
Novel Anti-Neuroinflammatory Properties of a Thiosemicarbazone–Pyridylhydrazone Copper(II) Complex
 , , , , , , , , and
, , , , , , , , and 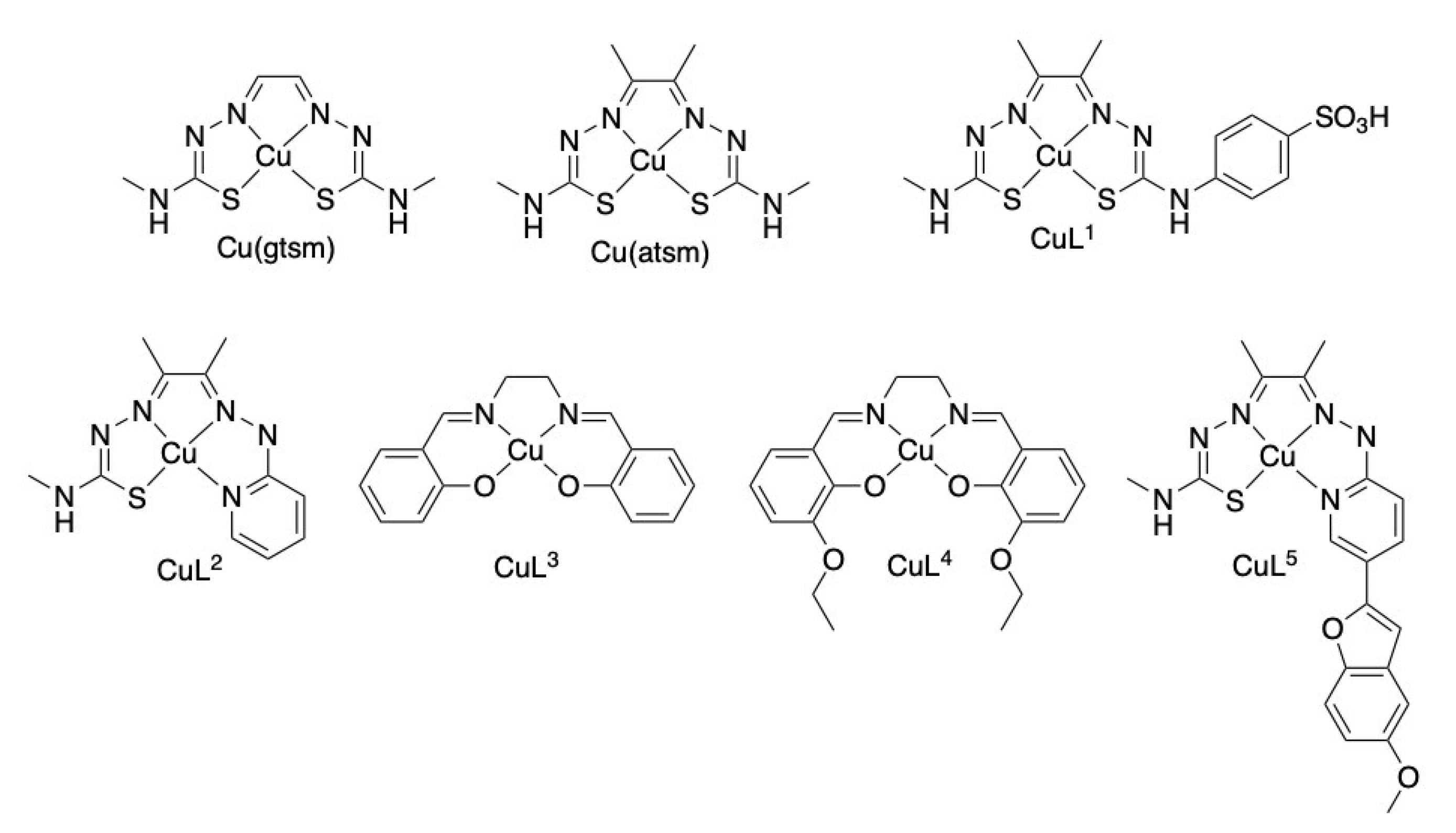{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
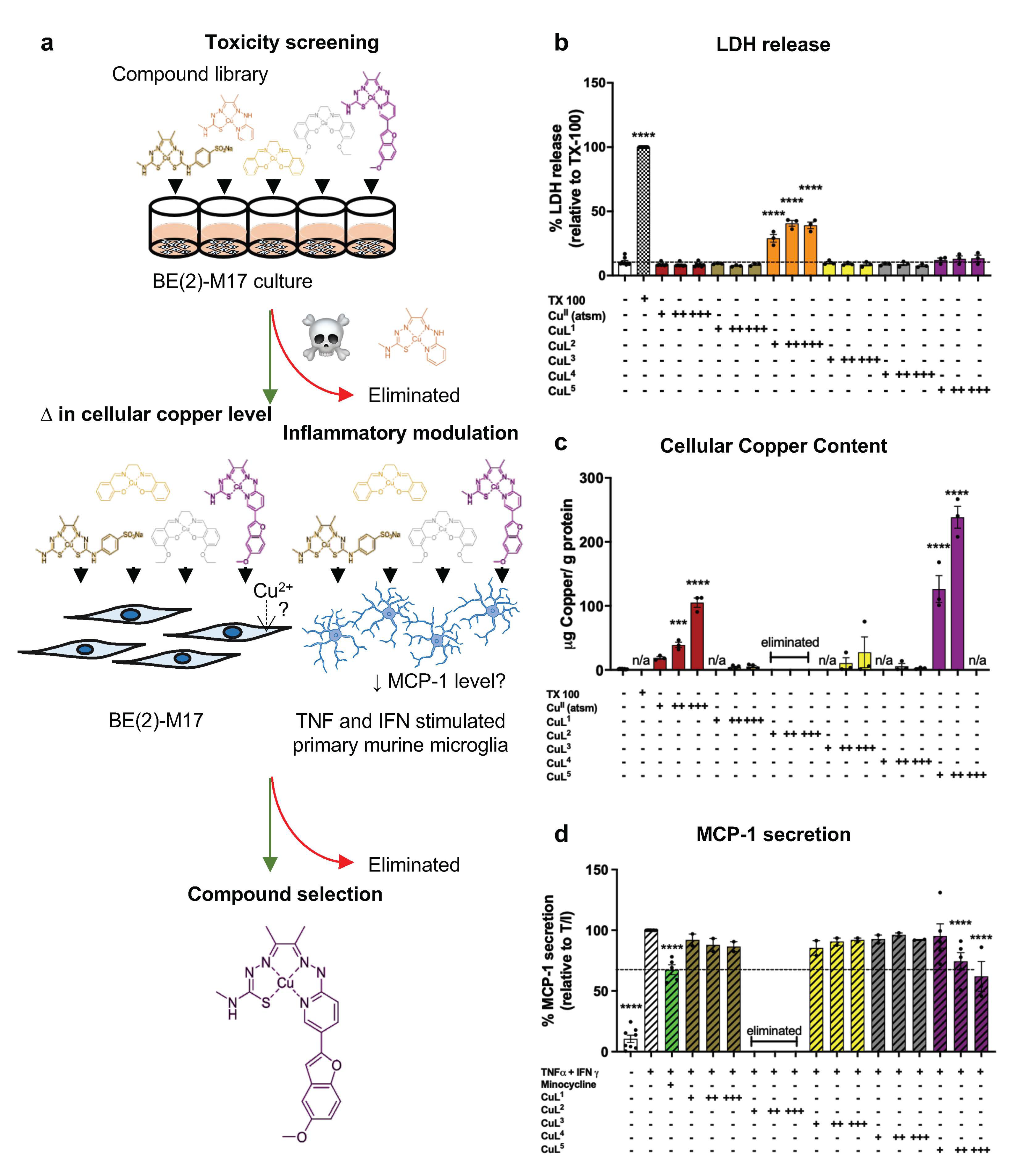
2.1. Cellular Toxicity and Copper Uptake of Copper(II) Complexes
2.2. CuL5 Inhibited MCP-1 Secretion by Murine Microglia
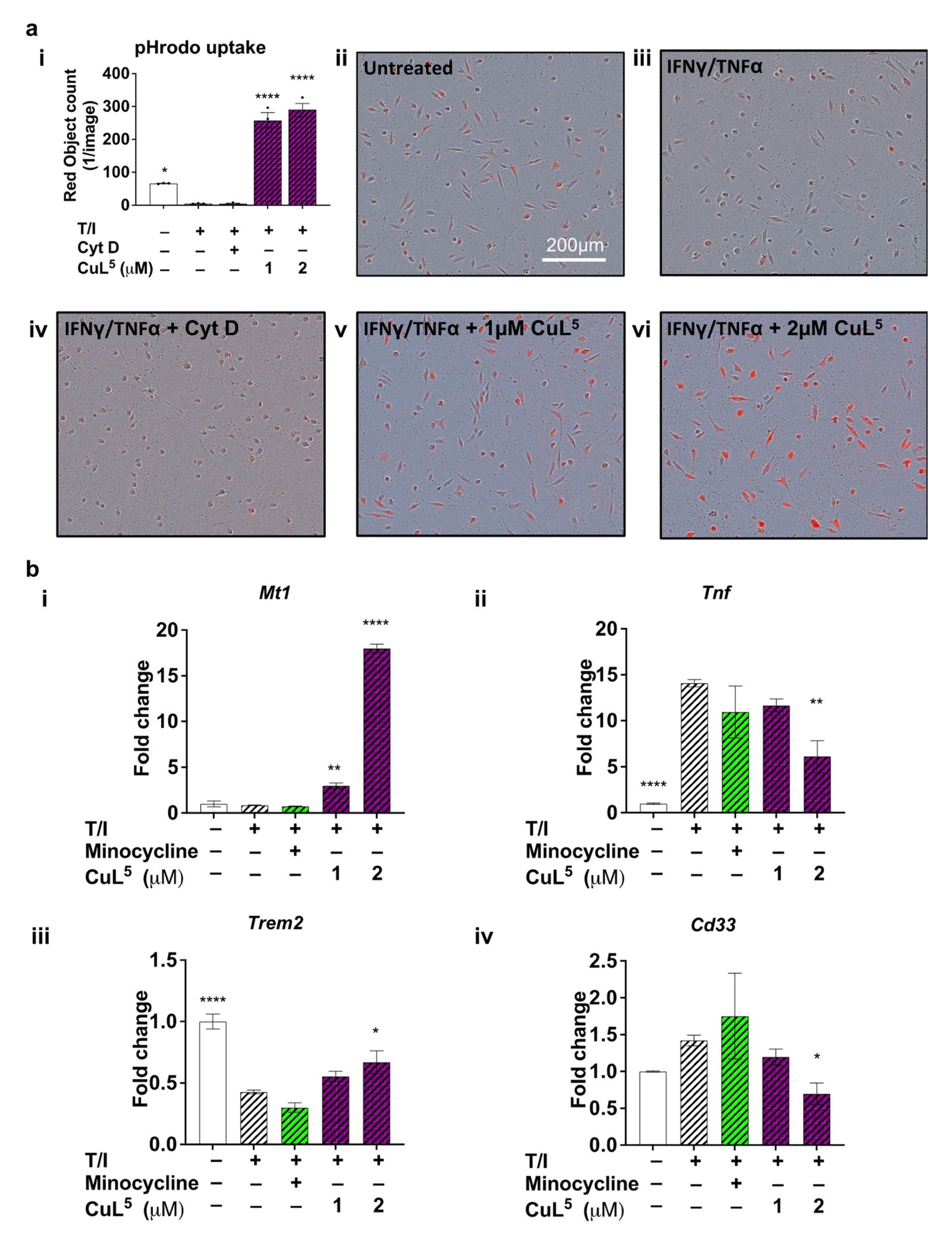
2.3. CuL5 Increased Phagocytic Activity of Microglia
2.4. CuL5 Inhibited Expression of the Pro-Inflammatory Cytokine, TNFa and Upregulated Metallothionein
2.5. CuL5 Modulated Expression of Late Onset AD risk Genes, Cd33 and Trem2
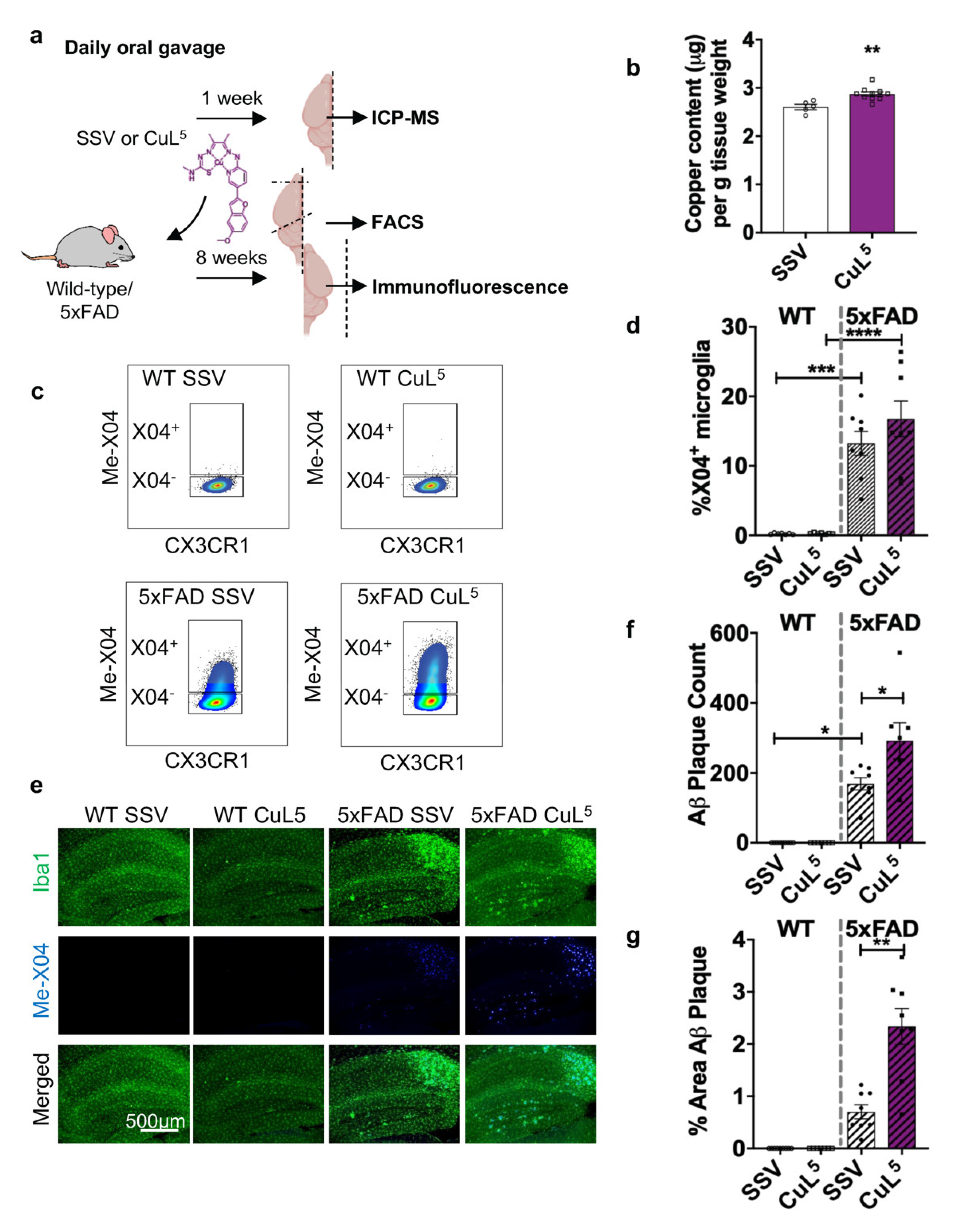
2.6. CuL5 Was Well Tolerated by Mice and Increased Copper Content in the Brain
2.7. CuL5 Treatment of 5xFAD Mice Increased Hippocampal Amyloid Load
2.8. CuL5 Induced Selective Improvement of the Cognitive Function of 5xFAD Mice
3. Discussion
3.1. Inflammation-Modulating Efficacy of CuL5
3.2. CuL5-Mediated Anti-Inflammatory Action May Be Linked to MT
3.3. Paradoxically, CuL5 Increases Phagocytic Function In Vitro but Induces Accumulated Amyloid In Vivo
3.4. CuL5 Improves Short-Term Memory Function in AD Model Mice
3.5. Summary of Findings, Limitations and Implications
4. Materials and Methods
4.1. Neuroblastoma Cell Line BE(2)-M17
4.2. Primary Cell Culture
4.2.1. Animals
4.2.2. Primary Mixed Glial Culture
4.2.3. Primary Microglia Culture
4.3. Copper(II) Complexes Synthesis
4.3.1. 2-Hydrazino-pyridinyl-5-(5′-methoxybenzofuran) (1)
4.3.2. Diacetyl-2-(2-hydrazino-pyridinyl-5-(5′-methoxybenzofuran))-(4-methyl-3-thiosemicarbazone) (H2L5)
4.3.3. [Copper(II)(Diacetyl-2-(2-hydrazino-pyridinyl-5-(5′-methoxybenzofuran))-(4-methyl-3-thiosemicarbazone))] (CuL5)
4.4. Cell Stimulation and/or Treatment
4.4.1. Preparation of Metal Complexes, Copper-Free Ligand and Inflammatory Mediators and Inhibitors
4.4.2. Stimulation and Treatment of Cell Cultures
4.5. 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide (MTT) Reduction Assay
4.6. LDH Cytotoxic Assay
4.7. Inductively Coupled Plasma Mass Spectrometry (ICP–MS)
4.8. ELISA
4.9. qRT-PCR
4.10. Phagocytosis Assay
4.11. Oral Treatment of 5xFAD with CuL5
4.12. Biochemical Analysis of Serum
4.13. Acute Microglia Isolation and Analysis
4.14. Immunohistology
4.15. Imaging and Image Analyses
4.16. Behavioral Battery
4.16.1. Nest-Building Test
4.16.2. igloo Test
4.16.3. Morris Water Maze Task
4.17. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Newcombe, E.A.; Camats-Perna, J.; Silva, M.L.; Valmas, N.; Huat, T.J.; Medeiros, R. Inflammation: The link between comorbidities, genetics, and Alzheimer’s disease. J. Neuroinflamm. 2018, 15, 276. [Google Scholar] [CrossRef] [PubMed]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a Common Feature of Neurodegenerative Disorders. Front. Pharmacol. 2019, 10, 1008. [Google Scholar] [CrossRef] [PubMed]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Pascoal, T.A.; Benedet, A.L.; Ashton, N.J.; Kang, M.S.; Therriault, J.; Chamoun, M.; Savard, M.; Lussier, F.Z.; Tissot, C.; Karikari, T.K.; et al. Microglial activation and tau propagate jointly across Braak stages. Nat. Med. 2021, 27, 1592–1599. [Google Scholar] [CrossRef]
