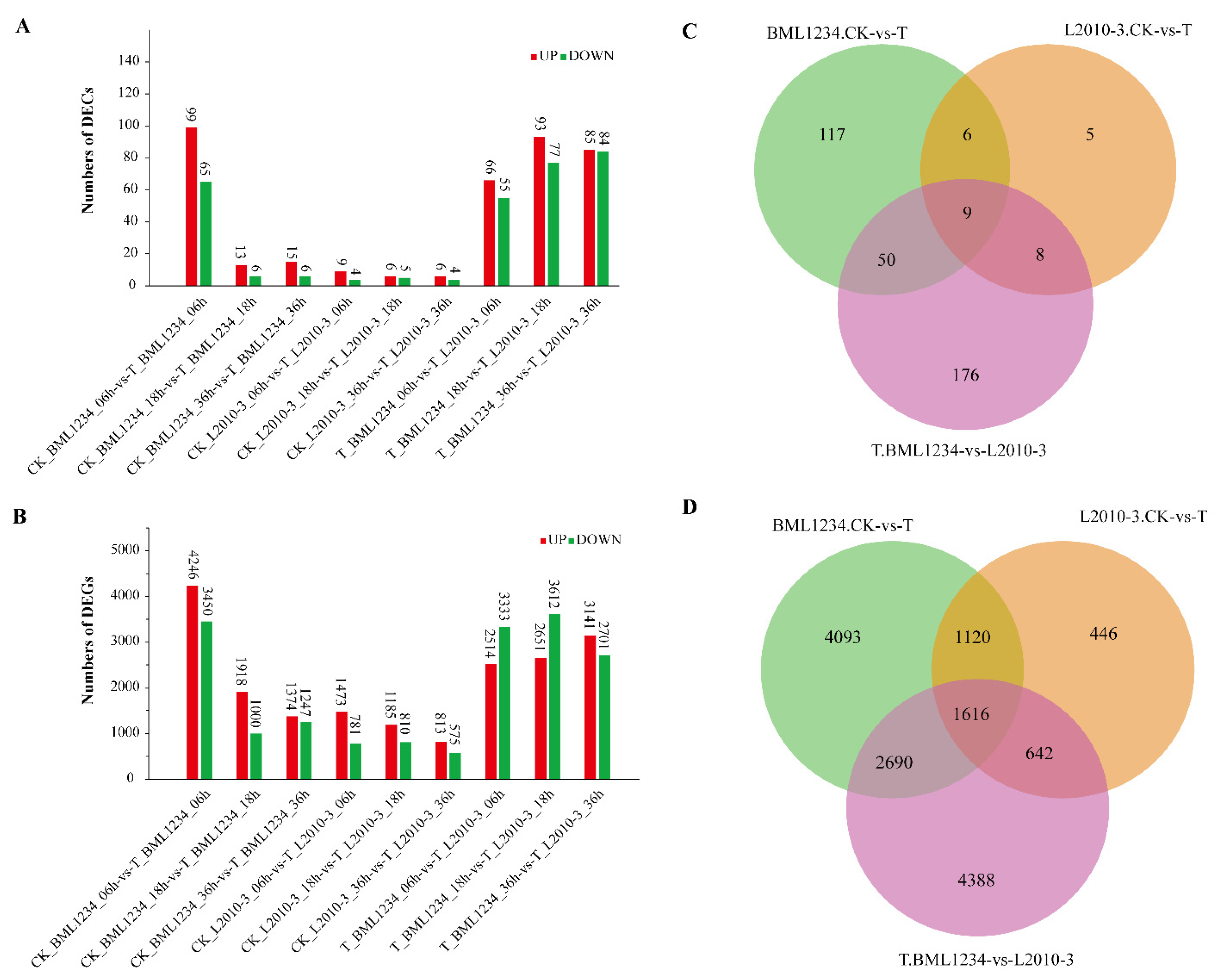
2.2. Differentially Expressed circRNAs and Genes Induced by Salt Stress
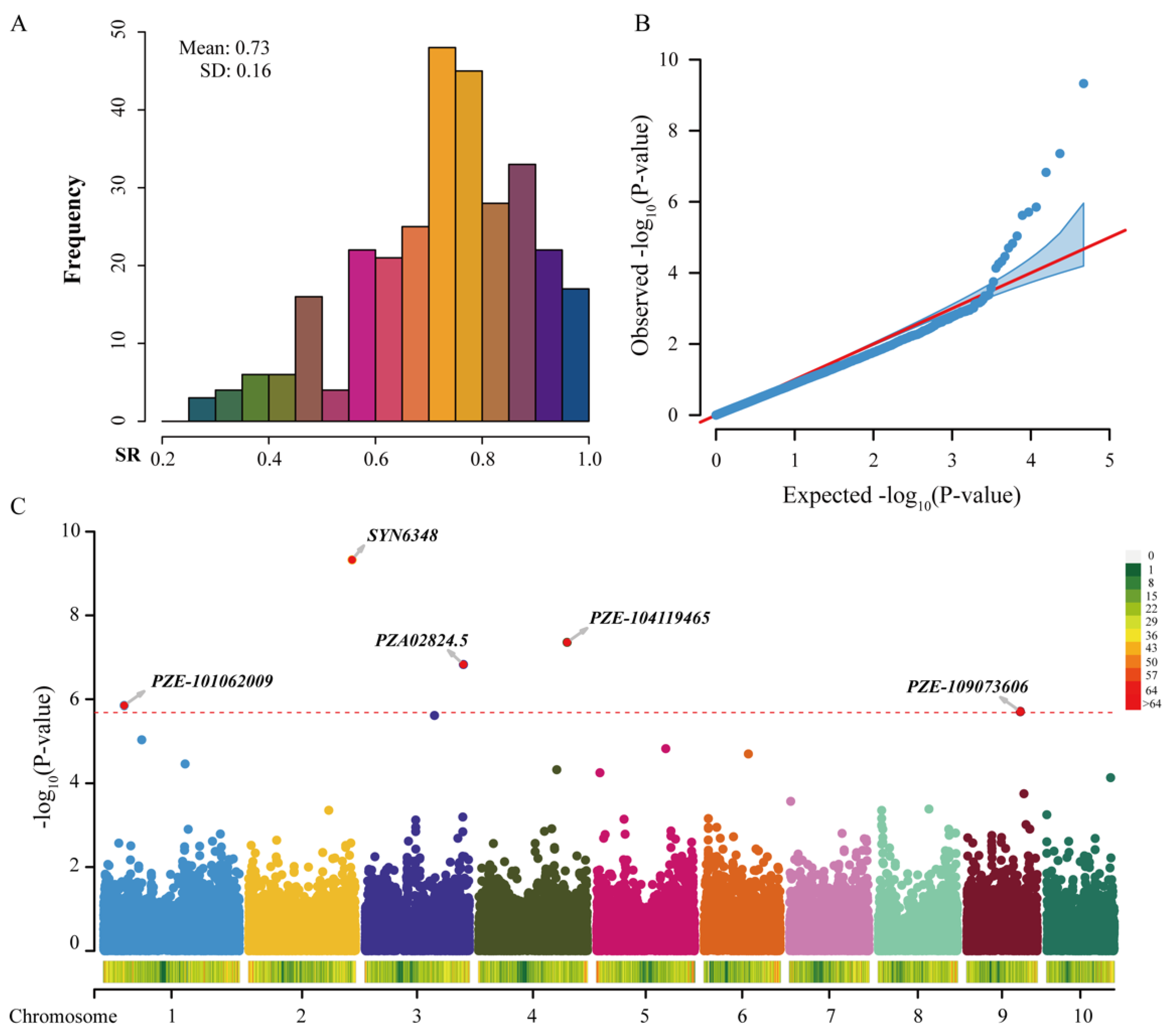
2.3. GWAS of Maize Salt Tolerance
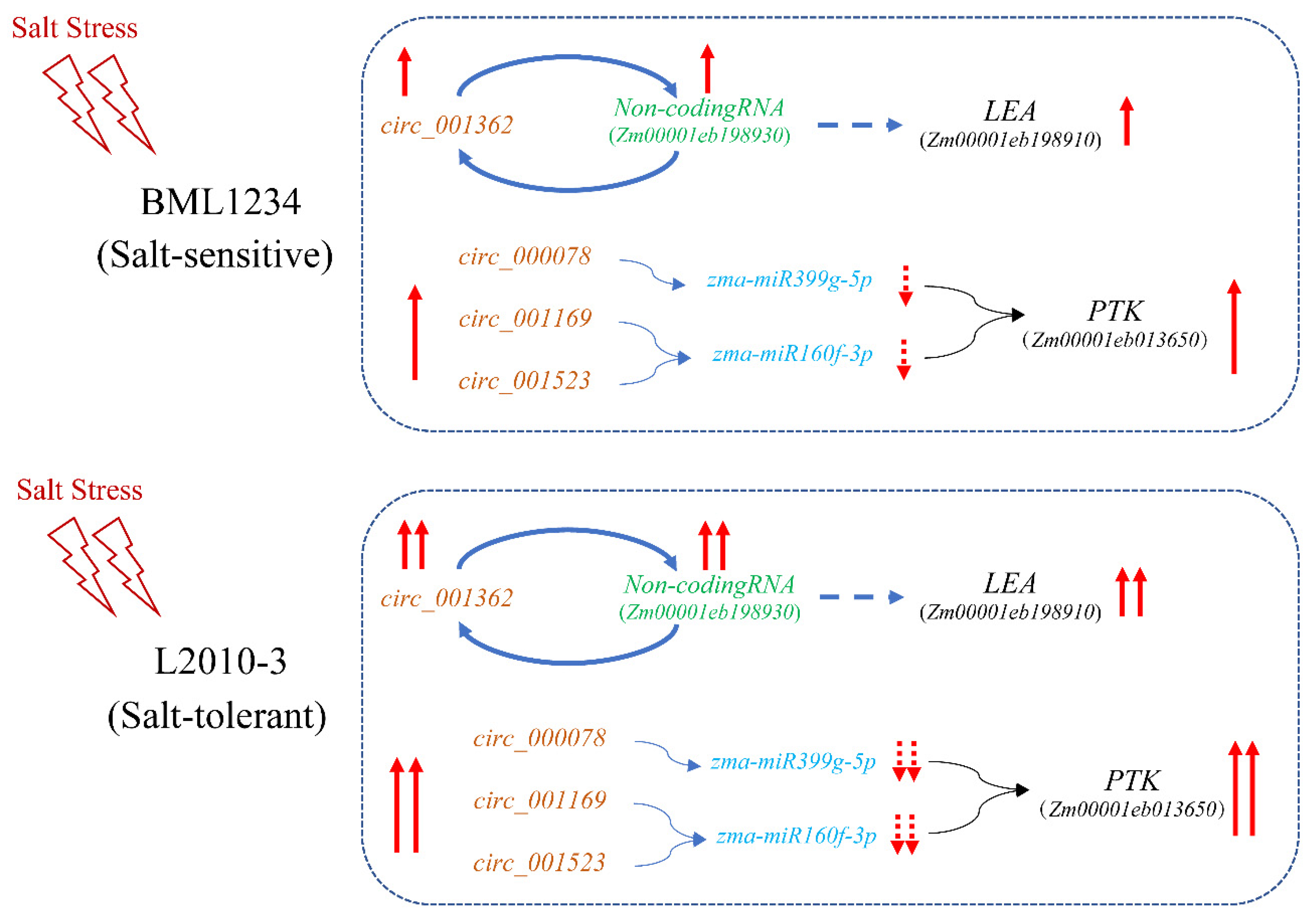
2.4. Combination of the GWAS and RNA-Seq to Identify circRNA-Mediated Hub Genes in Response to Salt Stress
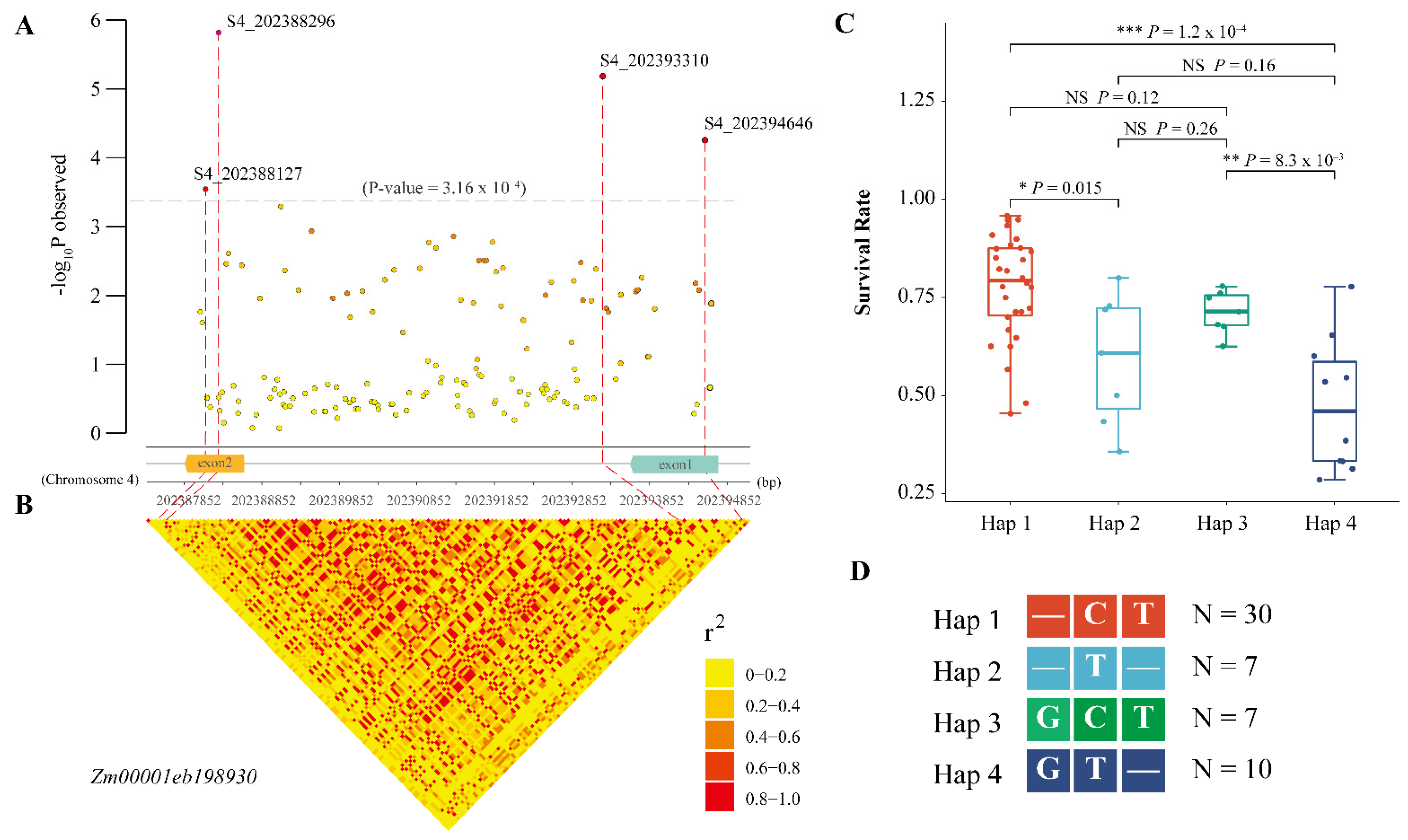
2.5. Intragenic Variations in Hub Genes Affecting the SR under Salt Stress
3. Discussion
3.1. circRNAs Play Important Roles in Response to Salt Stress in Maize
3.2. An Effective Method to Identify Causal Genes by Combining a GWAS and Transcriptome Analysis
3.3. circRNA-Mediated Regulatory Model under Salt Stress in Maize
4. Materials and Methods
4.1. Data Preparation for the Transcriptome Analysis
4.2. circRNA Identification and Differential Expression Analysis
4.3. Validation of the circRNAs
4.4. Target Gene Prediction of the circRNAs
4.5. Plant Materials, Phenotypic Data Collection, and Statistical Analysis
4.6. GWAS for the SR of Maize Seedlings under Salt Stress
4.7. Candidate Gene Association Study
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fang, S.; Hou, X.; Liang, X. Response Mechanisms of Plants Under Saline-Alkali Stress. Front. Plant Sci. 2021, 12, 667458. [Google Scholar] [CrossRef] [PubMed]
- Hanin, M.; Ebel, C.; Ngom, M.; Laplaze, L.; Masmoudi, K. New Insights on Plant Salt Tolerance Mechanisms and Their Potential Use for Breeding. Front. Plant Sci. 2016, 7, 1787. [Google Scholar] [CrossRef]
- Ma, L.; Zhang, M.; Chen, J.; Qing, C.; He, S.; Zou, C.; Yuan, G.; Yang, C.; Peng, H.; Pan, G.; et al. GWAS and WGCNA Uncover Hub Genes Controlling Salt Tolerance in Maize (Zea mays L.) Seedlings. Theor. Appl. Genet. 2021, 134, 3305–3318. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Qing, C.; Liu, P.; Zou, C.; Yuan, G.; Pan, G.; Shen, Y.; Ma, L. Joint GWAS and WGCNA Uncover the Genetic Control of Calcium Accumulation under Salt Treatment in Maize Seedlings. Physiol. Plant. 2022, 174, e13606. [Google Scholar] [CrossRef]
- Luo, X.; Wang, B.; Gao, S.; Zhang, F.; Terzaghi, W.; Dai, M. Genome-Wide Association Study Dissects the Genetic Bases of Salt Tolerance in Maize Seedlings. J. Integr. Plant Biol. 2019, 61, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Cao, Y.; Wang, Z.; Wang, Z.; Shi, J.; Liang, X.; Song, W.; Chen, Q.; Lai, J.; Jiang, C. A Retrotransposon in an HKT1 Family Sodium Transporter Causes Variation of Leaf Na+ Exclusion and Salt Tolerance in Maize. New Phytol. 2018, 217, 1161–1176. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Zhang, Y.; Li, J.; Zhang, P.; Chen, K.; Song, W.; Wang, X.; Yang, J.; Lu, X.; Lu, B.; et al. Molecular Dissection of Maize Seedling Salt Tolerance Using a Genome-Wide Association Analysis Method. Plant Biotechnol. J. 2021, 19, 1937–1951. [Google Scholar] [CrossRef]
