
Iron Metabolism and Ferroptosis in Physiological and Pathological Pregnancy

Abstract
:1. Introduction
2. Brief Understanding of Physiological Iron Metabolism
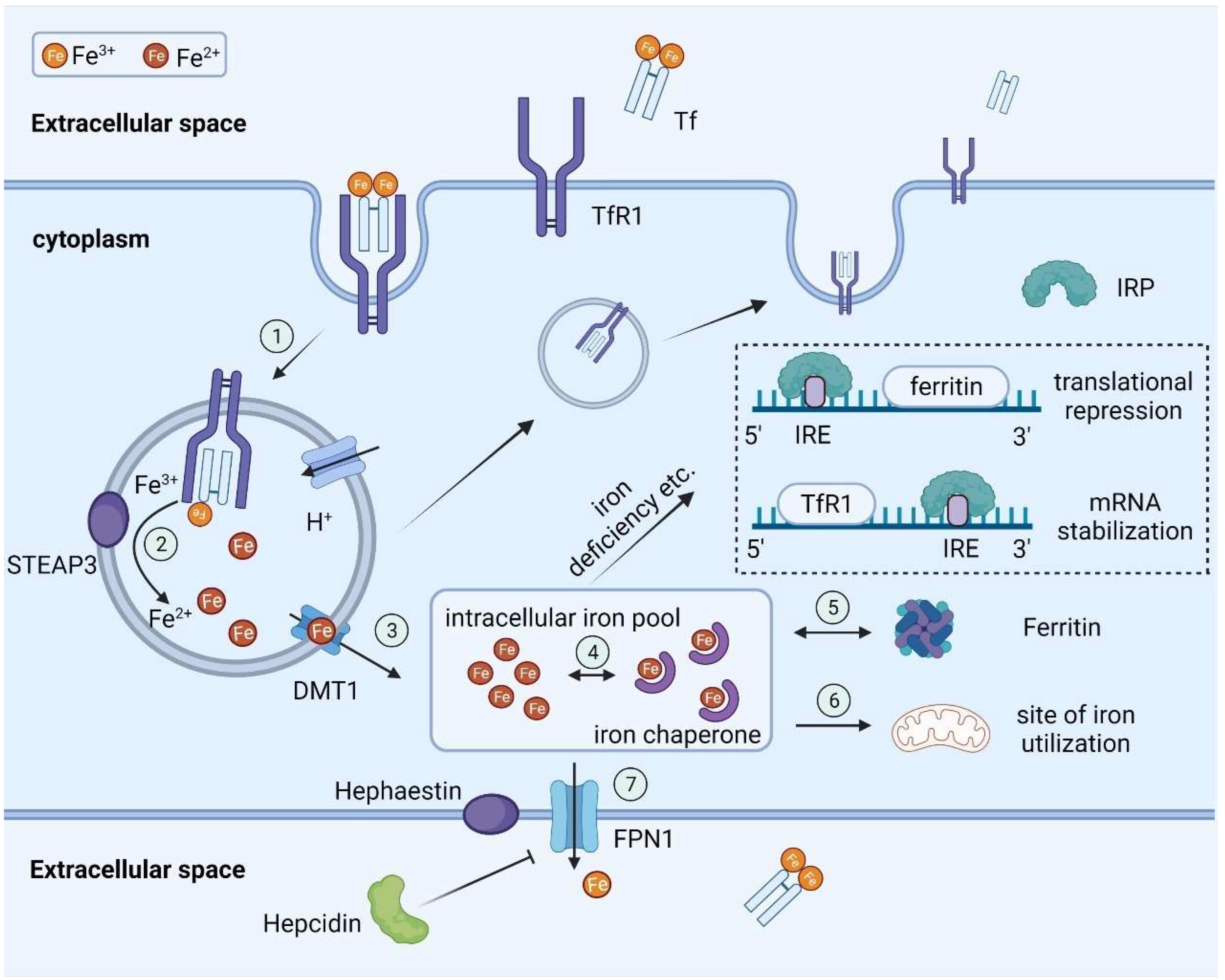
2.1. Basic Steps in Systemic Iron Transport
2.2. Regulation of Iron Homeostasis
3. Adaptions of Iron Metabolism in Physiological Pregnancy
3.1. Requirements of Iron during Pregnancy
3.2. Effects of Gestational Iron Deficiency on the Fetus
3.3. Iron Transport by Placenta
3.4. Regulation of Placental Iron Trafficking
4. Ferroptosis: An Iron-Related Programmed Cell Death Pathway
5. Role of Iron Metabolism and Ferroptosis in Pathological Pregnancy
5.1. In the View of Preeclampsia
5.2. In the View of Gestational Diabetes Mellitus
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
References
- Zimmermann, M.B.; Hurrell, R.F. Nutritional iron deficiency. Lancet 2007, 370, 511–520. [Google Scholar] [CrossRef]
- Cetin, I.; Berti, C.; Calabrese, S. Role of micronutrients in the periconceptional period. Hum. Reprod. Update 2009, 16, 80–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maltepe, E.; Fisher, S.J. Placenta: The Forgotten Organ. Annu. Rev. Cell Dev. Biol. 2015, 31, 523–552. [Google Scholar] [CrossRef] [PubMed]
- Sangkhae, V.; Fisher, A.L.; Chua, K.J.; Ruchala, P.; Ganz, T.; Nemeth, E. Maternal hepcidin determines embryo iron homeostasis in mice. Blood 2020, 136, 2206–2216. [Google Scholar] [CrossRef] [PubMed]
- Sangkhae, V.; Fisher, A.L.; Wong, S.; Koenig, M.D.; Tussing-Humphreys, L.; Chu, A.; Lelić, M.; Ganz, T.; Nemeth, E. Effects of maternal iron status on placental and fetal iron homeostasis. J. Clin. Investig. 2019, 130, 625–640. [Google Scholar] [CrossRef]
- Georgieff, M.K.; Krebs, N.F.; Cusick, S.E. The Benefits and Risks of Iron Supplementation in Pregnancy and Childhood. Annu. Rev. Nutr. 2019, 39, 121–146. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2020, 31, 107–125. [Google Scholar] [CrossRef]
- Gunshin, H.; MacKenzie, B.; Berger, U.V.; Gunshin, Y.; Romero, M.F.; Boron, W.F.; Nussberger, S.; Gollan, J.L.; Hediger, M.A. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 1997, 388, 482–488. [Google Scholar] [CrossRef]
- Canonne-Hergaux, F.; Zhang, A.-S.; Ponka, P.; Gros, P. Characterization of the iron transporter DMT1 (NRAMP2/DCT1) in red blood cells of normal and anemic mk/mkmice. Blood 2001, 98, 3823–3830. [Google Scholar] [CrossRef] [Green Version]
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005, 1, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Donovan, A.; Brownlie, A.; Zhou, Y.; Shepard, J.; Pratt, S.J.; Moynihan, J.; Paw, B.H.; Drejer, A.; Barut, B.; Zapata, A.; et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 2000, 403, 776–781. [Google Scholar] [CrossRef] [PubMed]
- Huebers, H.A.; Finch, C.A. The physiology of transferrin and transferrin receptors. Physiol. Rev. 1987, 67, 520–582. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zak, O.; Aisen, P.; Harrison, S.C.; Walz, T. Structure of the Human Transferrin Receptor-Transferrin Complex. Cell 2004, 116, 565–576. [Google Scholar] [CrossRef] [Green Version]
- Torti, F.M.; Torti, S.V. Regulation of ferritin genes and protein. Blood 2002, 99, 3505–3516. [Google Scholar] [CrossRef] [Green Version]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.V.; Ganz, T.; Kaplan, J. Hepcidin Regulates Cellular Iron Efflux by Binding to Ferroportin and Inducing Its Internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [Green Version]
- Drakesmith, H.; Nemeth, E.; Ganz, T. Ironing out Ferroportin. Cell Metab. 2015, 22, 777–787. [Google Scholar] [CrossRef] [Green Version]
- McKie, A.T.; Barrow, D.; Latunde-Dada, G.O.; Rolfs, A.; Sager, G.; Mudaly, E.; Mudaly, M.; Richardson, C.; Barlow, D.; Bomford, A.; et al. An Iron-Regulated Ferric Reductase Associated with the Absorption of Dietary Iron. Science 2001, 291, 1755–1759. [Google Scholar] [CrossRef]
- Yeh, K.; Yeh, M.; Glass, J. Interactions Between Ferroportin and Hephaestin in Rat Enterocytes Are Reduced After Iron Ingestion. Gastroenterology 2011, 141, 292–299.e1. [Google Scholar] [CrossRef]
- Lopez, A.; Cacoub, P.; Macdougall, I.C.; Peyrin-Biroulet, L. Iron deficiency anaemia. Lancet 2015, 387, 907–916. [Google Scholar] [CrossRef]
- Ohgami, R.S.; Campagna, D.R.; McDonald, A.; Fleming, M.D. The Steap proteins are metalloreductases. Blood 2006, 108, 1388–1394. [Google Scholar] [CrossRef]
- Bartnikas, T.B.; Andrews, N.C.; Fleming, M.D. Transferrin is a major determinant of hepcidin expression in hypotransferrinemic mice. Blood 2011, 117, 630–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Bencze, K.Z.; Stemmler, T.L.; Philpott, C.C. A Cytosolic Iron Chaperone That Delivers Iron to Ferritin. Science 2008, 320, 1207–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanoaica, L.; Darshan, D.; Richman, L.; Schümann, K.; Kühn, L.C. Intestinal Ferritin H Is Required for an Accurate Control of Iron Absorption. Cell Metab. 2010, 12, 273–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drakesmith, H.; Prentice, A.M.; Parducci, L.; Edwards, M.E.; Bennett, K.D.; Alm, T.; Elverland, E.; Tollefsrud, M.M.; Jørgensen, T.; Houmark-Nielsen, M.; et al. Hepcidin and the Iron-Infection Axis. Science 2012, 338, 768–772. [Google Scholar] [CrossRef] [Green Version]
