
Gut Microbiome and Metabolome Modulation by Maternal High-Fat Diet and Thermogenic Challenge

 , ,
, ,
Abstract
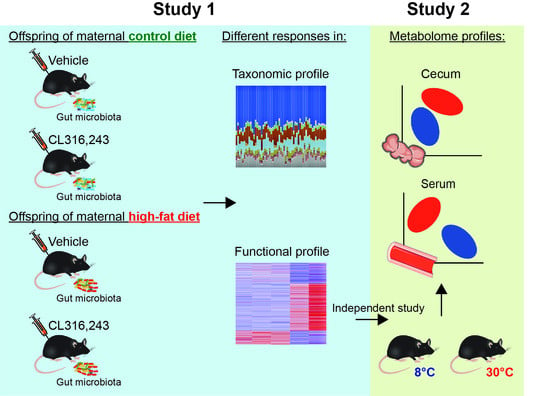
:
1. Introduction
2. Results
2.1. Gut Microbial Diversity
2.2. Taxonomic Profile Differences
2.3. Predicted Functional Profiles
2.4. Gut Microbial Diversity and Taxonomy from Mice Exposed to Different Temperatures
2.5. Cecum and Serum Metabolomic Profiling
3. Discussion
4. Materials and Methods
4.1. Experimental Design
4.1.1. Study 1
4.1.2. Study 2
4.2. Microbial Community Profiling Using 16S rRNA Amplicon Sequencing
4.3. Bioinformatics Analysis
4.4. Untargeted Metabolomics
4.5. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Heiss, C.N.; Olofsson, L.E. Gut Microbiota-Dependent Modulation of Energy Metabolism. J. Innate Immun. 2018, 10, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Aron-Wisnewsky, J.; Warmbrunn, M.V.; Nieuwdorp, M.; Clément, K. Metabolism and Metabolic Disorders and the Microbiome: The Intestinal Microbiota Associated with Obesity, Lipid Metabolism, and Metabolic Health—Pathophysiology and Therapeutic Strategies. Gastroenterology 2021, 160, 573–599. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef]
- Carmody, R.N.; Gerber, G.K.; Luevano, J.M., Jr.; Gatti, D.M.; Somes, L.; Svenson, K.L.; Turnbaugh, P.J. Diet Dominates Host Genotype in Shaping the Murine Gut Microbiota. Cell Host Microbe 2015, 17, 72–84. [Google Scholar] [CrossRef]
- Shankar, K.; Harrell, A.; Liu, X.; Gilchrist, J.M.; Ronis, M.J.J.; Badger, T.M. Maternal Obesity at Conception Programs Obesity in the Offspring. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2008, 294, R528–R538. [Google Scholar] [CrossRef]
- Wankhade, U.; Zhong, Y.; Kang, P.; Alfaro, M.; Chintapalli, S.V.; Thakali, K.M.; Shankar, K. Enhanced offspring predisposition to steatohepatitis with maternal high-fat diet is associated with epigenetic and microbiome alterations. PLoS ONE 2017, 12, e0175675. [Google Scholar] [CrossRef]
- Wankhade, U.D.; Zhong, Y.; Kang, P.; Alfaro, M.; Chintapalli, S.V.; Piccolo, B.D.; Mercer, K.E.; Andres, A.; Thakali, K.M.; Shankar, K. Maternal High-Fat Diet Programs Offspring Liver Steatosis in a Sexually Dimorphic Manner in Association with Changes in Gut Microbial Ecology in Mice. Sci. Rep. 2018, 8, 16502. [Google Scholar] [CrossRef]
- Fall, C.H.D.; Kumaran, K. Metabolic Programming in Early Life in Humans. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20180123. [Google Scholar] [CrossRef]
- Ellsworth, L.; Harman, E.; Padmanabhan, V.; Gregg, B. Lactational programming of glucose homeostasis: A window of opportunity. Reproduction 2018, 156, R23–R42. [Google Scholar] [CrossRef]
- Lecoutre, S.; Kwok, K.H.M.; Petrus, P.; Lambert, M.; Breton, C. Epigenetic Programming of Adipose Tissue in the Progeny of Obese Dams. Curr. Genom. 2019, 20, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.; Jellyman, J.; Han, G.; Beall, M.; Lane, R.H.; Ross, M.G. Maternal obesity and high-fat diet program offspring metabolic syndrome. Am. J. Obstet. Gynecol. 2014, 211, 237.e1–237.e13. [Google Scholar] [CrossRef] [PubMed]
- Bachman, E.S.; Dhillon, H.; Zhang, C.-Y.; Cinti, S.; Bianco, A.C.; Kobilka, B.K.; Lowell, B.B. βAR Signaling Required for Diet-Induced Thermogenesis and Obesity Resistance. Science 2002, 297, 843–845. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, C.; Stojanović, O.; Colin, D.J.; Suarez-Zamorano, N.; Tarallo, V.; Veyrat-Durebex, C.; Rigo, D.; Fabbiano, S.; Stevanović, A.; Hagemann, S.; et al. Gut Microbiota Orchestrates Energy Homeostasis during Cold. Cell 2015, 163, 1360–1374. [Google Scholar] [CrossRef]
