
The Role of Zinc in the Treatment of Wilson’s Disease

 , , , , and
, , , , and
Abstract
:
1. Introduction
2. Chronological Overview of Copper Disorder and WD
3. Mechanisms of Copper Toxicity
4. Different Mechanisms Postulated to Underlie Treatment
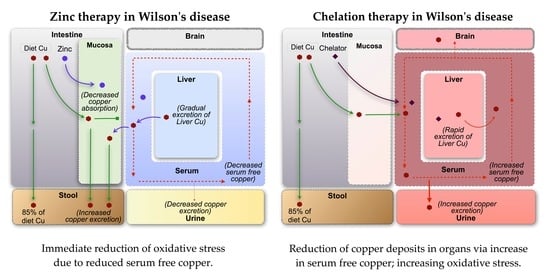
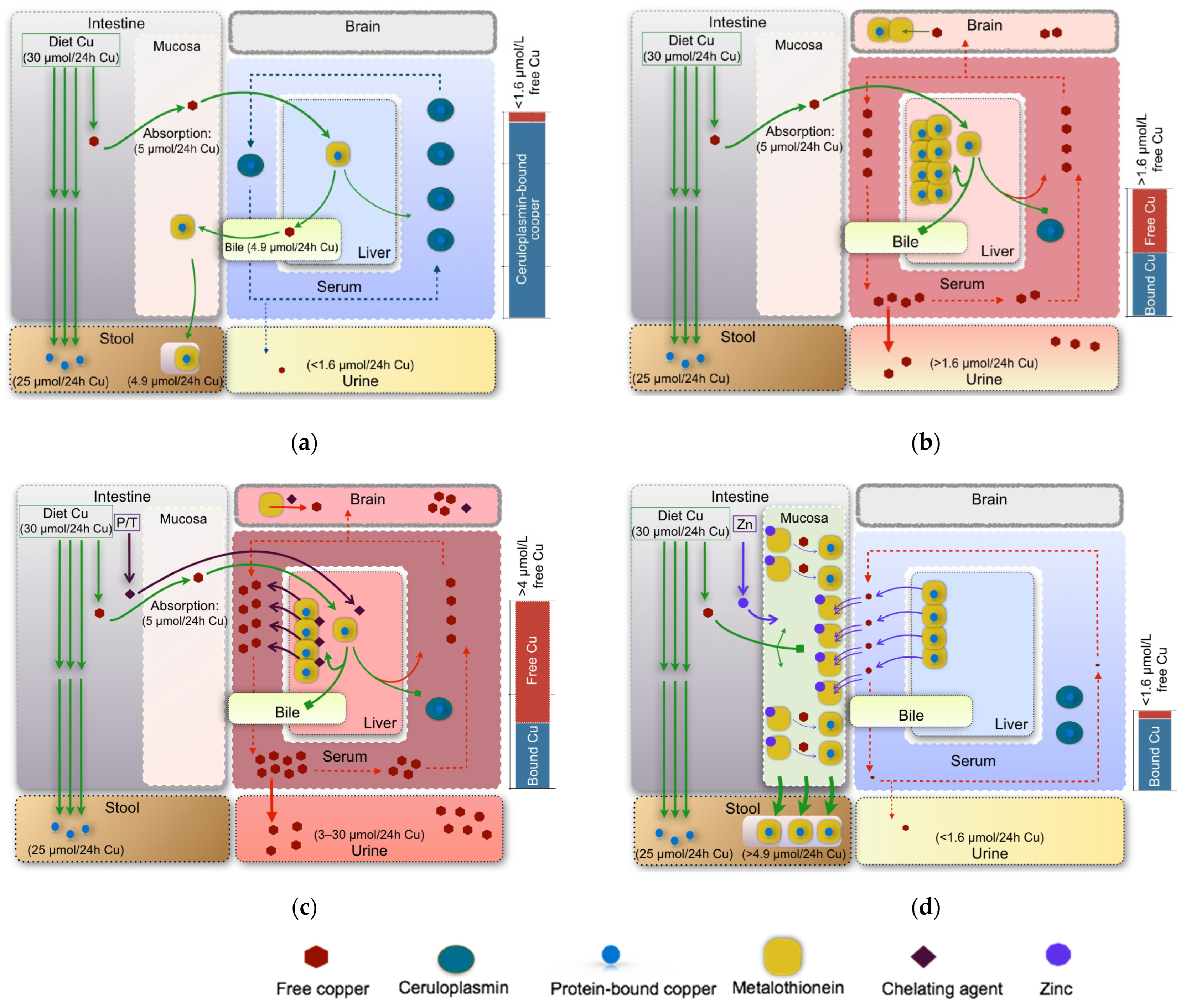
4.1. Copper Accumulation in Organs
4.2. Free Copper Intoxication/Oxidative Stress
5. The Challenge of Diagnosis
6. Effectiveness and Safety Profile of Treatment Options
7. Discussion
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Hoogenraad, T.U. Paradigm shift in treatment of Wilson’s disease: Zinc therapy now treatment of choice. Brain Dev. 2006, 28, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Shribman, S.; Poujois, A.; Bandmann, O.; Czlonkowska, A.; Warner, T.T. Wilson’s disease: Update on pathogenesis, biomarkers and treatments. J. Neurol. Neurosurg. Psychiatry 2021, 92, 1053–1061. [Google Scholar] [CrossRef] [PubMed]
- Osborn, S.B.; Walshe, J.M. Effects of penicillamine and dimercaprol on turnover of copper in patients with Wilson’s disease. Lancet 1958, 271, 70–73. [Google Scholar] [CrossRef]
- European Association for Study of Liver. EASL clinical practice guidelines: Wilson’s disease. J. Hepatol. 2012, 56, 671–685. [Google Scholar] [CrossRef]
- Dzieżyc, K.; Litwin, T.; Członkowska, A. Other organ involvement and clinical aspects of Wilson disease. Handb. Clin. Neurol. 2017, 142, 157–169. [Google Scholar] [CrossRef]
- De Cock, V.C.; Woimant, F.; Poujois, A. Sleep disorders in Wilson’s disease. Curr. Neurol. Neurosci. Rep. 2019, 19, 84. [Google Scholar] [CrossRef]
- Zheng, Z.-W.; Xu, M.-H.; Sun, C.-B.; Wu, Z.-Y.; Dong, Y. Acute-onset visual impairment in Wilson’s disease: A case report and literature review. Front. Neurol. 2022, 13, 911882. [Google Scholar] [CrossRef]
- Ferenci, P.; Stremmel, W.; Członkowska, A.; Szalay, F.; Viveiros, A.; Stättermayer, A.F.; Bruha, R.; Houwen, R.; Pop, T.L.; Stauber, R.; et al. Age and sex but not ATP7B genotype effectively influence the clinical phenotype of Wilson disease. Hepatology 2019, 69, 1464–1476. [Google Scholar] [CrossRef]
- Walshe, J.M.; Yealland, M. Wilson’s disease: The problem of delayed diagnosis. J. Neurol. Neurosurg. Psychiatry 1992, 55, 692–696. [Google Scholar] [CrossRef]
- Ferenci, P.; Ott, P. Wilson’s disease: Fatal when overlooked, curable when diagnosed. J. Hepatol. 2019, 71, 222–224. [Google Scholar] [CrossRef]
- Shribman, S.; Warner, T.T.; Dooley, J.S. Clinical presentations of Wilson disease. Ann. Transl. Med. 2019, 7, S60. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.F.; Dooley, J.S. ATP7B variant penetrance explains differences between genetic and clinical prevalence estimates for Wilson disease. Hum. Genet. 2020, 139, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Sandahl, T.D.; Laursen, T.L.; Munk, D.E.; Vilstrup, H.; Weiss, K.H.; Ott, P. The prevalence of Wilson’s disease: An update. Hepatology 2020, 71, 722–732. [Google Scholar] [CrossRef] [PubMed]
- Scheinberg, I.H.; Sternlieb, I. Wilson’s Disease; W B Saunders: Philadelphia, PA, USA, 1984. [Google Scholar]
- Yamaguchi, H.; Nagase, H.; Tokumoto, S.; Tomioka, K.; Nishiyama, M.; Takeda, H.; Ninchoji, T.; Nagano, C.; Iijima, K.; Nozu, K. Prevalence of Wilson disease based on genome databases in Japan. Pediatr. Int. 2021, 63, 918–922. [Google Scholar] [CrossRef]
- Gao, J.; Brackley, S.; Mann, J.P. The global prevalence of Wilson disease from next-generation sequencing data. Genet. Med. 2019, 21, 1155–1163. [Google Scholar] [CrossRef]
- Compston, A. Progressive lenticular degeneration: A familial nervous disease associated with cirrhosis of the liver, by S. A. Kinnier Wilson, (From the National Hospital, and the Laboratory of the National Hospital, Queen Square, London) Brain 1912: 34; 295–509. Brain 2009, 132, 1997–2001. [Google Scholar] [CrossRef]
- Cumings, J.N. THE copper and iron content of brain and liver in the normal and in hepato-lenticular degeneration. Brain 1948, 71, 410–415. [Google Scholar] [CrossRef]
- Porter, H. Copper excretion in the urine of normal individuals and of patients with hepatolenticular degeneration (Wilson’s disease). Arch. Biochem. Biophys. 1951, 31, 262–265. [Google Scholar] [CrossRef]
- Scheinberg, I.H.; Gitlin, D. Deficiency of ceruloplasmin in patients with hepatolenticular degeneration (Wilson’s disease). Science 1952, 116, 484–485. [Google Scholar] [CrossRef]