- Luo, M.; Zhao, Y.; Zhang, R.; Xing, J.; Duan, M.; Li, J.; Wang, N.; Wang, W.; Zhang, S.; Chen, Z.; et al. Mapping of a Major QTL for Salt Tolerance of Mature Field-Grown Maize Plants Based on SNP Markers. BMC Plant Biol. 2017, 17, 140. [Google Scholar] [CrossRef]
- Ma, L.; Wang, C.; Hu, Y.; Dai, W.; Liang, Z.; Zou, C.; Pan, G.; Lübberstedt, T.; Shen, Y. GWAS and Transcriptome Analysis Reveal MADS26 Involved in Seed Germination Ability in Maize. Theor. Appl. Genet. 2022, 135, 1717–1730. [Google Scholar] [CrossRef]
- Szabo, L.; Salzman, J. Detecting Circular RNAs: Bioinformatic and Experimental Challenges. Nat. Rev. Genet. 2016, 17, 679–692. [Google Scholar] [CrossRef] [Green Version]
- Verduci, L.; Tarcitano, E.; Strano, S.; Yarden, Y.; Blandino, G. CircRNAs: Role in Human Diseases and Potential Use as Biomarkers. Cell Death Dis. 2021, 12, 468. [Google Scholar] [CrossRef] [PubMed]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs Are a Large Class of Animal RNAs with Regulatory Potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Li, S.; Chen, M. Characterization and Function of Circular RNAs in Plants. Front. Mol. Biosci. 2020, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.-Y.; Chen, L.; Liu, C.; Zhu, Q.-H.; Fan, L. Widespread Noncoding Circular RNAs in Plants. New Phytol. 2015, 208, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Barrett, S.P.; Salzman, J. Circular RNAs: Analysis, Expression and Potential Functions. Development 2016, 143, 1838–1847. [Google Scholar] [CrossRef]
- Chen, L.-L. The Biogenesis and Emerging Roles of Circular RNAs. Nat. Rev. Mol. Cell Biol. 2016, 17, 205–211. [Google Scholar] [CrossRef]
- Zhu, Y.-X.; Jia, J.-H.; Yang, L.; Xia, Y.-C.; Zhang, H.-L.; Jia, J.-B.; Zhou, R.; Nie, P.-Y.; Yin, J.-L.; Ma, D.-F.; et al. Identification of Cucumber Circular RNAs Responsive to Salt Stress. BMC Plant Biol. 2019, 19, 164. [Google Scholar] [CrossRef]
- Yin, J.; Liu, Y.; Lu, L.; Zhang, J.; Chen, S.; Wang, B. Comparison of Tolerant and Susceptible Cultivars Revealed the Roles of Circular RNAs in Rice Responding to Salt Stress. Plant Growth Regul. 2022, 96, 243–254. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, M.; Wei, S.; Qin, F.; Zhao, H.; Suo, B. Identification of Circular RNAs and Their Targets in Leaves of Triticum aestivum L. under Dehydration Stress. Front. Plant Sci. 2017, 7, 2024. [Google Scholar] [CrossRef]