- Cribbs, D.H.; Berchtold, N.C.; Perreau, V.; Coleman, P.D.; Rogers, J.; Tenner, A.J.; Cotman, C.W. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: A microarray study. J. Neuroinflamm. 2012, 9, 179. [Google Scholar] [CrossRef]
- Santello, M.; Toni, N.; Volterra, A. Astrocyte function from information processing to cognition and cognitive impairment. Nat. Neurosci. 2019, 22, 154–166. [Google Scholar] [CrossRef]
- Perez-Catalan, N.A.; Doe, C.Q.; Ackerman, S.D. The role of astrocyte-mediated plasticity in neural circuit development and function. Neural Dev. 2021, 16, 1. [Google Scholar] [CrossRef]
- Hanisch, U.-K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef]
- Zhang, G.; Wang, Z.; Hu, H.; Zhao, M.; Sun, L. Microglia in Alzheimer’s Disease: A Target for Therapeutic Intervention. Front. Cell Neurosci. 2021, 15, 749587. [Google Scholar] [CrossRef]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.-C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429. [Google Scholar] [CrossRef] [Green Version]
- Wightman, D.P.; Jansen, I.E.; Savage, J.E.; Shadrin, A.A.; Bahrami, S.; Holland, D.; Rongve, A.; Børte, S.; Winsvold, B.S.; Drange, O.K.; et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat. Genet. 2021, 53, 1276–1282. [Google Scholar] [CrossRef]
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 2011, 43, 436–441. [Google Scholar] [CrossRef]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Tansey, K.E.; Cameron, D.; Hill, M.J. Genetic risk for Alzheimer’s disease is concentrated in specific macrophage and microglial transcriptional networks. Genome Med. 2018, 10, 14. [Google Scholar] [CrossRef]
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial Dysfunction and Defective β-Amyloid Clearance Pathways in Aging Alzheimer’s Disease Mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef]
- Schoch Kathleen, M.; Ezerskiy Lubov, A.; Morhaus Michaela, M.; Bannon Riley, N.; Sauerbeck Andrew, D.; Shabsovich, M.; Jafar-nejad, P.; Rigo, F.; Miller Timothy, M. Acute Trem2 reduction triggers increased microglial phagocytosis, slowing amyloid deposition in mice. Proc. Natl. Acad. Sci. USA 2021, 118, e2100356118. [Google Scholar] [CrossRef]
- Bhattacherjee, A.; Jung, J.; Zia, S.; Ho, M.; Eskandari-Sedighi, G.; St. Laurent, C.D.; McCord, K.A.; Bains, A.; Sidhu, G.; Sarkar, S.; et al. The CD33 short isoform is a gain-of-function variant that enhances Aβ1–42 phagocytosis in microglia. Mol. Neurodegener. 2021, 16, 19. [Google Scholar] [CrossRef]
- Wang, S.; Mustafa, M.; Yuede, C.M.; Salazar, S.V.; Kong, P.; Long, H.; Ward, M.; Siddiqui, O.; Paul, R.; Gilfillan, S.; et al. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J. Exp. Med. 2020, 217, e20200785. [Google Scholar] [CrossRef]