- Bearn, A.G.; Kunkel, H.G. Abnormalities of Copper Metabolism in Wilson’s Disease and Their Relationship to the Aminoaciduria. J. Clin. Investig. 1954, 33, 400–409. [Google Scholar] [CrossRef]
- Denny-Brown, D.; Porter, H. The effect of BAL (2,3-dimercaptopropanol) on hepatolenticular degeneration (Wilson’s disease). N. Engl. J. Med. 1951, 245, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Walshe, J. Wilson’s disease: New oral therapy. Lancet 1956, 267, 25–26. [Google Scholar] [CrossRef]
- Walshe, J. Management of penicillamine nephropathy in Wilson’s disease: A new chelating agent. Lancet 1969, 294, 1401–1402. [Google Scholar] [CrossRef]
- Walshe, J.M. Copper chelation in patients with Wilson’s disease. A comparison of penicillamine and triethylene tetramine dihydrochloride. Q. J. Med. 1973, 42, 441–452. [Google Scholar] [PubMed]
- Walshe, J. Treatment of Wilson’s disease with trientine (triethylene tetramine) dihydrochloride. Lancet 1982, 319, 643–647. [Google Scholar] [CrossRef]
- Walshe, J. Treatment of Wilson’s disease: The historical background. QJM Int. J. Med. 1996, 89, 553–556. [Google Scholar] [CrossRef]
- Weiss, K.H.; Kruse, C.; Manolaki, N.; Zuin, M.; Ferenci, P.; Van Scheppingen, D.; Wijnberg, L.; De Koning, C.E.; Dhawan, A. Multicentre, retrospective study to assess long-term outcomes of chelator based treatment with trientine in Wilson disease patients withdrawn from therapy with d-penicillamine. Eur. J. Gastroenterol. Hepatol. 2022, 34, 940–947. [Google Scholar] [CrossRef]
- Scheinberg, I.H.; Walshe, J.M. Orphan Diseases and Orphandrugs; Manchester University Press in Association with the Fulbright Commission: Manchester, NH, USA, 1986; pp. 76–86. [Google Scholar]
- Brewer, G.J.; Johnson, V.; Dick, R.D.; Kluin, K.J.; Fink, J.K.; Brunberg, J.A. Treatment of Wilson Disease with ammonium tetrathiomolybdate. II. Initial therapy in 33 neurologically affected patients and follow-up with zinc therapy. Arch. Neurol. 1996, 53, 1017–1025. [Google Scholar] [CrossRef]
- Weiss, K.H.; Askari, F.K.; Czlonkowska, A.; Ferenci, P.; Bronstein, J.M.; Bega, D.; Ala, A.; Nicholl, D.; Flint, S.; Olsson, L.; et al. Bis-choline tetrathiomolybdate in patients with Wilson’s disease: An open-label, multicentre, phase 2 study. Lancet Gastroenterol. Hepatol. 2017, 2, 869–876. [Google Scholar] [CrossRef]
- Weiss, K.H.; Członkowska, A.; Hedera, P.; Ferenci, P. WTX101—An investigational drug for the treatment of Wilson disease. Expert Opin. Investig. Drugs 2018, 27, 561–567. [Google Scholar] [CrossRef]
- Starzl, T.E.; Iwatsuki, S.; Van Thiel, D.H.; Gartner, J.C.; Zitelli, B.J.; Malatack, J.J.; Schade, R.R.; Shaw, B.W.; Hakala, T.R.; Rosenthal, J.T.; et al. Evolution of Liver Transplantation. Hepatology 1982, 2, 614–636. [Google Scholar] [CrossRef] [PubMed]
- Polson, R.J.; Rolles, K.; Calne, R.Y.; Williams, R.; Marsden, D. Reversal of severe neurological manifestations of Wilson’s disease following orthotopic liver transplantation. QJM Int. J. Med. 1987, 64, 685–691. [Google Scholar] [CrossRef]
- Milovanović, D.D.; Tomasević, R.; Bogdanović, G. Treatment of chronic Wilson’s disease in 2 patients using plasmapheresis-clinico-biochemical observations. Srp. Arh. Celok. Lek. 1998, 126, 327–334. [Google Scholar] [PubMed]
- Reynolds, H.V.; Talekar, C.R.; Bellapart, J.; Leggett, B.A.; Boots, R.J. Copper removal strategies for Wilson’s disease crisis in the ICU. Anaesth. Intensiv. Care 2014, 42, 253–257. [Google Scholar]
- Schouwink, G. De Hepato-Cerebral Degeneration (Met een Onderzoekvan de Zinknkstofwisseling); Van Der Wiel: Arnhem, The Netherlands, 1961. [Google Scholar]
- Hoogenraad, T.; Hamer, C.V.D.; Koevoet, R.; Korver, E.D.R. Oral zinc in Wilson’s disease. Lancet 1978, 312, 1262. [Google Scholar] [CrossRef]
- Hoogenraad, T.; Van Hattum, J.; Hamer, C.V.D. Management of Wilson’s disease with zinc sulphate: Experience in a series of 27 patients. J. Neurol. Sci. 1987, 77, 137–146. [Google Scholar] [CrossRef]
- Brewer, G.J.; Hill, G.M. The treatment of sickle cell anemia and Wilson’s disease with zinc. Prog. Clin. Biol. Res. 1983, 127, 97–113. [Google Scholar]
- Hill, G.M.; Brewer, G.J.; Prasad, A.S.; Hydrick, C.R.; Hartmann, D.E. Treatment of Wilson’s disease with zinc. I. oral Zinc therapy regimens. Hepatology 1987, 7, 522–528. [Google Scholar] [CrossRef]
- Hill, G.M.; Brewer, G.J.; Juni, J.; Prasad, A.S.; Dick, R.D. Treatment of Wilson’s disease with zinc. II. validation of oral 64copper with copper balance. Am. J. Med. Sci. 1986, 292, 344–349. [Google Scholar] [CrossRef]