- Fu, X.-Z.; Zhang, X.-Y.; Qiu, J.-Y.; Zhou, X.; Yuan, M.; He, Y.-Z.; Chun, C.-P.; Cao, L.; Ling, L.-L.; Peng, L.-Z. Whole-Transcriptome RNA Sequencing Reveals the Global Molecular Responses and CeRNA Regulatory Network of MRNAs, LncRNAs, MiRNAs and CircRNAs in Response to Copper Toxicity in Ziyang Xiangcheng (Citrus Junos Sieb. Ex Tanaka). BMC Plant Biol. 2019, 19, 509. [Google Scholar] [CrossRef] [Green Version]
- Li, G.-T.; Niu, Z.-M.; Zheng, Z.-Y.; Lv, J.-J.; Chen, Q.-Y.; Liu, J.-Q.; Wan, D.-S. Contrasting Origins, Expression Patterns and Functions of CircRNAs between Salt-Sensitive and Salt-Tolerant Poplars. Environ. Exp. Bot. 2021, 185, 104403. [Google Scholar] [CrossRef]
- Zhang, P.; Fan, Y.; Sun, X.; Chen, L.; Terzaghi, W.; Bucher, E.; Li, L.; Dai, M. A Large-Scale Circular RNA Profiling Reveals Universal Molecular Mechanisms Responsive to Drought Stress in Maize and Arabidopsis. Plant J. 2019, 98, 697–713. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Li, X.; Yan, Y.; Duan, M.-H.; Xu, J.-H. Identification, Characterization, and Functional Prediction of Circular RNAs in Maize. Mol. Genet. Genom. 2020, 295, 491–503. [Google Scholar] [CrossRef]
- Zhang, Z.; Mao, C.; Shi, Z.; Kou, X. The Amino Acid Metabolic and Carbohydrate Metabolic Pathway Play Important Roles during Salt-Stress Response in Tomato. Front. Plant Sci. 2017, 8, 1231. [Google Scholar] [CrossRef]
- Ding, D.; Zhang, L.; Wang, H.; Liu, Z.; Zhang, Z.; Zheng, Y. Differential Expression of MiRNAs in Response to Salt Stress in Maize Roots. Ann. Bot. 2009, 103, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Shan, T.; Fu, R.; Xie, Y.; Chen, Q.; Wang, Y.; Li, Z.; Song, X.; Li, P.; Wang, B. Regulatory Mechanism of Maize (Zea mays L.) MiR164 in Salt Stress Response. Russ. J. Genet. 2020, 56, 835–842. [Google Scholar] [CrossRef]
- Meng-Yu, H.A.O.; Ming-Lin, L.; Xue-Ju, Y. Construction of recombinant prokaryotic expression vector for Brassica juncea BjJ10-2 and identification of its function for high salinity tolerance. J. Nucl. Agric. Sci. 2011, 25, 247. [Google Scholar] [CrossRef]
- Luo, Z.; Szczepanek, A.; Abdel-Haleem, H. Genome-Wide Association Study (GWAS) Analysis of Camelina Seedling Germination under Salt Stress Condition. Agronomy 2020, 10, 1444. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, X.; Wan, W.; Zhang, H.; Liu, J.; Li, M.; Wang, H.; Xiao, J.; Wang, X. Identification and Characterization of the EXO70 Gene Family in Polyploid Wheat and Related Species. Int. J. Mol. Sci. 2018, 20, 60. [Google Scholar] [CrossRef]
- Sakuraba, Y.; Bülbül, S.; Piao, W.; Choi, G.; Paek, N.-C. Arabidopsis EARLY FLOWERING3 Increases Salt Tolerance by Suppressing Salt Stress Response Pathways. Plant J. 2017, 92, 1106–1120. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Q.; Gan, Z.; Wang, Y.; Lu, S.; Hou, Z.; Li, H.; Xiang, H.; Liu, B.; Kong, F.; Dong, L. The Soybean Gene J Contributes to Salt Stress Tolerance by Up-Regulating Salt-Responsive Genes. Front. Plant Sci. 2020, 11, 272. [Google Scholar] [CrossRef] [PubMed]