- Billesbølle, C.B.; Azumaya, C.M.; Kretsch, R.C.; Powers, A.S.; Gonen, S.; Schneider, S.; Arvedson, T.; Dror, R.O.; Cheng, Y.; Manglik, A. Structure of hepcidin-bound ferroportin reveals iron homeostatic mechanisms. Nature 2020, 586, 807–811. [Google Scholar] [CrossRef]
- Słomka, A.; Korbal, P.; Piekus, N.; Żekanowska, E. The use of cluster and principal component analysis in the estimation of iron status in term newborns. J. Matern. Neonatal Med. 2012, 26, 482–486. [Google Scholar] [CrossRef]
- Anderson, G.J.; Frazer, D.M. Current understanding of iron homeostasis. Am. J. Clin. Nutr. 2017, 106 (Suppl. 6), 1559S–1566S. [Google Scholar] [CrossRef] [Green Version]
- Shah, Y.M.; Matsubara, T.; Ito, S.; Yim, S.-H.; Gonzalez, F.J. Intestinal Hypoxia-Inducible Transcription Factors Are Essential for Iron Absorption following Iron Deficiency. Cell Metab. 2009, 9, 152–164. [Google Scholar] [CrossRef] [Green Version]
- Fisher, A.; Nemeth, E. Iron homeostasis during pregnancy. Am. J. Clin. Nutr. 2017, 106 (Suppl. 6), 1567S–1574S. [Google Scholar] [CrossRef]
- Georgieff, M.K. Iron deficiency in pregnancy. Am. J. Obstet. Gynecol. 2020, 223, 516–524. [Google Scholar] [CrossRef]
- Dewey, K.G.; Oaks, B.M. U-shaped curve for risk associated with maternal hemoglobin, iron status, or iron supplementation. Am. J. Clin. Nutr. 2017, 106, 1694S–1702S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.-H.; Chen, W.-H.; Su, C.-H.; Yu, H.-R.; Tain, Y.-L.; Huang, L.-T.; Sheen, J.-M. Maternal Iron Deficiency Programs Rat Offspring Hypertension in Relation to Renin—Angiotensin System and Oxidative Stress. Int. J. Mol. Sci. 2022, 23, 8294. [Google Scholar] [CrossRef] [PubMed]
- Monk, C.; Georgieff, M.K.; Xu, N.; Hao, X.; Bansal, R.; Gustafsson, H.; Spicer, J.; Peterson, B.S. Maternal prenatal iron status and tissue organization in the neonatal brain. Pediatr. Res. 2015, 79, 482–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCann, S.; Amadó, M.P.; Moore, S.E. The Role of Iron in Brain Development: A Systematic Review. Nutrients 2020, 12, 2001. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.V.; Kennedy, B.C.; Pisansky, M.T.; Won, K.-J.; Gewirtz, J.C.; Simmons, R.; Georgieff, M.K. Prenatal Choline Supplementation Diminishes Early-Life Iron Deficiency–Induced Reprogramming of Molecular Networks Associated with Behavioral Abnormalities in the Adult Rat Hippocampus. J. Nutr. 2016, 146, 484–493. [Google Scholar] [CrossRef] [Green Version]
- Radlowski, E.C.; Johnson, R.W. Perinatal iron deficiency and neurocognitive development. Front. Hum. Neurosci. 2013, 7, 585. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Ma, L.; Li, H.; Wang, X.; Wu, M.; Jing, J.; Chen, X.; Lan, R.; Tang, W.; Zhu, Y. Iron Supplementation Is Associated with Improvement of Motor Development, Hemoglobin Level, and Weight in Preterm Infants during the First Year of Life in China. Nutrients 2022, 14, 2624. [Google Scholar] [CrossRef]
- Cao, C.; Fleming, M.D. Localization and Kinetics of the Transferrin-Dependent Iron Transport Machinery in the Mouse Placenta. Curr. Dev. Nutr. 2021, 5, nzab025. [Google Scholar] [CrossRef]
- Bastin, J.; Drakesmith, H.; Rees, M.; Sargent, I.; Townsend, A. Localisation of proteins of iron metabolism in the human placenta and liver. Br. J. Haematol. 2006, 134, 532–543. [Google Scholar] [CrossRef]
- Chong, W.; Kwan, P.; Chan, L.; Chiu, P.; Cheung, T.; Lau, T.K. Expression of divalent metal transporter 1 (DMT1) isoforms in first trimester human placenta and embryonic tissues. Hum. Reprod. 2005, 20, 3532–3538. [Google Scholar] [CrossRef] [Green Version]
- Lakhal-Littleton, S. Advances in understanding the crosstalk between mother and fetus on iron utilization. Semin. Hematol. 2021, 58, 153–160. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, K.O. Maternal, fetal and placental regulation of placental iron trafficking. Placenta 2022, 125, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.E.; Jin, O.; Fujiwara, Y.; Kuo, F.; Andrews, N. Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nat. Genet. 1999, 21, 396–399. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-Q.; Bai, B.; Cao, X.-X.; Zhang, Y.-H.; Yan, H.; Zheng, Q.-Q.; Zhuang, G.-H. Divalent Metal Transporter 1 Expression and Regulation in Human Placenta. Biol. Trace Element Res. 2011, 146, 6–12. [Google Scholar] [CrossRef]