- Brewer, G.J.; Hill, G.M.; Dick, R.D.; Nostrant, T.T.; Sams, J.S.; Wells, J.J.; Prasad, A.S. Treatment of Wilson’s disease with zinc. III. prevention of reaccumulation of hepatic copper. J. Lab. Clin. Med. 1987, 109, 526–531. [Google Scholar]
- Brewer, G.J.; Hill, G.; Prasad, A.; Dick, R. The treatment of Wilson’s disease with zinc. IV. efficacy monitoring using urine and plasma copper. Proc. Soc. Exp. Biol. Med. 1987, 184, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Yuzbasiyan-Gurkan, V.; Brewer, G.J.; Abrams, G.D.; Main, B.; Giacherio, D. Treatment of Wilson’s disease with zinc. V. changes in serum levels of lipase, amylase, and alkaline phosphatase in patients with Wilson’s disease. J. Lab. Clin. Med. 1989, 114, 520–526. [Google Scholar] [PubMed]
- Brewer, G.J.; Yuzbasiyan-Gurkan, V.; Lee, D.Y.; Appelman, H. Treatment of Wilson’s disease with zinc. VI. Initial treatment studies. J. Lab. Clin. Med. 1989, 114, 633–638. [Google Scholar] [PubMed]
- Lee, D.Y.; Brewer, G.J.; Wang, Y.X. Treatment of Wilson’s disease with zinc. VII. protection of the liver from copper toxicity by zinc-induced metallothionein in a rat model. J. Lab. Clin. Med. 1989, 114, 639–645. [Google Scholar] [PubMed]
- Brewer, G.J.; Yuzbasiyan-Gurkan, V.; Dick, R. Zinc therapy of Wilson’s disease: VIII. dose response studies. J. Trace Elem. Exp. Med. 1990, 3, 227–234. [Google Scholar]
- Brewer, G.J.; Yuzbasiyan-Gurkan, V.; Johnson, V. Treatment of Wilson’s disease with zinc. IX: Response of serum lipids. J. Lab. Clin. Med. 1991, 118, 466–470. [Google Scholar]
- Yuzbasiyan-Gurkan, V.; Grider, A.; Nostrant, T.; Cousins, R.J.; Brewer, G.J. Treatment of Wilson’s disease with zinc: X. intestinal metallothionein induction. J. Lab. Clin. Med. 1992, 120, 380–386. [Google Scholar]
- Brewer, G.J.; Yuzbasiyan-Gurkan, V.; Johnson, V.; Dick, R.D.; Wang, Y. Treatment of Wilson’s disease with zinc: XI. interaction with other anticopper agents. J. Am. Coll. Nutr. 1993, 12, 26–30. [Google Scholar] [CrossRef]
- Brewer, G.J.; Yuzbasiyan-Gurkan, V.; Johnson, V.; Dick, R.D.; Wang, Y. Treatment of Wilson’s Disease with zinc XII: Dose regimen requirements. Am. J. Med. Sci. 1993, 305, 199–202. [Google Scholar] [CrossRef]
- Brewer, G.J.; Dick, R.D.; Yuzbasiyan-Gurkan, V.; Johnson, V.; Wang, Y. Treatment of Wilson’s disease with zinc. XIII: Therapy with zinc in presymptomatic patients from the time of diagnosis. J. Lab. Clin. Med. 1994, 123, 849–858. [Google Scholar]
- Brewer, G.J.; Johnson, V.; Kaplan, J. Treatment of Wilson’s disease with zinc: XIV studies of the effect of zinc on lymphocyte function. J. Lab. Clin. Med. 1997, 129, 649–652. [Google Scholar] [CrossRef]
- Brewer, G.J.; Dick, R.D.; Johnson, V.D.; Brunberg, J.A.; Kluin, K.J.; Fink, J.K. Treatment of Wilson’s disease with zinc: XV long-term follow-up studies. J. Lab. Clin. Med. 1998, 132, 264–278. [Google Scholar] [CrossRef]
- Brewer, G.J.; Dick, R.D.; Johnson, V.D.; Fink, J.K.; Kluin, K.J.; Daniels, S. Treatment of Wilson’s disease with zinc XVI: Treatment during the pediatric years. J. Lab. Clin. Med. 2001, 137, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J.; Johnson, V.D.; Dick, R.D.; Hedera, P.; Fink, J.K.; Kluin, K.J. Treatment of Wilson’s disease with zinc. XVII: Treatment during pregnancy. Hepatology 2000, 31, 364–370. [Google Scholar] [CrossRef]
- Askari, F.K.; Greenson, J.; Dick, R.D.; Johnson, V.D.; Brewer, G.J. Treatment of Wilson’s disease with zinc. XVIII. initial treatment of the hepatic decompensation presentation with trientine and zinc. J. Lab. Clin. Med. 2003, 142, 385–390. [Google Scholar] [CrossRef]
- Walshe, J.M. History of Wilson’s disease: 1912 to 2000. Mov. Disord. 2006, 21, 142–147. [Google Scholar] [CrossRef]
- Dooley, J.S.; Purchase, R. History of Wilson disease. In Wilson Disease; Elsevier: Amsterdam, The Netherlands, 2019; pp. 3–14. ISBN 9780128110775. [Google Scholar]
- Hoogenraad, T.U. Wilson’s disease. In Major Problems in Neurology, 1st ed.; van Gijn, J., Ed.; W B Saunders Co.: London, UK, 1996; Volume 30, ISBN 0702018422. [Google Scholar]
- Sensi, S.L.; Granzotto, A.; Siotto, M.; Squitti, R. Copper and zinc dysregulation in Alzheimer’s disease. Trends Pharmacol. Sci. 2018, 39, 1049–1063. [Google Scholar] [CrossRef]
- Husain, N.; Mahmood, R. Copper(II) generates ROS and RNS, impairs antioxidant system and damages membrane and DNA in human blood cells. Environ. Sci. Pollut. Res. Int. 2019, 26, 20654–20668. [Google Scholar] [CrossRef]