- Schapire, A.L.; Valpuesta, V.; Botella, M.A. TPR Proteins in Plant Hormone Signaling. Plant Signal. Behav. 2006, 1, 229–230. [Google Scholar] [CrossRef] [PubMed]
- Wei, K.; Han, P. Comparative Functional Genomics of the TPR Gene Family in Arabidopsis, Rice and Maize. Mol. Breed. 2017, 37, 152. [Google Scholar] [CrossRef]
- Zhou, X.; Zheng, Y.; Cai, Z.; Wang, X.; Liu, Y.; Yu, A.; Chen, X.; Liu, J.; Zhang, Y.; Wang, A. Identification and Functional Analysis of Tomato TPR Gene Family. Int. J. Mol. Sci. 2021, 22, 758. [Google Scholar] [CrossRef]
- Li, J.; Li, M.; Yao, S.; Cai, G.; Wang, X. Patatin-Related Phospholipase PPLAIIIγ Involved in Osmotic and Salt Tolerance in Arabidopsis. Plants 2020, 9, 650. [Google Scholar] [CrossRef]
- Cao, Y.; Zhang, Q.; Chen, Y.; Zhao, H.; Lang, Y.; Yu, C.; Yang, J. Identification of Differential Expression Genes in Leaves of Rice (Oryza sativa L.) in Response to Heat Stress by CDNA-AFLP Analysis. BioMed Res. Int. 2013, 2013, 576189. [Google Scholar] [CrossRef]
- Yang, L.; Jiang, T.; Fountain, J.C.; Scully, B.T.; Lee, R.D.; Kemerait, R.C.; Chen, S.; Guo, B. Protein Profiles Reveal Diverse Responsive Signaling Pathways in Kernels of Two Maize Inbred Lines with Contrasting Drought Sensitivity. Int. J. Mol. Sci. 2014, 15, 18892–18918. [Google Scholar] [CrossRef]
- Chen, Y.; Li, C.; Zhang, B.; Yi, J.; Yang, Y.; Kong, C.; Lei, C.; Gong, M. The Role of the Late Embryogenesis-Abundant (LEA) Protein Family in Development and the Abiotic Stress Response: A Comprehensive Expression Analysis of Potato (Solanum tuberosum). Genes 2019, 10, 148. [Google Scholar] [CrossRef]
- Che-Othman, M.H.; Millar, A.H.; Taylor, N.L. Connecting Salt Stress Signalling Pathways with Salinity-Induced Changes in Mitochondrial Metabolic Processes in C3 Plants. Plant Cell Environ. 2017, 40, 2875–2905. [Google Scholar] [CrossRef]
- Xu, D.; Duan, X.; Wang, B.; Hong, B.; Ho, T.H.D.; Wu, R. Expression of a Late Embryogenesis Abundant Protein Gene, HVA1, from Barley Confers Tolerance to Water Deficit and Salt Stress in Transgenic Rice. Plant Physiol. 1996, 110, 249–257. [Google Scholar] [CrossRef] [Green Version]
- McNEILL, H.; Knebel, A.; Arthur, J.S.C.; Cuenda, A.; Cohen, P. A Novel UBA and UBX Domain Protein That Binds Polyubiquitin and VCP and Is a Substrate for SAPKs. Biochem. J. 2004, 384, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Hundertmark, M.; Hincha, D.K. LEA (Late Embryogenesis Abundant) Proteins and Their Encoding Genes in Arabidopsis thaliana. BMC Genom. 2008, 9, 118. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Wei, Y.; Sun, K.; Wu, J.; Wang, Y.; Wu, K. The Maize AAA-Type Protein SKD1 Confers Enhanced Salt and Drought Stress Tolerance in Transgenic Tobacco by Interacting with Lyst-Interacting Protein 5. PLoS ONE 2013, 8, e69787. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Li, Z.; Wang, Q.; Guan, Y.; Li, X.; Huangfu, Y.; Meng, F.; Li, J.; Dai, S.; Liu, W. Phosphoproteomic Profiling Reveals Early Salt-Responsive Mechanisms in Two Foxtail Millet Cultivars. Front. Plant Sci. 2021, 12, 712257. [Google Scholar] [CrossRef]
- Udomchalothorn, T.; Maneeprasobsuk, S.; Bangyeekhun, E.; Boon-Long, P.; Chadchawan, S. The Role of the Bifunctional Enzyme, Fructose-6-Phosphate-2-Kinase/Fructose-2,6-Bisphosphatase, in Carbon Partitioning during Salt Stress and Salt Tolerance in Rice (Oryza sativa L.). Plant Sci. 2009, 176, 334–341. [Google Scholar] [CrossRef]
- Xia-Yu, G.; Meng, Z.; Ming-Dong, Z.; Ji-Rui, L.; Zhong-Wei, W.; Jian-Wu, L.; Bin, Z.; Zhi-Yong, A.; Hua-Feng, D. Comparative Transcriptomic Analysis of the Super Hybrid Rice Chaoyouqianhao under Salt Stress. BMC Plant Biol. 2022, 22, 233. [Google Scholar] [CrossRef]
- Lim, C.W.; Kim, J.-H.; Baek, W.; Kim, B.S.; Lee, S.C. Functional Roles of the Protein Phosphatase 2C, AtAIP1, in Abscisic Acid Signaling and Sugar Tolerance in Arabidopsis. Plant Sci. 2012, 187, 83–88. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, N.; Peng, X.; Shen, S. Identification of the PP2C Gene Family in Paper Mulberry (Broussonetia papyrifera) and Its Roles in the Regulation Mechanism of the Response to Cold Stress. Biotechnol. Lett. 2021, 43, 1089–1102. [Google Scholar] [CrossRef]
- Yu, X.; Han, J.; Li, L.; Zhang, Q.; Yang, G.; He, G. Wheat PP2C-A10 Regulates Seed Germination and Drought Tolerance in Transgenic Arabidopsis. Plant Cell Rep. 2020, 39, 635–651. [Google Scholar] [CrossRef]
- He, Z.; Wu, J.; Sun, X.; Dai, M. The Maize Clade A PP2C Phosphatases Play Critical Roles in Multiple Abiotic Stress Responses. Int. J. Mol. Sci. 2019, 20, 3573. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Ding, Y.; Yang, Y.; Song, C.; Wang, B.; Yang, S.; Guo, Y.; Gong, Z. Protein Kinases in Plant Responses to Drought, Salt, and Cold Stress. J. Integr. Plant Biol. 2021, 63, 53–78. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Liu, Y.; Zhang, H.; Wang, J.; Zinta, G.; Xie, S.; Zhu, W.; Nie, W.-F. Genome-Wide Identification of Circular RNAs in Response to Low-Temperature Stress in Tomato Leaves. Front. Genet. 2020, 11, 591806. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; Gao, S.; Zhang, H.Y.; Li, B.Y.; Zhong, H.X.; Wang, Y.K.; Hu, H.M.; Zhang, H.K.; Luo, B.W.; Zhang, X.; et al. Identification and Characterization of CircRNAs in Maize Seedlings under Deficient Nitrogen. Plant Biol. 2021, 23, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; Fu, L.; Wang, F.; Wu, D.; Shen, Q.; Zhang, G. GWAS and Transcriptomic Integrating Analysis Reveals Key Salt-Responding Genes Controlling Na+ Content in Barley Roots. Plant Physiol. Biochem. 2021, 167, 596–606. [Google Scholar] [CrossRef]