- Li, B.; Li, L.; Li, M.; Lam, S.M.; Wang, G.; Wu, Y.; Zhang, H.; Niu, C.; Zhang, X.; Liu, X.; et al. Microbiota Depletion Impairs Thermogenesis of Brown Adipose Tissue and Browning of White Adipose Tissue. Cell Rep. 2019, 26, 2720–2737.e5. [Google Scholar] [CrossRef]
- Lkhagva, E.; Chung, H.-J.; Hong, J.; Tang, W.H.W.; Lee, S.-I.; Hong, S.-T.; Lee, S. The regional diversity of gut microbiome along the GI tract of male C57BL/6 mice. BMC Microbiol. 2021, 21, 44. [Google Scholar] [CrossRef]
- Padmanabhan, V.; Cardoso, R.C.; Puttabyatappa, M. Developmental Programming, a Pathway to Disease. Endocrinology 2016, 157, 1328–1340. [Google Scholar] [CrossRef]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.-M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef]
- Muscogiuri, G.; Cantone, E.; Cassarano, S.; Tuccinardi, D.; Barrea, L.; Savastano, S.; Colao, A. Gut microbiota: A new path to treat obesity. Int. J. Obes. Suppl. 2019, 9, 10–19. [Google Scholar] [CrossRef]
- Chu, D.M.; Antony, K.M.; Ma, J.; Prince, A.L.; Showalter, L.; Moller, M.; Aagaard, K.M. The early infant gut microbiome varies in association with a maternal high-fat diet. Genome Med. 2016, 8, 77. [Google Scholar] [CrossRef] [Green Version]
- Ziętak, M.; Kovatcheva-Datchary, P.; Markiewicz, L.H.; Ståhlman, M.; Kozak, L.P.; Bäckhed, F. Altered Microbiota Contributes to Reduced Diet-Induced Obesity upon Cold Exposure. Cell Metab. 2016, 23, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Xie, R.; Sun, Y.; Wu, J.; Huang, S.; Jin, G.; Guo, Z.; Zhang, Y.; Liu, T.; Liu, X.; Cao, X.; et al. Maternal High Fat Diet Alters Gut Microbiota of Offspring and Exacerbates DSS-Induced Colitis in Adulthood. Front. Immunol. 2018, 9, 2608. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.P.B.; Raipuria, M.; Bahari, H.; Kaakoush, N.O.; Morris, M.J. Impacts of Diet and Exercise on Maternal Gut Microbiota Are Transferred to Offspring. Front. Endocrinol. 2018, 9, 716. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.C.; Isolauri, E.; Laitinen, K.; Salminen, S. Effect of mother’s weight on infant’s microbiota acquisition, composition, and activity during early infancy: A prospective follow-up study initiated in early pregnancy. Am. J. Clin. Nutr. 2010, 92, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- Mueller, N.; Shin, H.; Pizoni, A.; Werlang, I.C.; Matte, U.; Goldani, M.Z.; Goldani, H.A.S.; Dominguez-Bello, M.G. Birth mode-dependent association between pre-pregnancy maternal weight status and the neonatal intestinal microbiome. Sci. Rep. 2016, 6, 23133. [Google Scholar] [CrossRef]
- Soderborg, T.K.; Clark, S.; Mulligan, C.E.; Janssen, R.C.; Babcock, L.; Ir, D.; Young, B.; Krebs, N.; Lemas, D.J.; Johnson, L.K.; et al. The gut microbiota in infants of obese mothers increases inflammation and susceptibility to NAFLD. Nat. Commun. 2018, 9, 4462. [Google Scholar] [CrossRef]
- Galley, J.D.; Bailey, M.; Dush, C.K.; Schoppe-Sullivan, S.; Christian, L. Maternal Obesity Is Associated with Alterations in the Gut Microbiome in Toddlers. PLoS ONE 2014, 9, e113026. [Google Scholar] [CrossRef]