- Polishchuk, E.V.; Polishchuk, R.S. The emerging role of lysosomes in copper homeostasis. Metallomics 2016, 8, 853–862. [Google Scholar] [CrossRef]
- Xu, Q.; Qiu, H.; Chu, W.; Fu, Y.; Cai, S.; Min, H.; Sha, S. Copper ultrastructural localization, subcellular distribution, and phytotoxicity in hydrilla verticillata (L.f.) royle. Environ. Sci. Pollut. Res. Int. 2013, 20, 8672–8679. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chen, X.; Kroemer, G. Cuproptosis: A copper-triggered modality of mitochondrial cell death. Cell Res. 2022, 32, 417–418. [Google Scholar] [CrossRef] [PubMed]
- Zatta, P.; Drago, D.; Bolognin, S.; Sensi, S.L. Alzheimer’s disease, metal ions and metal homeostatic therapy. Trends Pharmacol. Sci. 2009, 30, 346–355. [Google Scholar] [CrossRef]
- Salustri, C.; Tecchio, F.; Zappasodi, F.; Tomasevic, L.; Ercolani, M.; Moffa, F.; Cassetta, E.; Rossini, P.M.; Squitti, R. Sensorimotor cortex reorganization in Alzheimer’s disease and metal dysfunction: A MEG Study. Int. J. Alzheimer’s Dis. 2013, 2013, 638312. [Google Scholar] [CrossRef] [PubMed]
- Dodani, S.C.; Firl, A.; Chan, J.; Nam, C.I.; Aron, A.T.; Onak, C.S.; Ramos-Torres, K.M.; Paek, J.; Webster, C.M.; Feller, M.B.; et al. Copper is an endogenous modulator of neural circuit spontaneous activity. Proc. Natl. Acad. Sci. USA 2014, 111, 16280–16285. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosi, N.; Rossi, L. Copper at synapse: Release, binding and modulation of neurotransmission. Neurochem. Int. 2015, 90, 36–45. [Google Scholar] [CrossRef]
- Sass-Kortsak, A.; Bearn, A.G. Hereditary Disorders of Copper Metabolism. In The Metabolic Basis of Inherited Disease; McGraw-Hill: New York, NY, USA, 1978; pp. 1098–1126. [Google Scholar]
- EFSA panel on dietetic products, nutrition and allergies (NDA) scientific opinion on dietary reference values for copper. EFSA J. 2015, 13, 4253. [CrossRef]
- Brewer, G. Wilson’s disease. In Harrison’s Principles of Internal Medicine 19th Edition and Harrison’s Manual of Medicine 19th Edition; Jameson, J.L., Fauci, A., Kasper, D., Hauser, S., Longo, D., Loscalzo, J., Eds.; McGraw-Hill Education/Medical: New York, NY, USA, 2017; ISBN 978-1260128857. [Google Scholar]
- Van Den Hamer, C.J.A.; Hoogenraad, T.U.; Klompjan, E.R.K. Persistence of the antagonistic influence of zinc on copper absorption after cessation of zinc supplementation for more than 5 days. Trace Elem. Med. 1984, 1, 88–90. [Google Scholar]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef]
- Litwin, T.; Gromadzka, G.; Szpak, G.; Jabłonka-Salach, K.; Bulska, E.; Członkowska, A. Brain metal accumulation in Wilson’s disease. J. Neurol. Sci. 2013, 329, 55–58. [Google Scholar] [CrossRef]
- Shribman, S.; Bocchetta, M.; Sudre, C.H.; Acosta-Cabronero, J.; Burrows, M.; Cook, P.; Thomas, D.L.; Gillett, G.T.; Tsochatzis, E.A.; Bandmann, O.; et al. Neuroimaging correlates of brain injury in Wilson’s disease: A multimodal, whole-brain MRI study. Brain 2022, 145, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.A.; Schilsky, M.L.; American Association for Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: An update. Hepatology 2008, 47, 2089–2111. [Google Scholar] [CrossRef] [PubMed]
- Ngwanou, D.H.; Couchonnal, E.; Parant, F.; Belmalih, A.; Guillaud, O.; Dumortier, J.; Bost, M.; Lachaux, A. Long-term urinary copper excretion and exchangeable copper in children with Wilson disease under chelation therapy. J. Pediatr. Gastroenterol. Nutr. 2022; online ahead of print. [Google Scholar] [CrossRef]
- Brewer, G.J.; Terry, C.A.; Aisen, A.M.; Hill, G.M. Worsening of neurologic syndrome in patients with Wilson’s disease with initial penicillamine therapy. Arch. Neurol. 1987, 44, 490–493. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J.; Askari, F.; Dick, R.B.; Sitterly, J.; Fink, J.K.; Carlson, M.; Kluin, K.J.; Lorincz, M.T. Treatment of Wilson’s disease with tetrathiomolybdate: V. control of free copper by tetrathiomolybdate and a comparison with trientine. Transl. Res. 2009, 154, 70–77. [Google Scholar] [CrossRef]
- Czlonkowska, A.; Litwin, T.; Karlinski, M.; Dzieżyc, K.; Chabik, G.; Czerska, M. D-penicillamine versus zinc sulfate as first-line therapy for Wilson’s disease. Eur. J. Neurol. 2014, 21, 599–606. [Google Scholar] [CrossRef]
- Brewer, G.J. Treatment of Wilson’s disease: Our patients deserve better. Expert Opin. Orphan Drugs 2014, 2, 1245–1248. [Google Scholar] [CrossRef]