- Yuan, Y.; Xing, H.; Zeng, W.; Xu, J.; Mao, L.; Wang, L.; Feng, W.; Tao, J.; Wang, H.; Zhang, H.; et al. Genome-Wide Association and Differential Expression Analysis of Salt Tolerance in Gossypium Hirsutum L at the Germination Stage. BMC Plant Biol. 2019, 19, 394. [Google Scholar] [CrossRef]
- Zhang, P.; Yuan, Z.; Wei, L.; Qiu, X.; Wang, G.; Liu, Z.; Fu, J.; Cao, L.; Wang, T. Overexpression of ZmPP2C55 Positively Enhances Tolerance to Drought Stress in Transgenic Maize Plants. Plant Sci. 2022, 314, 111127. [Google Scholar] [CrossRef]
- Hu, X.; Liu, L.; Xiao, B.; Li, D.; Xing, X.; Kong, X.; Li, D. Enhanced Tolerance to Low Temperature in Tobacco by Over-Expression of a New Maize Protein Phosphatase 2C, ZmPP2C2. J. Plant Physiol. 2010, 167, 1307–1315. [Google Scholar] [CrossRef]
- Li, X.; Cao, J. Late Embryogenesis Abundant (LEA) Gene Family in Maize: Identification, Evolution, and Expression Profiles. Plant Mol. Biol. Rep. 2016, 34, 15–28. [Google Scholar] [CrossRef]
- Figueras, M.; Pujal, J.; Saleh, A.; Save, R.; Pages, M.; Goday, A. Maize Rabl7 Overexpression in Arabidopsis Plants Promotes Osmotic Stress Tolerance. Ann. Appl. Biol. 2004, 144, 251–257. [Google Scholar] [CrossRef]
- Li, J.; Cui, J.; Dai, C.; Liu, T.; Cheng, D.; Luo, C. Whole-Transcriptome RNA Sequencing Reveals the Global Molecular Responses and CeRNA Regulatory Network of MRNAs, LncRNAs, MiRNAs and CircRNAs in Response to Salt Stress in Sugar Beet (Beta vulgaris). Int. J. Mol. Sci. 2021, 22, 289. [Google Scholar] [CrossRef]
- He, X.; Guo, S.; Wang, Y.; Wang, L.; Shu, S.; Sun, J. Systematic Identification and Analysis of Heat-Stress-Responsive LncRNAs, CircRNAs and MiRNAs with Associated Co-Expression and CeRNA Networks in Cucumber (Cucumis sativus L.). Physiol. Plant. 2020, 168, 736–754. [Google Scholar] [CrossRef] [PubMed]
- Chu, Q.; Bai, P.; Zhu, X.; Zhang, X.; Mao, L.; Zhu, Q.-H.; Fan, L.; Ye, C.-Y. Characteristics of Plant Circular RNAs. Brief. Bioinform. 2020, 21, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Hao, K.; Wang, Y.; Zhu, Z.; Wu, Y.; Chen, R.; Zhang, L. MiR160: An Indispensable Regulator in Plant. Front. Plant Sci. 2022, 13, 833322. [Google Scholar] [CrossRef]
- Pegler, J.L.; Oultram, J.M.J.; Grof, C.P.L.; Eamens, A.L. Molecular Manipulation of the MiR399/PHO2 Expression Module Alters the Salt Stress Response of Arabidopsis thaliana. Plants 2021, 10, 73. [Google Scholar] [CrossRef]