- Biber, K.; Bhattacharya, A.; Campbell, B.M.; Piro, J.R.; Rohe, M.; Staal, R.G.W.; Talanian, R.V.; Möller, T. Microglial Drug Targets in AD: Opportunities and Challenges in Drug Discovery and Development. Front. Pharmacol. 2019, 10, 840. [Google Scholar] [CrossRef] [Green Version]
- Mancuso, R.; Fryatt, G.; Cleal, M.; Obst, J.; Pipi, E.; Monzón-Sandoval, J.; Ribe, E.; Winchester, L.; Webber, C.; Nevado, A.; et al. CSF1R inhibitor JNJ-40346527 attenuates microglial proliferation and neurodegeneration in P301S mice. Brain 2019, 142, 3243–3264. [Google Scholar] [CrossRef]
- Liu, J.; Wang, F. Role of Neuroinflammation in Amyotrophic Lateral Sclerosis: Cellular Mechanisms and Therapeutic Implications. Front. Immunol. 2017, 8, 1005. [Google Scholar] [CrossRef]
- Soon, C.P.; Donnelly, P.S.; Turner, B.J.; Hung, L.W.; Crouch, P.J.; Sherratt, N.A.; Tan, J.L.; Lim, N.K.; Lam, L.; Bica, L.; et al. Diacetylbis(N(4)-methylthiosemicarbazonato) copper(II) (CuII(atsm)) protects against peroxynitrite-induced nitrosative damage and prolongs survival in amyotrophic lateral sclerosis mouse model. J. Biol. Chem. 2011, 286, 44035–44044. [Google Scholar] [CrossRef]
- Crouch, P.J.; Hung, L.W.; Adlard, P.A.; Cortes, M.; Lal, V.; Filiz, G.; Perez, K.A.; Nurjono, M.; Caragounis, A.; Du, T.; et al. Increasing Cu bioavailability inhibits Abeta oligomers and tau phosphorylation. Proc. Natl. Acad. Sci. USA 2009, 106, 381–386. [Google Scholar] [CrossRef]
- Hilton, J.B.; Mercer, S.W.; Lim, N.K.; Faux, N.G.; Buncic, G.; Beckman, J.S.; Roberts, B.R.; Donnelly, P.S.; White, A.R.; Crouch, P.J. Cu(II)(atsm) improves the neurological phenotype and survival of SOD1(G93A) mice and selectively increases enzymatically active SOD1 in the spinal cord. Sci. Rep. 2017, 7, 42292. [Google Scholar] [CrossRef]
- McAllum, E.J.; Lim, N.K.-H.; Hickey, J.L.; Paterson, B.M.; Donnelly, P.S.; Li, Q.-X.; Liddell, J.R.; Barnham, K.J.; White, A.R.; Crouch, P.J. Therapeutic effects of CuII (atsm) in the SOD1-G37R mouse model of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 586–590. [Google Scholar] [CrossRef]
- Roberts, B.R.; Lim, N.K.; McAllum, E.J.; Donnelly, P.S.; Hare, D.J.; Doble, P.A.; Turner, B.J.; Price, K.A.; Lim, S.C.; Paterson, B.M.; et al. Oral treatment with Cu(II)(atsm) increases mutant SOD1 in vivo but protects motor neurons and improves the phenotype of a transgenic mouse model of amyotrophic lateral sclerosis. J. Neurosci. 2014, 34, 8021–8031. [Google Scholar] [CrossRef]
- Hung, L.W.; Villemagne, V.L.; Cheng, L.; Sherratt, N.A.; Ayton, S.; White, A.R.; Crouch, P.J.; Lim, S.; Leong, S.L.; Wilkins, S.; et al. The hypoxia imaging agent CuII(atsm) is neuroprotective and improves motor and cognitive functions in multiple animal models of Parkinson’s disease. J. Exp. Med. 2012, 209, 837–854. [Google Scholar] [CrossRef]
- Donnelly, P.S.; Liddell, J.R.; Lim, S.; Paterson, B.M.; Cater, M.A.; Savva, M.S.; Mot, A.I.; James, J.L.; Trounce, I.A.; White, A.R.; et al. An impaired mitochondrial electron transport chain increases retention of the hypoxia imaging agent diacetylbis(4-methylthiosemicarbazonato)copperII. Proc. Natl. Acad. Sci. USA 2012, 109, 47–52. [Google Scholar] [CrossRef]