- Poujois, A.; Trocello, J.-M.; Djebrani-Oussedik, N.; Poupon, J.; Collet, C.; Girardot-Tinant, N.; Sobesky, R.; Habès, D.; Debray, D.; Vanlemmens, C.; et al. Exchangeable copper: A reflection of the neurological severity in Wilson’s disease. Eur. J. Neurol. 2017, 24, 154–160. [Google Scholar] [CrossRef]
- Gibbs, K.; Walshe, J.M. A study of the caeruloplasmin concentrations found in 75 patients with Wilson’s disease, their kin-ships and various control groups. Q. J. Med. 1979, 48, 447–463. [Google Scholar]
- Walshe, J.M. Clinical investigations standing committee of the association of clinical biochemists Wilson’s disease: The importance of measuring serum caeruloplasmin non-immunologically. Ann. Clin. Biochem. 2003, 40, 115–121. [Google Scholar] [CrossRef]
- Merle, U.; Eisenbach, C.; Weiss, K.H.; Tuma, S.; Stremmel, W. Serum ceruloplasmin oxidase activity is a sensitive and highly specific diagnostic marker for Wilson’s disease. J. Hepatol. 2009, 51, 925–930. [Google Scholar] [CrossRef]
- Ryan, A.; Nevitt, S.J.; Tuohy, O.; Cook, P. Biomarkers for diagnosis of Wilson’s disease. Cochrane Database Syst. Rev. 2019, 2019, CD012267. [Google Scholar] [CrossRef] [PubMed]
- Członkowska, A.; Tarnacka, B.; Möller, J.C.; Leinweber, B.; Bandmann, O.; Woimant, F.; Oertel, W.H. Unified Wilson’s disease rating scale—A proposal for the neurological scoring of Wilson’s disease patients. Neurol. Neurochir. Polska 2007, 41, 1–12. [Google Scholar] [CrossRef]
- Leinweber, B.; Möller, J.C.; Scherag, A.; Reuner, U.; Günther, P.; Lang, C.J.G.; Schmidt, H.H.J.; Schrader, C.; Bandmann, O.; Czlonkowska, A.; et al. Evaluation of the Unified Wilson’s Disease Rating Scale (UWDRS) in German patients with treated Wilson’s disease. Mov. Disord. 2008, 23, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, A.; Aggarwal, N.; Nagral, A.; Jankharia, G.; Bhatt, M. A novel Global Assessment Scale for Wilson’s Disease (GAS for WD). Mov. Disord. 2009, 24, 509–518. [Google Scholar] [CrossRef]
- Ferenci, P.; Steindl-Munda, P.; Vogel, W.; Jessner, W.; Gschwantler, M.; Stauber, R.; Datz, C.; Hackl, F.; Wrba, F.; Bauer, P.; et al. Diagnostic value of quantitative hepatic copper determination in patients with Wilson’s disease. Clin. Gastroenterol. Hepatol. 2005, 3, 811–818. [Google Scholar] [CrossRef]
- Merle, U.; Schaefer, M.; Ferenci, P.; Stremmel, W. Clinical presentation, diagnosis and long-term outcome of Wilson’s disease: A cohort study. Gut 2007, 56, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Steindl, P.; Ferenci, P.; Dienes, H.P.; Grimm, G.; Pabinger, I.; Madl, C.; Maier-Dobersberger, T.; Herneth, A.; Dragosics, B.; Meryn, S.; et al. Wilson’s disease in patients presenting with liver disease: A diagnostic challenge. Gastroenterology 1997, 113, 212–218. [Google Scholar] [CrossRef]
- El Balkhi, S.; Poupon, J.; Trocello, J.-M.; Leyendecker, A.; Massicot, F.; Galliot-Guilley, M.; Woimant, F. Determination of ultrafiltrable and exchangeable copper in plasma: Stability and reference values in healthy subjects. Anal. Bioanal. Chem. 2009, 394, 1477–1484. [Google Scholar] [CrossRef]
- Woimant, F.; Djebrani-Oussedik, N.; Poujois, A. New tools for Wilson’s disease diagnosis: Exchangeable copper fraction. Ann. Transl. Med. 2019, 7, S70. [Google Scholar] [CrossRef]
- Shribman, S.; Heller, C.; Burrows, M.; Heslegrave, A.; Swift, I.; Foiani, M.S.; Gillett, G.T.; Tsochatzis, E.A.; Rowe, J.B.; Gerhard, A.; et al. Plasma neurofilament light as a biomarker of neurological involvement in Wilson’s disease. Mov. Disord. 2021, 36, 503–508. [Google Scholar] [CrossRef]
- Collins, C.J.; Yi, F.; Dayuha, R.; Duong, P.; Horslen, S.; Camarata, M.; Coskun, A.K.; Houwen, R.H.; Pop, T.L.; Zoller, H.; et al. Direct measurement of ATP7B peptides is highly effective in the diagnosis of Wilson disease. Gastroenterology 2021, 160, 2367–2382.e1. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J.; Askari, F.; Lorincz, M.T.; Carlson, M.; Schilsky, M.; Kluin, K.J.; Hedera, P.; Moretti, P.; Fink, J.K.; Tankanow, R.; et al. Treatment of Wilson disease with ammonium tetrathiomolybdate: IV. Comparison of tetrathiomolybdate and trientine in a double-blind study of treatment of the neurologic presentation of Wilson disease. Arch. Neurol. 2006, 63, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.; Ala, A.; Askari, F.K.; Czlonkowska, A.; Hilgers, R.; Poujois, A.; Roberts, E.A.; Sandahl, T.D.; Weiss, K.H.; Ferenci, P.; et al. Designing clinical trials in Wilson’s disease. Hepatology 2021, 74, 3460–3471. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Bai, L.; Hou, W.; Hu, Z.; Chen, X.; Zhao, J.; Liang, C.; Zhang, W.; Duan, Z.; Zheng, S. Comparison of the effectiveness and safety of d-penicillamine and zinc salt treatment for symptomatic Wilson disease: A systematic review and meta-analysis. Front. Pharmacol. 2022, 13, 847436. [Google Scholar] [CrossRef]