- Liu, P.; Zhang, Y.; Zou, C.; Yang, C.; Pan, G.; Ma, L.; Shen, Y. Integrated Analysis of Long Non-Coding RNAs and MRNAs Reveals the Regulatory Network of Maize Seedling Root Responding to Salt Stress. BMC Genom. 2022, 23, 50. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, P.; Qing, C.; Yang, C.; Shen, Y.; Ma, L. Comparative Transcriptome Analyses of Maize Seedling Root Responses to Salt Stress. PeerJ 2021, 9, e10765. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Gao, Y.; Wang, J.; Zhao, F. CIRI: An Efficient and Unbiased Algorithm for de Novo Circular RNA Identification. Genome Biol. 2015, 16, 4. [Google Scholar] [CrossRef]
- Zheng, Y.; Ji, P.; Chen, S.; Hou, L.; Zhao, F. Reconstruction of Full-Length Circular RNAs Enables Isoform-Level Quantification. Genome Med. 2019, 11, 2. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Zhao, F. Visualization of Circular RNAs and Their Internal Splicing Events from Transcriptomic Data. Bioinformatics 2020, 36, 2934–2935. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.H.-O.; Shi, C.H.; Wang, B.; Chow, S.H.-C.; Chung, G.T.-Y.; Lung, R.W.-M.; Tan, K.-E.; Lim, Y.-Y.; Tsang, A.C.-M.; Lo, K.-W.; et al. Quantifying Full-Length Circular RNAs in Cancer. Genome Res. 2021, 31, 2340–2353. [Google Scholar] [CrossRef]
- Risso, D.; Ngai, J.; Speed, T.P.; Dudoit, S. Normalization of RNA-Seq Data Using Factor Analysis of Control Genes or Samples. Nat. Biotechnol. 2014, 32, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Betel, D.; Koppal, A.; Agius, P.; Sander, C.; Leslie, C. Comprehensive Modeling of MicroRNA Targets Predicts Functional Non-Conserved and Non-Canonical Sites. Genome Biol. 2010, 11, R90. [Google Scholar] [CrossRef]
- Bo, X.; Wang, S. TargetFinder: A Software for Antisense Oligonucleotide Target Site Selection Based on MAST and Secondary Structures of Target MRNA. Bioinformatics 2005, 21, 1401–1402. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, H.; Li, L.; Lan, H.; Ren, Z.; Liu, D.; Wu, L.; Liu, H.; Jaqueth, J.; Li, B.; et al. Characterizing the Population Structure and Genetic Diversity of Maize Breeding Germplasm in Southwest China Using Genome-Wide SNP Markers. BMC Genom. 2016, 17, 697. [Google Scholar] [CrossRef]
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z. Iterative Usage of Fixed and Random Effect Models for Powerful and Efficient Genome-Wide Association Studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X.; et al. RMVP: A Memory-Efficient, Visualization-Enhanced, and Parallel-Accelerated Tool for Genome-Wide Association Study. Genom. Proteom. Bioinform. 2021, 19, 619–628. [Google Scholar] [CrossRef]