- Sankar, S.A.; Lagier, J.C.; Pontarotti, P.; Raoult, D.; Fournier, P.E. The human gut microbiome, a taxonomic conundrum. Syst. Appl. Microbiol. 2015, 38, 276–286. [Google Scholar] [CrossRef]
- Crovesy, L.; Masterson, D.; Rosado, E.L. Profile of the gut microbiota of adults with obesity: A systematic review. Eur. J. Clin. Nutr. 2020, 74, 1251–1262. [Google Scholar] [CrossRef]
- Mariat, D.; Firmesse, O.; Levenez, F.; Guimarăes, V.D.; Sokol, H.; Doré, J.; Corthier, G.; Furet, J.-P. The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol. 2009, 9, 123. [Google Scholar] [CrossRef]
- Magne, F.; Gotteland, M.; Gauthier, L.; Zazueta, A.; Pesoa, S.; Navarrete, P.; Balamurugan, R. The Firmicutes/Bacteroidetes Ratio: A Relevant Marker of Gut Dysbiosis in Obese Patients? Nutrients 2020, 12, 1474. [Google Scholar] [CrossRef] [PubMed]
- Börnigen, D.; Morgan, X.C.; Franzosa, E.A.; Ren, B.; Xavier, R.J.; Garrett, W.S.; Huttenhower, C. Functional profiling of the gut microbiome in disease-associated inflammation. Genome Med. 2013, 5, 65. [Google Scholar] [CrossRef] [PubMed]
- Mann, P.E.; Huynh, K.; Widmer, G. Maternal high fat diet and its consequence on the gut microbiome: A rat model. Gut Microbes 2018, 9, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ping, X.; Zhang, Y.; Li, G.; Zhang, T.; Chen, G.; Ma, X.; Wang, D.; Xu, L. Comparative Transcriptome Profiling of Cold Exposure and β3-AR Agonist CL316,243-Induced Browning of White Fat. Front. Physiol. 2021, 12, 667698. [Google Scholar] [CrossRef]
- Langille, M.G.; Meehan, C.J.; Koenig, J.E.; Dhanani, A.S.; Rose, R.A.; Howlett, S.E.; Beiko, R.G. Microbial shifts in the aging mouse gut. Microbiome 2014, 2, 50. [Google Scholar] [CrossRef]
- Dey, N.; Wagner, V.E.; Blanton, L.V.; Cheng, J.; Fontana, L.; Haque, R.; Ahmed, T.; Gordon, J.I. Regulators of Gut Motility Revealed by a Gnotobiotic Model of Diet-Microbiome Interactions Related to Travel. Cell 2015, 163, 95–107. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Bäckhed, F.; Fulton, L.; Gordon, J.I. Diet-Induced Obesity Is Linked to Marked but Reversible Alterations in the Mouse Distal Gut Microbiome. Cell Host Microbe 2008, 3, 213–223. [Google Scholar] [CrossRef]
- Finelli, C.; Padula, M.C.; Martelli, G.; Tarantino, G. Could the improvement of obesity-related co-morbidities depend on modified gut hormones secretion? World J. Gastroenterol. 2014, 20, 16649–16664. [Google Scholar] [CrossRef]
- Worthmann, A.; John, C.; Rühlemann, M.C.; Baguhl, M.; Heinsen, F.-A.; Schaltenberg, N.; Heine, M.; Schlein, C.; Evangelakos, I.; Mineo, C.; et al. Cold-induced conversion of cholesterol to bile acids in mice shapes the gut microbiome and promotes adaptive thermogenesis. Nat. Med. 2017, 23, 839–849. [Google Scholar] [CrossRef]
- Wahlström, A.; Sayin, S.I.; Marschall, H.-U.; Bäckhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B.; Bajaj, J.S. Bile Acids and the Gut Microbiome. Curr. Opin. Gastroenterol. 2014, 30, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, D.R.; Garge, N.; Zhang, X.; Sun, W.; O’Connell, T.M.; Bunger, M.K.; Bultman, S.J. The Microbiome and Butyrate Regulate Energy Metabolism and Autophagy in the Mammalian Colon. Cell Metab. 2011, 13, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate Improves Insulin Sensitivity and Increases Energy Expenditure in Mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yi, C.-X.; Katiraei, S.; Kooijman, S.; Zhou, E.; Chung, C.K.; Gao, Y.; van den Heuvel, J.K.; Meijer, O.C.; Berbée, J.F.P.; et al. Butyrate reduces appetite and activates brown adipose tissue via the gut-brain neural circuit. Gut 2018, 67, 1269–1279. [Google Scholar] [CrossRef] [PubMed]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef]