- Gunshin, H.; Fujiwara, Y.; Custodio, A.O.; DiRenzo, C.; Robine, S.; Andrews, N.C. Slc11a2 is required for intestinal iron absorption and erythropoiesis but dispensable in placenta and liver. J. Clin. Investig. 2005, 115, 1258–1266. [Google Scholar] [CrossRef] [Green Version]
- Hojyo, S.; Fukada, T.; Shimoda, S.; Ohashi, W.; Bin, B.-H.; Koseki, H.; Hirano, T. The Zinc Transporter SLC39A14/ZIP14 Controls G-Protein Coupled Receptor-Mediated Signaling Required for Systemic Growth. PLoS ONE 2011, 6, e18059. [Google Scholar] [CrossRef] [Green Version]
- Gálvez-Peralta, M.; He, L.; Jorge-Nebert, L.F.; Wang, B.; Miller, M.L.; Eppert, B.L.; Afton, S.; Nebert, D.W. ZIP8 Zinc Transporter: Indispensable Role for Both Multiple-Organ Organogenesis and Hematopoiesis In Utero. PLoS ONE 2012, 7, e36055. [Google Scholar] [CrossRef] [Green Version]
- Fuqua, B.K.; Lu, Y.; Darshan, D.; Frazer, D.M.; Wilkins, S.J.; Wolkow, N.; Bell, A.G.; Hsu, J.; Yu, C.C.; Chen, H.; et al. The Multicopper Ferroxidase Hephaestin Enhances Intestinal Iron Absorption in Mice. PLoS ONE 2014, 9, e98792. [Google Scholar] [CrossRef] [Green Version]
- Vulpe, C.; Kuo, Y.-M.; Murphy, T.L.; Cowley, L.; Askwith, C.; Libina, N.; Gitschier, J.; Anderson, G. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat. Genet. 1999, 21, 195–199. [Google Scholar] [CrossRef]
- Guller, S.; Buhimschi, C.S.; Ma, Y.Y.; Huang, S.T.J.; Yang, L.; Kuczynski, E.; Zambrano, E.; Lockwood, C.J.; Buhimschi, I.A. Placental expression of ceruloplasmin in pregnancies complicated by severe preeclampsia. Lab. Investig. 2008, 88, 1057–1067. [Google Scholar] [CrossRef] [Green Version]
- Harris, Z.L.; Durley, A.P.; Man, T.K.; Gitlin, J.D. Targeted gene disruption reveals an essential role for ceruloplasmin in cellular iron efflux. Proc. Natl. Acad. Sci. USA 1999, 96, 10812–10817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenawi, M.; Rouger, E.; Island, M.; Leroyer, P.; Robin, F.; Rémy, S.; Tesson, L.; Anegon, I.; Nay, K.; Derbré, F.; et al. Ceruloplasmin deficiency does not induce macrophagic iron overload: Lessons from a new rat model of hereditary aceruloplasminemia. FASEB J. 2019, 33, 13492–13502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surekha, M.V.; Sujatha, T.; Gadhiraju, S.; Kumar, P.U.; Kotturu, S.K.; Sharada, K.; Bhaskar, V. Impact of maternal iron deficiency anaemia on the expression of the newly discovered multi-copper ferroxidase, Zyklopen, in term placentas. J. Obstet. Gynaecol. 2021, 42, 74–82. [Google Scholar] [CrossRef]
- Helman, S.L.; Wilkins, S.J.; McKeating, D.R.; Perkins, A.V.; Whibley, P.E.; Cuffe, J.S.M.; Simmons, D.G.; Fuqua, B.K.; Vulpe, C.D.; Wallace, D.F.; et al. The Placental Ferroxidase Zyklopen Is Not Essential for Iron Transport to the Fetus in Mice. J. Nutr. 2021, 151, 2541–2550. [Google Scholar] [CrossRef]
- Keel, S.B.; Doty, R.T.; Yang, Z.; Quigley, J.G.; Chen, J.; Knoblaugh, S.; Kingsley, P.D.; De Domenico, I.; Vaughn, M.B.; Kaplan, J.; et al. A Heme Export Protein Is Required for Red Blood Cell Differentiation and Iron Homeostasis. Science 2008, 319, 825–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlaming, M.L.; Lagas, J.S.; Schinkel, A.H. Physiological and pharmacological roles of ABCG2 (BCRP): Recent findings in Abcg2 knockout mice. Adv. Drug Deliv. Rev. 2009, 61, 14–25. [Google Scholar] [CrossRef]
- Jonker, J.W.; Buitelaar, M.; Wagenaar, E.; van der Valk, M.A.; Scheffer, G.L.; Scheper, R.J.; Plösch, T.; Kuipers, F.; Elferink, R.P.J.O.; Rosing, H.; et al. The breast cancer resistance protein protects against a major chlorophyll-derived dietary phototoxin and protoporphyria. Proc. Natl. Acad. Sci. USA 2002, 99, 15649–15654. [Google Scholar] [CrossRef] [Green Version]
- Herz, J.; Clouthier, D.E.; Hammer, R.E. LDL receptor-related protein internalizes and degrades uPA-PAI-1 complexes and is essential for embryo implantation. Cell 1992, 71, 411–421. [Google Scholar] [CrossRef]
- Inoue, R.; Irie, Y.; Akagi, R. Role of heme oxygenase-1 in human placenta on iron supply to fetus. Placenta 2020, 103, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wong, R.; Kalish, F.; Nayak, N.; Stevenson, D. Effect of Heme Oxygenase-1 Deficiency on Placental Development. Placenta 2009, 30, 861–868. [Google Scholar] [CrossRef] [Green Version]
- Liuzzi, J.P.; Aydemir, F.; Nam, H.; Knutson, M.D.; Cousins, R.J. Zip14 (Slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13612–13617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, N.; Gao, J.; Enns, C.A.; Knutson, M.D. ZRT/IRT-like Protein 14 (ZIP14) Promotes the Cellular Assimilation of Iron from Transferrin. J. Biol. Chem. 2010, 285, 32141–32150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, Y.; Ohba, K.-I.; Ohta, H. Participation of metal transporters in cadmium transport from mother rat to fetus. J. Toxicol. Sci. 2012, 37, 1035–1044. [Google Scholar] [CrossRef] [Green Version]