- Southon, A.; Szostak, K.; Acevedo, K.M.; Dent, K.A.; Volitakis, I.; Belaidi, A.A.; Barnham, K.J.; Crouch, P.J.; Ayton, S.; Donnelly, P.S.; et al. Cu(II) (atsm) inhibits ferroptosis: Implications for treatment of neurodegenerative disease. Br. J. Pharm. 2020, 177, 656–667. [Google Scholar] [CrossRef]
- Rowe, D.; Mathers, S.; Noel, K.; Rosenfeld, C. CuATSM Phase 2a Study Confirms Disease-Modifying Effects in Patients with Sporadic ALS Observed in the Phase 1 Study (1338). Neurology 2020, 94, 1338. [Google Scholar]
- Evans, A.; Rowe, D.; Lee, W.; Noel, K.; Rosenfeld, C. Preliminary Evidence of CuATSM Treatment Benefit in Parkinson’s Disesase. In Proceedings of the XXIV World Congress on Parkinson’s Disease and Related Disorders, Montreal, QC, Canada, 16–19 June 2019; pp. 16–19. [Google Scholar]
- Choo, X.Y.; Liddell, J.R.; Huuskonen, M.T.; Grubman, A.; Moujalled, D.; Roberts, J.; Kysenius, K.; Patten, L.; Quek, H.; Oikari, L.E.; et al. Cu(II)(atsm) Attenuates Neuroinflammation. Front. Neurosci. 2018, 12, 668. [Google Scholar] [CrossRef] [PubMed]
- Huuskonen, M.T.; Tuo, Q.Z.; Loppi, S.; Dhungana, H.; Korhonen, P.; McInnes, L.E.; Donnelly, P.S.; Grubman, A.; Wojciechowski, S.; Lejavova, K.; et al. The Copper bis(thiosemicarbazone) Complex Cu(II)(atsm) is Protective Against Cerebral Ischemia Through Modulation of the Inflammatory Milieu. Neurotherapeutics 2017, 14, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Chung, R.S.; Leung, Y.K.; Butler, C.W.; Chen, Y.; Eaton, E.D.; Pankhurst, M.W.; West, A.K.; Guillemin, G.J. Metallothionein treatment attenuates microglial activation and expression of neurotoxic quinolinic acid following traumatic brain injury. Neurotox Res. 2009, 15, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, J.; Aschner, M.; Zatta, P.; Vašák, M. Roles of the metallothionein family of proteins in the central nervous system. Brain Res. Bull. 2001, 55, 133–145. [Google Scholar] [CrossRef]
- Pankhurst, M.W.; Bennett, W.; Kirkcaldie, M.T.; West, A.K.; Chung, R.S. Increased circulating leukocyte numbers and altered macrophage phenotype correlate with the altered immune response to brain injury in metallothionein (MT)-I/II null mutant mice. J. Neuroinflamm. 2011, 8, 172. [Google Scholar] [CrossRef] [PubMed]
- Buncic, G.; Hickey, J.L.; Schieber, C.; White, J.M.; Crouch, P.J.; White, A.R.; Xiao, Z.; Wedd, A.G.; Donnelly, P.S. Water-soluble Bis(thiosemicarbazonato)copper(II) Complexes. Aust. J. Chem. 2011, 64, 244–252. [Google Scholar] [CrossRef]
- Cowley, A.R.; Dilworth, J.R.; Donnelly, P.S.; White, J.M. Copper Complexes of Thiosemicarbazone−Pyridylhydrazine (thynic) Hybrid Ligands: A New Versatile Potential Bifunctional Chelator for Copper Radiopharmaceuticals. Inorg. Chem. 2006, 45, 496–498. [Google Scholar] [CrossRef]
- McInnes, L.E.; Noor, A.; Kysenius, K.; Cullinane, C.; Roselt, P.; McLean, C.A.; Chiu, F.C.K.; Powell, A.K.; Crouch, P.J.; White, J.M.; et al. Potential Diagnostic Imaging of Alzheimer’s Disease with Copper-64 Complexes That Bind to Amyloid-β Plaques. Inorg. Chem. 2019, 58, 3382–3395. [Google Scholar] [CrossRef]