- Hsiao, E.Y.; McBride, S.W.; Hsien, S.; Sharon, G.; Hyde, E.R.; McCue, T.; Codelli, J.A.; Chow, J.; Reisman, S.E.; Petrosino, J.F.; et al. Microbiota Modulate Behavioral and Physiological Abnormalities Associated with Neurodevelopmental Disorders. Cell 2013, 155, 1451–1463. [Google Scholar] [CrossRef]
- Koh, A.; Molinaro, A.; Ståhlman, M.; Khan, M.T.; Schmidt, C.; Mannerås-Holm, L.; Wu, H.; Carreras, A.; Jeong, H.; Olofsson, L.E.; et al. Microbially Produced Imidazole Propionate Impairs Insulin Signaling through mTORC1. Cell 2018, 175, 947–961.e17. [Google Scholar] [CrossRef]
- Lu, X.; Solmonson, A.; Lodi, A.; Nowinski, S.M.; Sentandreu, E.; Riley, C.L.; Mills, E.M.; Tiziani, S. The early metabolomic response of adipose tissue during acute cold exposure in mice. Sci. Rep. 2017, 7, 3455. [Google Scholar] [CrossRef]
- Baskin, A.S.; Linderman, J.D.; Brychta, R.J.; McGehee, S.; Anflick-Chames, E.; Cero, C.; Johnson, J.W.; O’Mara, A.E.; Fletcher, L.A.; Leitner, B.P.; et al. Regulation of Human Adipose Tissue Activation, Gallbladder Size, and Bile Acid Metabolism by A β3-Adrenergic Receptor Agonist. Diabetes 2018, 67, 2113–2125. [Google Scholar] [CrossRef]
- Arch, J.R.S. Challenges in β3-adrenoceptor agonist drug development. Ther. Adv. Endocrinol. Metab. 2011, 2, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2014, 30, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Rideout, J.R.; He, Y.; Navas-Molina, J.A.; Walters, W.A.; Ursell, L.K.; Gibbons, S.M.; Chase, J.; McDonald, D.; Gonzalez, A.; Robbins-Pianka, A.; et al. Subsampled open-reference clustering creates consistent, comprehensive OTU definitions and scales to billions of sequences. PeerJ 2014, 2, e545. [Google Scholar] [CrossRef]
- Knights, D.; Kuczynski, J.; Charlson, E.S.; Zaneveld, J.; Mozer, M.C.; Collman, R.G.; Bushman, F.D.; Knight, R.T.; Kelley, S.T. Bayesian community-wide culture-independent microbial source tracking. Nat. Methods 2011, 8, 761–763. [Google Scholar] [CrossRef]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef]
- Mercer, K.E.; Yeruva, L.; Pack, L.; Graham, J.L.; Stanhope, K.L.; Chintapalli, S.V.; Wankhade, U.D.; Shankar, K.; Havel, P.J.; Adams, S.H.; et al. Xenometabolite signatures in the UC Davis type 2 diabetes mellitus rat model revealed using a metabolomics platform enriched with microbe-derived metabolites. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 319, G157–G169. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, B.D.; Mercer, K.E.; Bhattacharyya, S.; Bowlin, A.K.; Saraf, M.K.; Pack, L.; Chintapalli, S.V.; Shankar, K.; Adams, S.; Badger, T.M.; et al. Early Postnatal Diets Affect the Bioregional Small Intestine Microbiome and Ileal Metabolome in Neonatal Pigs. J. Nutr. 2017, 147, 1499–1509. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Konietschke, F.; Placzek, M.; Schaarschmidt, F.; Hothorn, L.A. nparcomp: An R Software Package for Nonparametric Multiple Comparisons and Simultaneous Confidence Intervals. J. Stat. Softw. 2015, 64, 1–17. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. R Package Version 2.5-3. 2018. Available online: https://cran.r-project.org/web/packages/vegan/index.html (accessed on 1 May 2022).