- Nam, H.; Wang, C.-Y.; Zhang, L.; Zhang, W.; Hojyo, S.; Fukada, T.; Knutson, M.D. ZIP14 and DMT1 in the liver, pancreas, and heart are differentially regulated by iron deficiency and overload: Implications for tissue iron uptake in iron-related disorders. Haematologica 2013, 98, 1049–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.Y.; Jenkitkasemwong, S.; Duarte, S.; Sparkman, B.K.; Shawki, A.; Mackenzie, B.; Knutson, M.D. ZIP8 Is an Iron and Zinc Transporter Whose Cell-surface Expression Is Up-regulated by Cellular Iron Loading. J. Biol. Chem. 2012, 287, 34032–34043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; He, L.; Dong, H.; Dalton, T.P.; Nebert, D.W. Generation of a Slc39a8 hypomorph mouse: Markedly decreased ZIP8 Zn2+/(HCO3-)2 transporter expression. Biochem. Biophys. Res. Commun. 2011, 410, 289–294. [Google Scholar] [CrossRef] [Green Version]
- Vashchenko, G.; MacGillivray, R.T.A. Multi-Copper Oxidases and Human Iron Metabolism. Nutrients 2013, 5, 2289–2313. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Attieh, Z.K.; Syed, B.A.; Kuo, Y.; Stevens, V.; Fuqua, B.K.; Andersen, H.S.; Naylor, C.E.; Evans, R.W.; Gambling, L.; et al. Identification of Zyklopen, a New Member of the Vertebrate Multicopper Ferroxidase Family, and Characterization in Rodents and Human Cells. J. Nutr. 2010, 140, 1728–1735. [Google Scholar] [CrossRef] [Green Version]
- Jenkitkasemwong, S.; Wang, C.-Y.; Coffey, R.; Zhang, W.; Chan, A.; Biel, T.; Kim, J.-S.; Hojyo, S.; Fukada, T.; Knutson, M.D. SLC39A14 Is Required for the Development of Hepatocellular Iron Overload in Murine Models of Hereditary Hemochromatosis. Cell Metab. 2015, 22, 138–150. [Google Scholar] [CrossRef] [Green Version]
- Knutson, M.D. Non-transferrin-bound iron transporters. Free Radic. Biol. Med. 2018, 133, 101–111. [Google Scholar] [CrossRef]
- Ji, C.; Kosman, D.J. Molecular mechanisms of non-transferrin-bound and transferring-bound iron uptake in primary hippocampal neurons. J. Neurochem. 2015, 133, 668–683. [Google Scholar] [CrossRef] [Green Version]
- Martini, S.; Austin, T.; Aceti, A.; Faldella, G.; Corvaglia, L. Free radicals and neonatal encephalopathy: Mechanisms of injury, biomarkers, and antioxidant treatment perspectives. Pediatr. Res. 2019, 87, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Thornburg, K.L.; Faber, J.J. The steady state concentration gradients of an electron-dense marker (ferritin in the three-layered hemochorial placenta of the rabbit. J. Clin. Investig. 1976, 58, 912–925. [Google Scholar] [CrossRef] [PubMed]
- Lamparelli, R.D.V.; Friedman, B.M.; Macphail, A.P.; Bothwell, T.H.; Phillips, J.I.; Baynes, R.D. The fate of intravenously injected tissue ferritin in pregnant guinea-pigs. Br. J. Haematol. 1989, 72, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.J.; Johnson, P.M.; Ogbimi, A.O.; Tappin, J.A. Characterization and localization of human placental ferritin. Biochem. J. 1979, 182, 763–769. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Fang, C.J.; Ryan, J.C.; Niemi, E.C.; Lebrón, J.A.; Björkman, P.J.; Arase, H.; Torti, F.M.; Torti, S.V.; Nakamura, M.C.; et al. Binding and uptake of H-ferritin are mediated by human transferrin receptor-1. Proc. Natl. Acad. Sci. USA 2010, 107, 3505–3510. [Google Scholar] [CrossRef] [Green Version]
- Li, J.Y.; Paragas, N.; Ned, R.M.; Qiu, A.; Viltard, M.; Leete, T.; Drexler, I.R.; Chen, X.; Sanna-Cherchi, S.; Mohammed, F.; et al. Scara5 Is a Ferritin Receptor Mediating Non-Transferrin Iron Delivery. Dev. Cell 2009, 16, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Jaacks, L.M.; Young, M.F.; Essley, B.V.; McNanley, T.J.; Cooper, E.M.; Pressman, E.K.; McIntyre, A.W.; Orlando, M.S.; Abkowitz, J.L.; Guillet, R.; et al. Placental Expression of the Heme Transporter, Feline Leukemia Virus Subgroup C Receptor, Is related to Maternal Iron Status in Pregnant Adolescents. J. Nutr. 2011, 141, 1267–1272. [Google Scholar] [CrossRef] [Green Version]
- Cao, C.; Pressman, E.K.; Cooper, E.M.; Guillet, R.; Westerman, M.; O’Brien, K.O. Placental heme receptor LRP1 correlates with the heme exporter FLVCR1 and neonatal iron status. Reproduction 2014, 148, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Best, C.M.; Pressman, E.K.; Cao, C.; Cooper, E.; Guillet, R.; Yost, O.L.; Galati, J.; Kent, T.R.; O’Brien, K.O. Maternal iron status during pregnancy compared with neonatal iron status better predicts placental iron transporter expression in humans. FASEB J. 2016, 30, 3541–3550. [Google Scholar] [CrossRef] [Green Version]
- Gambling, L.; Danzeisen, R.; Gair, S.; Lea, R.G.; Charania, Z.; Solanky, N.; Joory, K.D.; Srai, S.K.S.; McArdle, H.J. Effect of iron deficiency on placental transfer of iron and expression of iron transport proteins in vivo and in vitro. Biochem. J. 2001, 356, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-Q.; Yan, H.; Bai, B. Change in iron transporter expression in human term placenta with different maternal iron status. Eur. J. Obstet. Gynecol. Reprod. Biol. 2008, 140, 48–54. [Google Scholar] [CrossRef]