- Meda, L.; Bernasconi, S.; Bonaiuto, C.; Sozzani, S.; Zhou, D.; Otvos, L.; Mantovani, A.; Rossi, F.; Cassatella, M.A. Beta-amyloid (25-35) peptide and IFN-gamma synergistically induce the production of the chemotactic cytokine MCP-1/JE in monocytes and microglial cells. J. Immunol. 1996, 157, 1213–1218. [Google Scholar]
- Choi, Y.; Kim, H.-S.; Shin, K.Y.; Kim, E.-M.; Kim, M.; Kim, H.-S.; Park, C.H.; Jeong, Y.H.; Yoo, J.; Lee, J.-P.; et al. Minocycline Attenuates Neuronal Cell Death and Improves Cognitive Impairment in Alzheimer’s Disease Models. Neuropsychopharmacology 2007, 32, 2393. [Google Scholar] [CrossRef]
- Kriz, J.; Nguyen, M.D.; Julien, J.-P. Minocycline Slows Disease Progression in a Mouse Model of Amyotrophic Lateral Sclerosis. Neurobiol. Dis. 2002, 10, 268–278. [Google Scholar] [CrossRef] [Green Version]
- Nizami, S.; Hall-Roberts, H.; Warrier, S.; Cowley, S.A.; Di Daniel, E. Microglial inflammation and phagocytosis in Alzheimer’s disease: Potential therapeutic targets. Br. J. Pharm. 2019, 176, 3515–3532. [Google Scholar] [CrossRef]
- Gabandé-Rodríguez, E.; Keane, L.; Capasso, M. Microglial phagocytosis in aging and Alzheimer’s disease. J. Neurosci. Res. 2020, 98, 284–298. [Google Scholar] [CrossRef]
- Ribes, S.; Ebert, S.; Regen, T.; Agarwal, A.; Tauber, S.C.; Czesnik, D.; Spreer, A.; Bunkowski, S.; Eiffert, H.; Hanisch, U.K.; et al. Toll-like receptor stimulation enhances phagocytosis and intracellular killing of nonencapsulated and encapsulated Streptococcus pneumoniae by murine microglia. Infect. Immun. 2010, 78, 865–871. [Google Scholar] [CrossRef]
- Richards, M.P. Recent developments in trace element metabolism and function: Role of metallothionein in copper and zinc metabolism. J. Nutr. 1989, 119, 1062–1070. [Google Scholar] [CrossRef]
- Butler, C.A.; Thornton, P.; Brown, G.C. CD33M inhibits microglial phagocytosis, migration and proliferation, but the Alzheimer’s disease-protective variant CD33m stimulates phagocytosis and proliferation, and inhibits adhesion. J. Neurochem. 2021, 158, 297–310. [Google Scholar] [CrossRef]
- Kim, S.-M.; Mun, B.-R.; Lee, S.-J.; Joh, Y.; Lee, H.-Y.; Ji, K.-Y.; Choi, H.-R.; Lee, E.-H.; Kim, E.-M.; Jang, J.-H.; et al. TREM2 promotes Aβ phagocytosis by upregulating C/EBPα-dependent CD36 expression in microglia. Sci. Rep. 2017, 7, 11118. [Google Scholar] [CrossRef]
- Herrup, K. The case for rejecting the amyloid cascade hypothesis. Nat. Neurosci. 2015, 18, 794–799. [Google Scholar] [CrossRef]
- Hemonnot, A.-L.; Hua, J.; Ulmann, L.; Hirbec, H. Microglia in Alzheimer Disease: Well-Known Targets and New Opportunities. Front. Aging Neurosci. 2019, 11, 233. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Onuska, K.M. The Dual Role of Microglia in the Progression of Alzheimer’s Disease. J. Neurosci. 2020, 40, 1608. [Google Scholar] [CrossRef]