- Appenzeller-Herzog, C.; Mathes, T.; Heeres, M.L.S.; Weiss, K.H.; Houwen, R.H.J.; Ewald, H. Comparative effectiveness of common therapies for Wilson disease: A systematic review and meta-analysis of controlled studies. Liver Int. 2019, 39, 2136–2152. [Google Scholar] [CrossRef]
- Efficacy and Safety of ALXN1840 (Administered for 48 Weeks Versus Standard of Care in Participants with Wilson Disease)—ClinicalTrials.gov Identifier: NCT03403205. Available online: https://clinicaltrials.gov/ct2/show/NCT03403205 (accessed on 8 July 2022).
- Lowette, K.F.; Desmet, K.; Witters, P.; Laleman, W.; Verslype, C.; Nevens, F.; Fevery, J.; Cassiman, D.M. Wilson’s disease: Long-term follow-up of a cohort of 24 patients treated with D-penicillamine. Eur. J. Gastroenterol. Hepatol. 2010, 22, 564–571. [Google Scholar] [CrossRef]
- Kleine, R.T.; Mendes, R.; Pugliese, R.; Miura, I.; Danesi, V.; Porta, G. Wilson’s disease: An analysis of 28 Brazilian children. Clinics 2012, 67, 231–235. [Google Scholar] [CrossRef]
- Weiss, K.H.; Gotthardt, D.N.; Klemm, D.; Merle, U.; Ferenci–Foerster, D.; Schaefer, M.; Ferenci, P.; Stremmel, W. Zinc monotherapy is not as effective as chelating agents in treatment of Wilson disease. Gastroenterology 2011, 140, 1189–1198.e1. [Google Scholar] [CrossRef]
- Durand, F.; Bernuau, J.; Giostra, E.; Mentha, G.; Shouval, D.; Degott, C.; Benhamou, J.P.; Valla, D. Wilson’s disease with severe hepatic insufficiency: Beneficial effects of early administration of D-penicillamine. Gut 2001, 48, 849–852. [Google Scholar] [CrossRef]
- Brewer, G.J. Zinc and tetrathiomolybdate for the treatment of Wilson’s disease and the potential efficacy of anticopper therapy in a wide variety of diseases. Metallomics 2009, 1, 199–206. [Google Scholar] [CrossRef]
- Brewer, G.J.; Turkay, A.; Yuzbasiyan-Gurkan, V. Development of Neurologic Symptoms in a Patient With Asymptomatic Wilson’s Disease Treated With Penicillamine. Arch. Neurol. 1994, 51, 304–305. [Google Scholar] [CrossRef] [PubMed]
- Litwin, T.; Dzieżyc, K.; Karliński, M.; Chabik, G.; Czepiel, W.; Członkowska, A. Early neurological worsening in patients with Wilson’s disease. J. Neurol. Sci. 2015, 355, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Firwana, B.; Ibrahim, N.; Taftaf, R.; Saz, A.S.; Sonbol, M.B.; Hasan, R.; Gluud, C. Medical therapeutic agents for Wilson’s disease. Cochrane Database Syst. Rev. 2011, 3, CD009057. [Google Scholar] [CrossRef]
- Medici, V.; Trevisan, C.P.; Bigotto, M.A.; D’Incà, R.; Martines, D.; Pont, E.D.; Sturniolo, G.C. Adverse reaction after tetrathiomolybdate treatment for Wilson’s disease: A case report. Mov. Disord. 2006, 21, 2030–2032. [Google Scholar] [CrossRef] [PubMed]
- Medici, V.; Trevisan, C.P.; D’Incà, R.; Barollo, M.; Zancan, L.; Fagiuoli, S.; Martines, D.; Irato, P.; Sturniolo, G.C. Diagnosis and management of Wilson’s disease: Results of a single center experience. J. Clin. Gastroenterol. 2006, 40, 936–941. [Google Scholar] [CrossRef]
- Dell’era, L.; Boati, E.; Nebbia, G.; Corona, F. Wilson’s disease treated with penicillamine and lupus erythematosus: Related or distinct entities? Minerva Pediatr. 2012, 64, 55–57. [Google Scholar]
- Samal, S.; Sable, M. Elastosis perforans serpiginosa: A D-penicillamine induced dermatoses in a patient with Wilson s disease. Autops. Case Rep. 2020, 10, e2020167. [Google Scholar] [CrossRef]
- Na, S.Y.; Choi, M.; Kim, M.J.; Lee, J.H.; Cho, S. Penicillamine-induced elastosis perforans serpiginosa and cutis laxa in a patient with Wilson’s disease. Ann. Dermatol. 2010, 22, 468–471. [Google Scholar] [CrossRef]
- Siafakas, C.G.; Jonas, M.M.; Alexander, S.; Herrin, J.; Furuta, G. Early onset of nephrotic syndrome after treatment with D-penicillamine in a patient with Wilson’s disease. Am. J. Gastroenterol. 1998, 93, 2544–2546. [Google Scholar] [CrossRef]
- Poulas, K.; Koutsouraki, E.; Kordas, G.; Kokla, A.; Tzartos, S.J. Anti-MuSK- and anti-AChR-positive myasthenia gravis induced by d-penicillamine. J. Neuroimmunol. 2012, 250, 94–98. [Google Scholar] [CrossRef]