- Muckenthaler, M.U.; Galy, B.; Hentze, M.W. Systemic Iron Homeostasis and the Iron-Responsive Element/Iron-Regulatory Protein (IRE/IRP) Regulatory Network. Annu. Rev. Nutr. 2008, 28, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Yoshinaga, M.; Nakatsuka, Y.; Vandenbon, A.; Ori, D.; Uehata, T.; Tsujimura, T.; Suzuki, Y.; Mino, T.; Takeuchi, O. Regnase-1 Maintains Iron Homeostasis via the Degradation of Transferrin Receptor 1 and Prolyl-Hydroxylase-Domain-Containing Protein 3 mRNAs. Cell Rep. 2017, 19, 1614–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milman, N. Iron and pregnancy—A delicate balance. Ann. Hematol. 2006, 85, 559–565. [Google Scholar] [CrossRef]
- Young, M.F.; Griffin, I.; Pressman, E.; McIntyre, A.W.; Cooper, E.; McNanley, T.; Harris, Z.L.; Westerman, M.; O’Brien, K.O. Maternal Hepcidin Is Associated with Placental Transfer of Iron Derived from Dietary Heme and Nonheme Sources. J. Nutr. 2011, 142, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Delaney, K.M.; Cao, C.; Guillet, R.; Pressman, E.K.; O’Brien, K.O. Fetal iron uptake from recent maternal diet and the maternal RBC iron pool. Am. J. Clin. Nutr. 2022, 115, 1069–1079. [Google Scholar] [CrossRef]
- Sangkhae, V.; Nemeth, E. Regulation of the Iron Homeostatic Hormone Hepcidin. Adv. Nutr. Int. Rev. J. 2017, 8, 126–136. [Google Scholar] [CrossRef]
- Van Santen, S.; Kroot, J.J.; Zijderveld, G.; Wiegerinck, E.T.; Spaanderman, M.E.; Swinkels, D.W. The iron regulatory hormone hepcidin is decreased in pregnancy: A prospective longitudinal study. Clin. Chem. Lab. Med. 2013, 51, 1395–1401. [Google Scholar] [CrossRef]
- Millard, K.N.; Frazer, D.; Wilkins, S.J.; Anderson, G. Changes in the expression of intestinal iron transport and hepatic regulatory molecules explain the enhanced iron absorption associated with pregnancy in the rat. Gut 2004, 53, 655–660. [Google Scholar] [CrossRef] [Green Version]
- Nicolas, G.; Bennoun, M.; Porteu, A.; Mativet, S.; Beaumont, C.; Grandchamp, B.; Sirito, M.; Sawadogo, M.; Kahn, A.; Vaulont, S. Severe iron deficiency anemia in transgenic mice expressing liver hepcidin. Proc. Natl. Acad. Sci. USA 2002, 99, 4596–4601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kämmerer, L.; Mohammad, G.; Wolna, M.; Robbins, P.A.; Lakhal-Littleton, S. Fetal liver hepcidin secures iron stores in utero. Blood 2020, 136, 1549–1557. [Google Scholar] [CrossRef] [PubMed]
- Mireku, M.O.; Davidson, L.L.; Boivin, M.J.; Zoumenou, R.; Massougbodji, A.; Cot, M.; Bodeau-Livinec, F. Prenatal Iron Deficiency, Neonatal Ferritin, and Infant Cognitive Function. Pediatrics 2016, 138, e20161319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, G.; Xu, G.; Zhou, M.; Jiang, Y.; Richards, B.; Clark, K.M.; Kaciroti, N.; Georgieff, M.K.; Zhang, Z.; Tardif, T.; et al. Prenatal Iron Supplementation Reduces Maternal Anemia, Iron Deficiency, and Iron Deficiency Anemia in a Randomized Clinical Trial in Rural China, but Iron Deficiency Remains Widespread in Mothers and Neonates. J. Nutr. 2015, 145, 1916–1923. [Google Scholar] [CrossRef]
- Mazgaj, R.; Lipiński, P.; Edison, E.S.; Msc, A.B.; Staroń, R.; Haberkiewicz, O.; Lenartowicz, M.; Smuda, E.; Jończy, A.; Starzyński, R.R. Marginally reduced maternal hepatic and splenic ferroportin under severe nutritional iron deficiency in pregnancy maintains systemic iron supply. Am. J. Hematol. 2021, 96, 659–670. [Google Scholar] [CrossRef]
- Zhang, D.; Hughes, R.M.; Ollivierre-Wilson, H.; Ghosh, M.C.; Rouault, T.A. A Ferroportin Transcript that Lacks an Iron-Responsive Element Enables Duodenal and Erythroid Precursor Cells to Evade Translational Repression. Cell Metab. 2009, 9, 461–473. [Google Scholar] [CrossRef] [Green Version]
- Roberts, H.; Woodman, A.G.; Baines, K.J.; Jeyarajah, M.J.; Bourque, S.L.; Renaud, S.J. Maternal Iron Deficiency Alters Trophoblast Differentiation and Placental Development in Rat Pregnancy. Endocrinology 2021, 162, bqab215. [Google Scholar] [CrossRef]
- Barad, A.; Guillet, R.; Pressman, E.K.; Katzman, P.J.; Miller, R.K.; Darrah, T.H.; O’Brien, K.O. Placental Iron Content Is Lower than Previously Estimated and Is Associated with Maternal Iron Status in Women at Greater Risk of Gestational Iron Deficiency and Anemia. J. Nutr. 2021, 152, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Yagoda, N.; Von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS–RAF–MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 865–869. [Google Scholar] [CrossRef] [Green Version]
- Imai, H.; Matsuoka, M.; Kumagai, T.; Sakamoto, T.; Koumura, T. Lipid Peroxidation-Dependent Cell Death Regulated by GPx4 and Ferroptosis. Apoptotic Non-Apoptotic Cell Death 2016, 403, 143–170. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Central Sci. 2018, 4, 387–396. [Google Scholar] [CrossRef]