- Takata, K.; Ginhoux, F.; Shimohama, S. Roles of microglia in Alzheimer’s disease and impact of new findings on microglial heterogeneity as a target for therapeutic intervention. Biochem. Pharmacol. 2021, 192, 114754. [Google Scholar] [CrossRef]
- Decourt, B.; Lahiri, D.K.; Sabbagh, M.N. Targeting Tumor Necrosis Factor Alpha for Alzheimer’s Disease. Curr. Alzheimer Res. 2017, 14, 412–425. [Google Scholar] [CrossRef]
- Zheng, C.; Zhou, X.-W.; Wang, J.-Z. The dual roles of cytokines in Alzheimer’s disease: Update on interleukins, TNF-α, TGF-β and IFN-γ. Transl. Neurodegener. 2016, 5, 7. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef]
- Grubman, A.; Choo, X.Y.; Chew, G.; Ouyang, J.F.; Sun, G.; Croft, N.P.; Rossello, F.J.; Simmons, R.; Buckberry, S.; Landin, D.V.; et al. Transcriptional signature in microglia associated with Aβ plaque phagocytosis. Nat. Commun. 2021, 12, 3015. [Google Scholar] [CrossRef]
- Walker, D.G.; Whetzel, A.M.; Serrano, G.; Sue, L.I.; Beach, T.G.; Lue, L.-F. Association of CD33 polymorphism rs3865444 with Alzheimer’s disease pathology and CD33 expression in human cerebral cortex. Neurobiol. Aging 2015, 36, 571–582. [Google Scholar] [CrossRef]
- Okuzono, Y.; Sakuma, H.; Miyakawa, S.; Ifuku, M.; Lee, J.; Das, D.; Banerjee, A.; Zhao, Y.; Yamamoto, K.; Ando, T.; et al. Reduced TREM2 activation in microglia of patients with Alzheimer’s disease. FEBS Open Bio 2021, 11, 3063–3080. [Google Scholar] [CrossRef]
- Lue, L.F.; Schmitz, C.T.; Serrano, G.; Sue, L.I.; Beach, T.G.; Walker, D.G. TREM2 Protein Expression Changes Correlate with Alzheimer’s Disease Neurodegenerative Pathologies in Post-Mortem Temporal Cortices. Brain Pathol. 2015, 25, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s Disease Risk Gene CD33 Inhibits Microglial Uptake of Amyloid Beta. Neuron 2013, 78, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Stratoulias, V.; Venero, J.L.; Tremblay, M.; Joseph, B. Microglial subtypes: Diversity within the microglial community. Embo J. 2019, 38, e101997. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Waeytens, A.; de Vos, M.; Laukens, D. Evidence for a Potential Role of Metallothioneins in Inflammatory Bowel Diseases. Mediat. Inflamm. 2009, 2009, 729172. [Google Scholar] [CrossRef]
- Kim, J.-H.; Nam, Y.-P.; Jeon, S.-M.; Han, H.-S.; Suk, K. Amyloid neurotoxicity is attenuated by metallothionein: Dual mechanisms at work. J. Neurochem. 2012, 121, 751–762. [Google Scholar] [CrossRef]
- Manso, Y.; Comes, G.; López-Ramos, J.C.; Belfiore, M.; Molinero, A.; Giralt, M.; Carrasco, J.; Adlard, P.A.; Bush, A.I.; Delgado-García, J.M.; et al. Overexpression of Metallothionein-1 Modulates the Phenotype of the Tg2576 Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 51, 81–95. [Google Scholar] [CrossRef]
- Pokusa, M.; Kráľová Trančíková, A. The Central Role of Biometals Maintains Oxidative Balance in the Context of Metabolic and Neurodegenerative Disorders. Oxid. Med. Cell. Longev. 2017, 2017, 8210734. [Google Scholar] [CrossRef]