- Seessle, J.; Gotthardt, D.N.; Schafer, M.; Gohdes, A.; Pfeiffenberger, J.; Ferenci, P.; Stremmel, W.; Weiss, K.H. Concomitant immune-related events in Wilson disease: Implications for monitoring chelator therapy. J. Inherit. Metab. Dis. 2016, 39, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Singh, A.P.; Wheeler, N.; Galindo, C.L.; Kim, J.-J. Safety profile of D-penicillamine: A comprehensive pharmacovigilance analysis by FDA adverse event reporting system. Expert Opin. Drug Saf. 2021, 20, 1443–1450. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J. Penicillamine should not be used as initial therapy in Wilson’s disease. Mov. Disord. 1999, 14, 551–554. [Google Scholar] [CrossRef]
- Rustgi, V.K.; Gupta, K.; Tait, C.; Bhurwal, A.; Kabaria, S.; Catalano, C.; Li, Y.; Minacapelli, C.D. Wilson’s disease: An analysis of health care use and cost burden of commercially insured adults in the United States. Hepatol. Commun. 2021, 6, 389–398. [Google Scholar] [CrossRef]
- Schilsky, M.L.; Roberts, E.A.; Hahn, S.; Askari, F. Costly choices for treating Wilson’s disease. Hepatology 2015, 61, 1106–1108. [Google Scholar] [CrossRef] [PubMed]
- Weiss, K.H.; Thurik, F.; Gotthardt, D.N.; Schäfer, M.; Teufel, U.; Wiegand, F.; Merle, U.; Ferenci–Foerster, D.; Maieron, A.; Stauber, R.; et al. Efficacy and safety of oral chelators in treatment of patients with Wilson disease. Clin. Gastroenterol. Hepatol. 2013, 11, 1028–1035.e2. [Google Scholar] [CrossRef] [PubMed]
- Samanci, B.; Sahin, E.; Bilgic, B.; Tufekcioglu, Z.; Gurvit, H.; Emre, M.; Demir, K.; Hanagasi, H.A. Neurological features and outcomes of Wilson’s disease: A single-center experience. Neurol. Sci. 2021, 42, 3829–3834. [Google Scholar] [CrossRef] [PubMed]
- Kalita, J.; Kumar, V.; Chandra, S.; Kumar, B.; Misra, U.K. Worsening of Wilson disease following penicillamine therapy. Eur. Neurol. 2014, 71, 126–131. [Google Scholar] [CrossRef]
- Czlonkowska, A.; Gajda, J.; Rodo, M. Effects of long-term treatment in Wilson’s disease with d-penicillamine and zinc sulphate. J. Neurol. 1996, 243, 269–273. [Google Scholar] [CrossRef]
- Marcellini, M.; Di Ciommo, V.; Callea, F.; De Vito, R.; Comparcola, D.; Sartorelli, M.; Carelli, F.; Nobili, V. Treatment of Wilson’s disease with zinc from the time of diagnosis in pediatric patients: A single-hospital, 10-year follow-up study. J. Lab. Clin. Med. 2005, 145, 139–143. [Google Scholar] [CrossRef]
- Mizuochi, T.; Kimura, A.; Shimizu, N.; Nishiura, H.; Matsushita, M.; Yoshino, M. Zinc monotherapy from time of diagnosis for young pediatric patients with presymptomatic Wilson disease. J. Pediatr. Gastroenterol. Nutr. 2011, 53, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Eda, K.; Mizuochi, T.; Iwama, I.; Inui, A.; Etani, Y.; Araki, M.; Hara, S.; Kumagai, H.; Hagiwara, S.-I.; Murayama, K.; et al. Zinc monotherapy for young children with presymptomatic Wilson disease: A multicenter study in Japan. J. Gastroenterol. Hepatol. 2018, 33, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Jacquelet, E.; Poujois, A.; Pheulpin, M.; Demain, A.; Tinant, N.; Gastellier, N.; Woimant, F. Adherence to treatment, a challenge even in treatable metabolic rare diseases: A cross sectional study of Wilson’s disease. J. Inherit. Metab. Dis. 2021, 44, 1481–1488. [Google Scholar] [CrossRef] [PubMed]
- Masełbas, W.; Członkowska, A.; Litwin, T.; Niewada, M. Persistence with treatment for Wilson disease: A retrospective study. BMC Neurol. 2019, 19, 278. [Google Scholar] [CrossRef]
- Lee, V.D.; Northup, P.G.; Berg, C.L. Resolution of decompensated cirrhosis from Wilson’s disease with zinc monotherapy: A potential therapeutic option? Clin. Gastroenterol. Hepatol. 2006, 4, 1069–1071. [Google Scholar] [CrossRef] [PubMed]
- Linn, F.H.H.; Houwen, R.H.J.; Van Hattum, J.; Van Der Kleij, S.; Van Erpecum, K.J. Long-term exclusive zinc monotherapy in symptomatic Wilson disease: Experience in 17 patients. Hepatology 2009, 50, 1442–1452. [Google Scholar] [CrossRef] [PubMed]
- Avan, A.; Kianifar, H.R.; Hoogenraad, T.U. Initial zinc therapy in a Wilson’s disease patient with acute liver failure and copper intoxication: A clinical observation. Mov. Disord. 2013, 28, 990. [Google Scholar]
- Dzieżyc, K.; Litwin, T.; Członkowska, A. Neurological deterioration or symptom progression in Wilson’s disease after starting zinc sulphate treatment—A case report. Glob. Drugs Ther. 2016, 2, 1–3. [Google Scholar] [CrossRef]
- Lang, C.J.G.; Rabas-Kolominsky, P.; Engelhardt, A.; Kobras, G.; Konig, H.J. Fatal deterioration of Wilson’s disease after institution of oral zinc therapy. Arch. Neurol. 1993, 50, 1007–1008. [Google Scholar] [CrossRef]