- Wenzel, S.E.; Tyurina, Y.Y.; Zhao, J.; St Croix, C.M.S.; Dar, H.H.; Mao, G.; Tyurin, V.A.; Anthonymuthu, T.S.; Kapralov, A.A.; Amoscato, A.A.; et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell 2017, 171, 628–641.e26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef] [Green Version]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., III; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Hu, F.; Feng, H.; Linkermann, A.; Min, W.; Stockwell, B.R. Determination of the Subcellular Localization and Mechanism of Action of Ferrostatins in Suppressing Ferroptosis. ACS Chem. Biol. 2018, 13, 1013–1020. [Google Scholar] [CrossRef]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Li, J.; Kang, R.; Klionsky, D.J.; Tang, D. Ferroptosis: Machinery and regulation. Autophagy 2020, 17, 2054–2081. [Google Scholar] [CrossRef]
- Ives, C.W.; Sinkey, R.; Rajapreyar, I.; Tita, A.T.; Oparil, S. Preeclampsia—Pathophysiology and Clinical Presentations: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 76, 1690–1702. [Google Scholar] [CrossRef]
- Serdar, Z.; Gur, E.; Develioglu, O. Serum iron and copper status and oxidative stress in severe and mild preeclampsia. Cell Biochem. Funct. 2006, 24, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Geetha, N.S.; Bobby, Z.; Dorairajan, G.; Jacob, S.E. Increased hepcidin levels in preeclampsia: A protective mechanism against iron overload mediated oxidative stress? J. Matern. Neonatal Med. 2020, 35, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, I.A.; Jaleel, A.; Al Kadri, H.M.F.; Al Saeed, W.; Tamimi, W. Iron status parameters in preeclamptic women. Arch. Gynecol. Obstet. 2010, 284, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.J.; Iqbal, K.; Kozai, K. Hypoxia and Placental Development. Birth Defects Res. 2017, 109, 1309–1329. [Google Scholar] [CrossRef]
- Schoots, M.H.; Gordijn, S.J.; Scherjon, S.A.; van Goor, H.; Hillebrands, J.-L. Oxidative stress in placental pathology. Placenta 2018, 69, 153–161. [Google Scholar] [CrossRef]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.P.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422.e21. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Guo, D.; Gu, H.; Zhang, L.; Lv, S. Selenium and Preeclampsia: A Systematic Review and Meta-analysis. Biol. Trace Element Res. 2015, 171, 283–292. [Google Scholar] [CrossRef]
- Zhang, H.; He, Y.; Wang, J.-X.; Chen, M.-H.; Xu, J.-J.; Jiang, M.-H.; Feng, Y.-L.; Gu, Y.-F. miR-30-5p-mediated ferroptosis of trophoblasts is implicated in the pathogenesis of preeclampsia. Redox Biol. 2019, 29, 101402. [Google Scholar] [CrossRef]
- Mistry, H.D.; Wilson, V.; Ramsay, M.M.; Symonds, M.E.; Pipkin, F.B. Reduced Selenium Concentrations and Glutathione Peroxidase Activity in Preeclamptic Pregnancies. Hypertension 2008, 52, 881–888. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.; Lin, Y.; Li, J.; Liu, M.; Wang, J.; Li, X.; Liu, J.; Jia, X.; Jing, Z.; Huang, Z.; et al. Evaluation of Glutathione Peroxidase 4 role in Preeclampsia. Sci. Rep. 2016, 6, 33300. [Google Scholar] [CrossRef]
- Mistry, H.; Kurlak, L.; Williams, P.; Ramsay, M.; Symonds, M.; Pipkin, F.B. Differential expression and distribution of placental glutathione peroxidases 1, 3 and 4 in normal and preeclamptic pregnancy. Placenta 2010, 31, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Imai, H.; Hirao, F.; Sakamoto, T.; Sekine, K.; Mizukura, Y.; Saito, M.; Kitamoto, T.; Hayasaka, M.; Hanaoka, K.; Nakagawa, Y. Early embryonic lethality caused by targeted disruption of the mouse PHGPx gene. Biochem. Biophys. Res. Commun. 2003, 305, 278–286. [Google Scholar] [CrossRef]
- Beharier, O.; Tyurin, V.A.; Goff, J.P.; Guerrero-Santoro, J.; Kajiwara, K.; Chu, T.; Tyurina, Y.Y.; Croix, C.M.S.; Wallace, C.T.; Parry, S.; et al. PLA2G6 guards placental trophoblasts against ferroptotic injury. Proc. Natl. Acad. Sci. USA 2020, 117, 27319–27328. [Google Scholar] [CrossRef] [PubMed]
- Ramanadham, S.; Ali, T.; Ashley, J.; Bone, R.; Hancock, W.D.; Lei, X. Calcium-independent phospholipases A2 and their roles in biological processes and diseases. J. Lipid Res. 2015, 56, 1643–1668. [Google Scholar] [CrossRef] [Green Version]
- McIntyre, H.D.; Catalano, P.; Zhang, C.; Desoye, G.; Mathiesen, E.R.; Damm, P. Gestational diabetes mellitus. Nat. Rev. Dis. Prim. 2019, 5, 47. [Google Scholar] [CrossRef]
- Qiu, C.; Zhang, C.; Gelaye, B.; Enquobahrie, D.A.; Frederick, I.O.; Williams, M.A. Gestational Diabetes Mellitus in Relation to Maternal Dietary Heme Iron and Nonheme Iron Intake. Diabetes Care 2011, 34, 1564–1569. [Google Scholar] [CrossRef] [Green Version]