- Dai, H.; Wang, L.; Li, L.; Huang, Z.; Ye, L. Metallothionein 1: A New Spotlight on Inflammatory Diseases. Front. Immunol. 2021, 12, 739918. [Google Scholar] [CrossRef]
- Kamphuis, W.; Kooijman, L.; Schetters, S.; Orre, M.; Hol, E.M. Transcriptional profiling of CD11c-positive microglia accumulating around amyloid plaques in a mouse model for Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 1847–1860. [Google Scholar] [CrossRef]
- Podleśny-Drabiniok, A.; Marcora, E.; Goate, A.M. Microglial Phagocytosis: A Disease-Associated Process Emerging from Alzheimer’s Disease Genetics. Trends Neurosci. 2020, 43, 965–979. [Google Scholar] [CrossRef]
- Triboulet, S.; Aude-Garcia, C.; Carrière, M.; Diemer, H.; Proamer, F.; Habert, A.; Chevallet, M.; Collin-Faure, V.; Strub, J.M.; Hanau, D.; et al. Molecular responses of mouse macrophages to copper and copper oxide nanoparticles inferred from proteomic analyses. Mol. Cell Proteom. 2013, 12, 3108–3122. [Google Scholar] [CrossRef]
- Ogundeji, A.O.; Porotloane, B.F.; Pohl, C.H.; Kendrekar, P.S.; Sebolai, O.M. Copper Acyl Salicylate Has Potential as an Anti-Cryptococcus Antifungal Agent. Antimicrob. Agents Chemother. 2018, 62, e02345-17. [Google Scholar] [CrossRef] [Green Version]
- Kaur, P.; Johnson, A.; Northcote-Smith, J.; Lu, C.; Suntharalingam, K. Immunogenic Cell Death of Breast Cancer Stem Cells Induced by an Endoplasmic Reticulum-Targeting Copper(II) Complex. ChemBioChem 2020, 21, 3618–3624. [Google Scholar] [CrossRef]
- Sasaguri, H.; Nilsson, P.; Hashimoto, S.; Nagata, K.; Saito, T.; de Strooper, B.; Hardy, J.; Vassar, R.; Winblad, B.; Saido, T.C. APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 2017, 36, 2473–2487. [Google Scholar] [CrossRef]
- Franco, R.; Cedazo-Minguez, A. Successful therapies for Alzheimer’s disease: Why so many in animal models and none in humans? Front. Pharm. 2014, 5, 146. [Google Scholar] [CrossRef]
- Karran, E.; Hardy, J. A critique of the drug discovery and phase 3 clinical programs targeting the amyloid hypothesis for Alzheimer disease. Ann. Neurol. 2014, 76, 185–205. [Google Scholar] [CrossRef]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.G.; et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef]
- Farlow, M.; Arnold, S.E.; van Dyck, C.H.; Aisen, P.S.; Snider, B.J.; Porsteinsson, A.P.; Friedrich, S.; Dean, R.A.; Gonzales, C.; Sethuraman, G.; et al. Safety and biomarker effects of solanezumab in patients with Alzheimer’s disease. Alzheimers Dement. 2012, 8, 261–271. [Google Scholar] [CrossRef]
- Bohrmann, B.; Baumann, K.; Benz, J.; Gerber, F.; Huber, W.; Knoflach, F.; Messer, J.; Oroszlan, K.; Rauchenberger, R.; Richter, W.F.; et al. Gantenerumab: A novel human anti-Aβ antibody demonstrates sustained cerebral amyloid-β binding and elicits cell-mediated removal of human amyloid-β. J. Alzheimer’s Dis. 2012, 28, 49–69. [Google Scholar] [CrossRef]