- Camarata, M.A.; Ala, A.; Schilsky, M.L. Zinc maintenance therapy for Wilson disease: A comparison between zinc acetate and alternative zinc preparations. Hepatol. Commun. 2019, 3, 1151–1158. [Google Scholar] [CrossRef]
- Mohr, I.; Weiss, K.H. Current anti-copper therapies in management of Wilson disease. Ann. Transl. Med. 2019, 7, S69. [Google Scholar] [CrossRef] [PubMed]
- Kathawala, M.; Hirschfield, G.M. Insights into the management of Wilson’s disease. Ther. Adv. Gastroenterol. 2017, 10, 889–905. [Google Scholar] [CrossRef] [PubMed]
- Walshe, J.M. The pattern of urinary copper excretion and its response to treatment in patients with Wilson’s disease. QJM Int. J. Med. 2011, 104, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, M.W.; Karim, M.B.; Rukunuzzaman, M. Penicillamine challenge test in the diagnosis of Wilson’s disease. Mymensingh Med. J. 2014, 23, 489–495. [Google Scholar]
- Pfeiffenberger, J.; Lohse, C.M.; Gotthardt, D.; Rupp, C.; Weiler, M.; Teufel, U.; Weiss, K.H.; Gauss, A. Long-term evaluation of urinary copper excretion and non-caeruloplasmin associated copper in Wilson disease patients under medical treatment. J. Inherit. Metab. Dis. 2019, 42, 371–380. [Google Scholar] [CrossRef]
- Klaassen, C.D.; Lehman-McKeeman, L.D. Induction of metallothionein. J. Am. Coll. Toxicol. 1989, 8, 1315–1321. [Google Scholar] [CrossRef]
- Nagral, A.; Sarma, M.S.; Matthai, J.; Kukkle, P.L.; Devarbhavi, H.; Sinha, S.; Alam, S.; Bavdekar, A.; Dhiman, R.K.; Eapen, C.E.; et al. Wilson’s disease: Clinical practice guidelines of the Indian national association for study of the liver, the Indian society of pediatric gastroenterology, hepatology and nutrition, and the movement disorders society of India. J. Clin. Exp. Hepatol. 2019, 9, 74–98. [Google Scholar] [CrossRef]
- Shribman, S.; Marjot, T.; Sharif, A.; Vimalesvaran, S.; Ala, A.; Alexander, G.; Dhawan, A.; Dooley, J.; Gillett, G.T.; Kelly, D.; et al. Investigation and management of Wilson’s disease: A practical guide from the British association for the study of the liver. Lancet Gastroenterol. Hepatol. 2022, 7, 560–575. [Google Scholar] [CrossRef]
- Socha, P.; Janczyk, W.; Dhawan, A.; Baumann, U.; D’Antiga, L.; Tanner, S.; Iorio, R.; Vajro, P.; Houwen, R.; Fischler, B.; et al. Wilson’s disease in children: A position paper by the hepatology committee of the european society for paediatric gastroenterology, hepatology and nutrition. J. Pediatr. Gastroenterol. Nutr. 2018, 66, 334–344. [Google Scholar] [CrossRef]
- Roberts, E.A. Zinc toxicity: From “no, never” to “hardly ever”. Gastroenterology 2011, 140, 1132–1135. [Google Scholar] [CrossRef]
- Brewer, G.J. Zinc acetate for the treatment of Wilson’s disease. Expert Opin. Pharmacother. 2001, 2, 1473–1477. [Google Scholar] [CrossRef] [PubMed]
- Ferner, R.; Hughes, D. The problem of orphan drugs. BMJ 2010, 341, c6456. [Google Scholar] [CrossRef] [PubMed]
- Habka, D.; Mann, D.; Landes, R.; Soto-Gutierrez, A. Future economics of liver transplantation: A 20-year cost modeling forecast and the prospect of bioengineering autologous liver grafts. PLoS ONE 2015, 10, e0131764. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Criteria | Hepatic and Neurologic WD [94] | Hepatic WD [95] | Neurologic WD [95] |
|---|---|---|---|
| Serum ceruloplasmin (<200 mg/L) | 88% | 59% | 85% |
| Urine copper (>1.6 μmol/24 h) | 87% | 90% | 78% |
| Liver copper content (>4 μmol/g) | 90% | 93% | |
| Histological signs of liver damage | 73% | ||
| Kayser–Fleischer rings (with slit lamp examination) | 66% | 41% | 90% |
| Mutations in ATP7B on 1 or 2 alleles | 85% | ||
| Non-ceruloplasmin bound copper (>4 μmol/L) | 86% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Avan, A.; Członkowska, A.; Gaskin, S.; Granzotto, A.; Sensi, S.L.; Hoogenraad, T.U. The Role of Zinc in the Treatment of Wilson’s Disease. Int. J. Mol. Sci. 2022, 23, 9316. https://doi.org/10.3390/ijms23169316
Avan A, Członkowska A, Gaskin S, Granzotto A, Sensi SL, Hoogenraad TU. The Role of Zinc in the Treatment of Wilson’s Disease. International Journal of Molecular Sciences. 2022; 23(16):9316. https://doi.org/10.3390/ijms23169316
Chicago/Turabian StyleAvan, Abolfazl, Anna Członkowska, Susan Gaskin, Alberto Granzotto, Stefano L. Sensi, and Tjaard U. Hoogenraad. 2022. "The Role of Zinc in the Treatment of Wilson’s Disease" International Journal of Molecular Sciences 23, no. 16: 9316. https://doi.org/10.3390/ijms23169316