- Bowers, K.; Yeung, E.; Williams, M.A.; Qi, L.; Tobias, D.K.; Hu, F.B.; Zhang, C. A Prospective Study of Prepregnancy Dietary Iron Intake and Risk for Gestational Diabetes Mellitus. Diabetes Care 2011, 34, 1557–1563. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.; Chan, B.; Lam, K.; Tam, S.; Lao, T.; Chan, K. Iron supplement in pregnancy and development of gestational diabetes-a randomised placebo-controlled trial. BJOG 2009, 116, 789–798. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, M.; Zhong, C.; Huang, L.; Zhang, Y.; Chen, R.; Zhou, X.; Xu, S.; Li, Q.; Cui, W.; et al. Association between maternal plasma ferritin concentration, iron supplement use, and the risk of gestational diabetes: A prospective cohort study. Am. J. Clin. Nutr. 2021, 114, 1100–1106. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, S.; Zhong, C.; Li, Q.; Wu, M.; Zhang, G.; Chen, R.; Liu, C.; Wu, J.; Huang, L.; et al. Periconceptional iron supplementation and risk of gestational diabetes mellitus: A prospective cohort study. Diabetes Res. Clin. Pract. 2021, 176, 108853. [Google Scholar] [CrossRef]
- Petry, C.; Ong, K.; Hughes, I.; Dunger, D. Associations between Maternal Iron Supplementation in Pregnancy and Changes in Offspring Size at Birth Reflect Those of Multiple Micronutrient Supplementation. Nutrients 2021, 13, 2480. [Google Scholar] [CrossRef] [PubMed]
- Khambalia, A.Z.; Aimone, A.; Nagubandi, P.; Roberts, C.L.; McElduff, A.; Morris, J.M.; Powell, K.L.; Tasevski, V.; Nassar, N. High maternal iron status, dietary iron intake and iron supplement use in pregnancy and risk of gestational diabetes mellitus: A prospective study and systematic review. Diabet. Med. 2016, 33, 1211–1221. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Zhao, J.; Lu, M.; Gu, Y.; Zhu, Y.; Chen, D.; Fu, J. Expression of Hepcidin and Ferroportin in the Placenta, and Ferritin and Transferrin Receptor 1 Levels in Maternal and Umbilical Cord Blood in Pregnant Women with and without Gestational Diabetes. Int. J. Environ. Res. Public Health 2016, 13, 766. [Google Scholar] [CrossRef] [Green Version]
- Rawal, S.; Hinkle, S.; Bao, W.; Zhu, Y.; Grewal, J.; Albert, P.S.; Weir, N.L.; Tsai, M.; Zhang, C. A longitudinal study of iron status during pregnancy and the risk of gestational diabetes: Findings from a prospective, multiracial cohort. Diabetologia 2016, 60, 249–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowers, K.A.; Olsen, S.F.; Bao, W.; Halldorsson, T.I.; Strøm, M.; Zhang, C. Plasma Concentrations of Ferritin in Early Pregnancy Are Associated with Risk of Gestational Diabetes Mellitus in Women in the Danish National Birth Cohort. J. Nutr. 2016, 146, 1756–1761. [Google Scholar] [CrossRef] [Green Version]
- Khambalia, A.; Collins, C.E.; Roberts, C.L.; Morris, J.M.; Powell, K.L.; Tasevski, V.; Nassar, N. Iron deficiency in early pregnancy using serum ferritin and soluble transferrin receptor concentrations are associated with pregnancy and birth outcomes. Eur. J. Clin. Nutr. 2015, 70, 358–363. [Google Scholar] [CrossRef] [Green Version]
- Swaminathan, S.; Fonseca, V.A.; Alam, M.G.; Shah, S.V. The Role of Iron in Diabetes and Its Complications. Diabetes Care 2007, 30, 1926–1933. [Google Scholar] [CrossRef] [Green Version]
- Erbağcı, M.O.; Tuna, G.; Köse, S.; Dal-Bekar, N.E.; Akış, M.; Kant, M.; Altunyurt, S.; Işlekel, G.H. Association between early oxidative DNA damage and iron status in women with gestational diabetes mellitus. Reprod. Toxicol. 2021, 103, 171–180. [Google Scholar] [CrossRef]
- Zaugg, J.; Melhem, H.; Huang, X.; Wegner, M.; Baumann, M.; Surbek, D.; Körner, M.; Albrecht, C. Gestational diabetes mellitus affects placental iron homeostasis: Mechanism and clinical implications. FASEB J. 2020, 34, 7311–7329. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Li, M.; Li, H. High glucose suppresses the viability and proliferation of HTR-8/SVneo cells through regulation of the miR-137/PRKAA1/IL-6 axis. Int. J. Mol. Med. 2018, 42, 799–810. [Google Scholar] [CrossRef] [Green Version]
- Han, D.; Jiang, L.; Gu, X.; Huang, S.; Pang, J.; Wu, Y.; Yin, J.; Wang, J. SIRT3 deficiency is resistant to autophagy-dependent ferroptosis by inhibiting the AMPK/mTOR pathway and promoting GPX4 levels. J. Cell. Physiol. 2020, 235, 8839–8851. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Protein (Gene) | Function | Localization in Human Placenta | Disruption and Phenotype |
|---|---|---|---|
| TfR1 (TFRC) | TBI and ferritin uptake | On the apical membrane of STB [39] | Trfc−/− was embryonic lethal at E12.5; Trfc+/− mice showed severe anemia [43] |
| DMT1 (DMT1; SLC11A2) | Intracellular iron trafficking | Prominently near maternal side; rarely on fetal side; scatter staining in stroma [44] | Slc11a2−/− mice were pale at birth; not iron deficient in all tissues comparing to wildtype littermates; Slc11a2+/− mice were viable without visible abnormalities [45] |
| ZIP14 (SLC39A14) | NTBI uptake and intracellular iron trafficking | Not available | SLC39A14−/− mice were viable with growth retardation; iron relating parameters not reported [46] |