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.-É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the Gap between Raw Spectra and Functional Insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef] [PubMed]
- Jewison, T.; Su, Y.; Disfany, F.M.; Liang, Y.; Knox, C.; Maciejewski, A.; Poelzer, J.; Huynh, J.; Zhou, Y.; Arndt, D.; et al. SMPDB 2.0: Big Improvements to the Small Molecule Pathway Database. Nucleic Acids Res. 2014, 42, D478–D484. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Level | Index | Treatment 1 | p-Value 2 | |||||
|---|---|---|---|---|---|---|---|---|
| CCV | CCCL | HCV | HCCL | Maternal | CL316,243 | Interaction | ||
| Phylum | Observed OTUs | 5.80 ± 0.37 | 6.00 ± 0.55 | 6.00 ± 0.01 | 5.75 ± 0.25 | 0.95 | 1.00 | 0.58 |
| Pielou’s evenness | 0.72 ± 0.04 | 0.76 ± 0.03 | 0.81 ± 0.03 | 0.76 ± 0.02 | 0.14 | 0.92 | 0.14 | |
| Shannon index | 1.81 ± 0.09 | 1.95 ± 0.11 | 2.10 ± 0.06 | 1.92 ± 0.01 | 0.15 | 0.96 | 0.09 | |
| Genus | Observed OTUs | 21.8 ± 0.97 | 23.0 ± 1.18 | 21.3 ± 0.85 | 23.5 ± 0.29 | 0.98 | 0.10 | 0.59 |
| Pielou’s evenness | 0.57 ± 0.01 | 0.61 ± 0.02 | 0.60 ± 0.01 | 0.59 ± 0.01 | 0.48 | 0.17 | 0.06 | |
| Shannon index | 2.51 ± 0.07 | 2.76 ± 0.10 | 2.66 ± 0.04 | 2.70 ± 0.02 | 0.52 | 0.04 | 0.14 | |
| Treatment 1 | ||||
|---|---|---|---|---|
| Level | Index | 8 °C | 30 °C | p-Value 2 |
| Phylum | Observed OTUs | 4.60 ± 0.24 | 5.00 ± 0.01 | 0.14 |
| Shannon index | 0.71 ± 0.07 | 1.09 ± 0.14 | 0.04 | |
| Pielou’s evenness | 0.32 ± 0.03 | 0.47 ± 0.06 | 0.06 | |
| Genus | Observed OTUs | 26.4 ± 0.51 | 26.6 ± 1.33 | 0.89 |
| Shannon index | 2.70 ± 0.23 | 2.80 ± 0.17 | 0.74 | |
| Pielou’s evenness | 0.57 ± 0.05 | 0.59 ± 0.03 | 0.73 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paz, H.A.; Pilkington, A.-C.; Zhong, Y.; Chintapalli, S.V.; Sikes, J.; Lan, R.S.; Shankar, K.; Wankhade, U.D. Gut Microbiome and Metabolome Modulation by Maternal High-Fat Diet and Thermogenic Challenge. Int. J. Mol. Sci. 2022, 23, 9658. https://doi.org/10.3390/ijms23179658
Paz HA, Pilkington A-C, Zhong Y, Chintapalli SV, Sikes J, Lan RS, Shankar K, Wankhade UD. Gut Microbiome and Metabolome Modulation by Maternal High-Fat Diet and Thermogenic Challenge. International Journal of Molecular Sciences. 2022; 23(17):9658. https://doi.org/10.3390/ijms23179658
Chicago/Turabian StylePaz, Henry A., Anna-Claire Pilkington, Ying Zhong, Sree V. Chintapalli, James Sikes, Renny S. Lan, Kartik Shankar, and Umesh D. Wankhade. 2022. "Gut Microbiome and Metabolome Modulation by Maternal High-Fat Diet and Thermogenic Challenge" International Journal of Molecular Sciences 23, no. 17: 9658. https://doi.org/10.3390/ijms23179658