- Gao, X.; Starmer, J.; Martin, E.R. A Multiple Testing Correction Method for Genetic Association Studies Using Correlated Single Nucleotide Polymorphisms. Genet. Epidemiol. 2008, 32, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Guan, Z.; Li, Z.; Liu, P.; Ma, L.; Zhang, Y.; Pan, L.; He, S.; Zhang, Y.; Li, P.; et al. A Combination of Linkage Mapping and GWAS Brings New Elements on the Genetic Basis of Yield-Related Traits in Maize across Multiple Environments. Theor. Appl. Genet. 2020, 133, 2881–2895. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Cao, Y. Genetic Dissection of Grain Yield of Maize and Yield-Related Traits Through Association Mapping and Genomic Prediction. Front. Plant Sci. 2021, 12, 690059. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for Association Mapping of Complex Traits in Diverse Samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Ding, S.; Wang, H.; Qin, F. IntAssoPlot: An R Package for Integrated Visualization of Genome-Wide Association Study Results With Gene Structure and Linkage Disequilibrium Matrix. Front. Genet. 2020, 11, 260. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Markers | Chr | Position | Allele | Effect | SE | PVE | p-Value |
|---|---|---|---|---|---|---|---|
| SYN6348 | 2 | 229,479,599 | A/G | −0.0396 | 0.0067 | 4.41% | 4.71 × 10−10 |
| PZE-104119465 | 4 | 196,370,737 | G/A | 0.0362 | 0.0072 | 5.79% | 4.39 × 10−8 |
| PZA02824.5 | 3 | 218,898,682 | G/A | 0.0424 | 0.0090 | 1.83% | 1.48 × 10−7 |
| PZE-101062009 | 1 | 45,633,273 | A/C | −0.0288 | 0.0067 | 4.40% | 1.41 × 10−6 |
| PZE-109073606 | 9 | 118,923,900 | C/A | −0.0347 | 0.0083 | 0.75% | 1.95 × 10−6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, P.; Zhu, Y.; Liu, H.; Liang, Z.; Zhang, M.; Zou, C.; Yuan, G.; Gao, S.; Pan, G.; Shen, Y.; et al. A Combination of a Genome-Wide Association Study and a Transcriptome Analysis Reveals circRNAs as New Regulators Involved in the Response to Salt Stress in Maize. Int. J. Mol. Sci. 2022, 23, 9755. https://doi.org/10.3390/ijms23179755
Liu P, Zhu Y, Liu H, Liang Z, Zhang M, Zou C, Yuan G, Gao S, Pan G, Shen Y, et al. A Combination of a Genome-Wide Association Study and a Transcriptome Analysis Reveals circRNAs as New Regulators Involved in the Response to Salt Stress in Maize. International Journal of Molecular Sciences. 2022; 23(17):9755. https://doi.org/10.3390/ijms23179755
Chicago/Turabian StyleLiu, Peng, Yuxiao Zhu, Hao Liu, Zhenjuan Liang, Minyan Zhang, Chaoying Zou, Guangsheng Yuan, Shibin Gao, Guangtang Pan, Yaou Shen, and et al. 2022. "A Combination of a Genome-Wide Association Study and a Transcriptome Analysis Reveals circRNAs as New Regulators Involved in the Response to Salt Stress in Maize" International Journal of Molecular Sciences 23, no. 17: 9755. https://doi.org/10.3390/ijms23179755