| ZIP8 (SLC39A8) | NTBI uptake and intracellular iron trafficking | Not available | SLC39A8neo/neo newborns were pale and growth was stunted with diminished iron uptake [47] |
| Hephaestin (HEPH) | Ferroxidases | Not available | Mice with global or intestine-specific knockout of Heph were viable with microcytic anemia due to reduced intestinal iron absorption [48,49] |
| Ceruloplasmin (CP) | Ferroxidases | Intervillous space [50] | Although normal at birth, Cp−/− mice showed progressive accumulation of iron in liver and spleen, but not in macrophages [51,52] |
| Zyklopen (ZP, HEPHL1) | Ferroxidases | Cytoplasm of STB [53]; maternal decidua [54] | Zp−/− showed increased placenta size with no change in fetal iron transfer [54] |
| SCARA5 (SCARA5) | Ferritin uptake | Not available | Not available |
| FLVCR1 (FLVCR1) | Heme uptake | Not available | FLVCR1−/− was embryonic lethal at E7.5 due to impaired erythropoiesis [55] |
| BCRP (ABCG2) | Heme uptake | On the apical membrane of STB [56] | ABCG2−/− mice were viable; iron relating parameters not reported [57] |
| LRP1 (LRP1) | Heme uptake | Not available | LRP1−/− was embryonic lethal at E12.5 [58] |
| HO-1 (HO1) | Heme iron metabolism | In STB and cytotrophoblasts [59] | HO1−/− decreased embryo viability; HO1+/− led to placental dysfunction; iron relating parameters not reported [60] |
| FPN (FPN, SLC40A1) | Iron export | On the basolateral membrane of STB | SLC40A1−/− causes embryo lethality before E7.5; Meox2-Cre; Fpnflox/flox mice, in which FPN was only expressed in placenta, were viable with anemia and cellular iron accumulation [10] |
| Factors | Study | Research Design | Comparison Groups | Adjusted RR/OR (95% CI) |
|---|---|---|---|---|
| Dietary heme iron intake | Qiu et al. (2011) [126] | Prospective cohort; 3158 pregnant women | Heme iron intake levels (≥1.52 vs. <0.48 mg per day) | 3.31 (1.02–10.72) |
| Bowers et al. (2011) [127] | Prospective study; 13,475 pregnant women | Median heme iron intake levels (1.60 vs. 0.66 mg per day) | 1.58 (1.21–2.08) | |
| Dietary non-heme iron intake | Qiu et al. (2011) [126] | Prospective cohort; 3158 pregnant women | Non-heme iron intake levels (≥12.98 vs. <0.10 mg per day) | 0.61 (0.31–1.18) |
| Bowers et al. (2011) [127] | Prospective study; 13,475 pregnant women | Median heme iron intake levels (45.33 vs. 7.58 mg per day) | 0.97 (0.78–1.20) | |
| Iron supple-mentation | Bowers et al. (2011) [127] | Prospective study; 13,475 pregnant women | Median Iron supplementation levels (60.00 vs. 0 mg per day) | 1.04 (0.84–1.28) |
| Chan et al. (2009) [128] | RCT; 1164 pregnant women with Hb level between 8–14 g/dl | 60 mg daily iron supplementation vs. placebo group | 1.04 (0.70–1.53) | |
| Zhang et al. (2021) [129] | Prospective cohort; 2117 pregnant women | >60 mg daily iron supplementation during the second trimester vs. non-users | 1.43 (1.06, 1.92) | |
| Zhang et al. (2021) [130] | Prospective cohort; 5101 pregnant women | >30 mg daily iron supplementation for more than 3 months vs. non-users | 1.53 (1.21–1.93) | |
| Serum ferritin | Rawal et al. (2017) [134] | Prospective case–control study; 107 women with GDM and 214 controls | Highest vs. lowest quartile of serum ferritin level | 2.43 (1.12–5.28) |
| Bowers et al. (2016) [135] | Prospective case–control study; 350 women with GDM and 349 controls | Highest vs. lowest quartile of serum ferritin level | 2.22 (1.23–4.01) | |
| Khambalia et al. (2016) [136] | Prospective cohort study; 4420 pregnant women | Serum ferritin level <12 μg/L vs. normal | 0.43 (0.23–0.78) | |
| Serum sTfR | Rawal et al. [134] | Prospective case–control study; 107 women with GDM and 214 controls | Highest vs. lowest quartile of serum sTfR level | 1.00 (0.45–2.20) |
| Bowers et al. [135] | Prospective case–control study; 350 women with GDM and 349 controls | Highest vs. lowest quartile of serum sTfR level | 1.48 (0.82–2.70) | |
| Khambalia et al. (2016) [136] | Prospective cohort study; 4420 pregnant women | Serum sTfR level >21 nmol/L vs. normal | 1.25 (0.82–1.92) | |
| Serum hepcidin | Rawal et al. (2017) [134] | Prospective case–control study; 107 women with GDM and 214 controls | Highest vs. lowest quartile of serum hepcidin level | 2.61 (1.07–6.36) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Lu, Y.; Jin, L. Iron Metabolism and Ferroptosis in Physiological and Pathological Pregnancy. Int. J. Mol. Sci. 2022, 23, 9395. https://doi.org/10.3390/ijms23169395
Zhang Y, Lu Y, Jin L. Iron Metabolism and Ferroptosis in Physiological and Pathological Pregnancy. International Journal of Molecular Sciences. 2022; 23(16):9395. https://doi.org/10.3390/ijms23169395
Chicago/Turabian StyleZhang, Yijun, Yun Lu, and Liping Jin. 2022. "Iron Metabolism and Ferroptosis in Physiological and Pathological Pregnancy" International Journal of Molecular Sciences 23, no. 16: 9395. https://doi.org/10.3390/ijms23169395