- Rinne, J.O.; Brooks, D.J.; Rossor, M.N.; Fox, N.C.; Bullock, R.; Klunk, W.E.; Mathis, C.A.; Blennow, K.; Barakos, J.; Okello, A.A.; et al. 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer’s disease treated with bapineuzumab: A phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol. 2010, 9, 363–372. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Ferreira, S.; Lourenco, M.; Oliveira, M.; de Felice, F. Soluble amyloid-b oligomers as synaptotoxins leading to cognitive impairment in Alzheimer’s disease. Front. Cell. Neurosci. 2015, 9, 191. [Google Scholar] [CrossRef] [Green Version]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef]
- Borlikova, G.G.; Trejo, M.; Mably, A.J.; Mc Donald, J.M.; Sala Frigerio, C.; Regan, C.M.; Murphy, K.J.; Masliah, E.; Walsh, D.M. Alzheimer brain-derived amyloid β-protein impairs synaptic remodeling and memory consolidation. Neurobiol. Aging 2013, 34, 1315–1327. [Google Scholar] [CrossRef]
- Kittelberger, K.A.; Piazza, F.; Tesco, G.; Reijmers, L.G. Natural amyloid-β oligomers acutely impair the formation of a contextual fear memory in mice. PLoS ONE 2012, 7, e29940. [Google Scholar] [CrossRef]
- Balducci, C.; Beeg, M.; Stravalaci, M.; Bastone, A.; Sclip, A.; Biasini, E.; Tapella, L.; Colombo, L.; Manzoni, C.; Borsello, T.; et al. Synthetic amyloid-β oligomers impair long-term memory independently of cellular prion protein. Proc. Natl. Acad. Sci. USA 2010, 107, 2295–2300. [Google Scholar] [CrossRef]
- Freir, D.B.; Fedriani, R.; Scully, D.; Smith, I.M.; Selkoe, D.J.; Walsh, D.M.; Regan, C.M. Aβ oligomers inhibit synapse remodelling necessary for memory consolidation. Neurobiol. Aging 2011, 32, 2211–2218. [Google Scholar] [CrossRef]
- Gureviciene, I.; Gurevicius, K.; Mugantseva, E.; Kislin, M.; Khiroug, L.; Tanila, H. Amyloid Plaques Show Binding Capacity of Exogenous Injected Amyloid-β. J. Alzheimer’s Dis. 2017, 55, 147–157. [Google Scholar] [CrossRef]
- Kass, B.; Schemmert, S.; Zafiu, C.; Pils, M.; Bannach, O.; Kutzsche, J.; Bujnicki, T.; Willbold, D. Aβ oligomer concentration in mouse and human brain and its drug-induced reduction ex vivo. Cell Rep. Med. 2022, 3, 100630. [Google Scholar] [CrossRef]
- Atwood, C.S.; Scarpa, R.C.; Huang, X.; Moir, R.D.; Jones, W.D.; Fairlie, D.P.; Tanzi, R.E.; Bush, A.I. Characterization of Copper Interactions with Alzheimer Amyloid β Peptides. J. Neurochem. 2000, 75, 1219–1233. [Google Scholar] [CrossRef] [PubMed]
- Vorhees, C.V.; Williams, M.T. Morris water maze: Procedures for assessing spatial and related forms of learning and memory. Nat. Protoc. 2006, 1, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Vorhees, C.V.; Williams, M.T. Assessing Spatial Learning and Memory in Rodents. ILAR J. 2014, 55, 310–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deacon, R.M.; Croucher, A.; Rawlins, J.N. Hippocampal cytotoxic lesion effects on species-typical behaviours in mice. Behav. Brain Res. 2002, 132, 203–213. [Google Scholar] [CrossRef]
- Deacon, R.M.; Penny, C.; Rawlins, J.N. Effects of medial prefrontal cortex cytotoxic lesions in mice. Behav. Brain Res. 2003, 139, 139–155. [Google Scholar] [CrossRef]
- Kolb, B.; Whishaw, I.Q. Neonatal frontal lesions in hamsters impair species-typical behaviors and reduce brain weight and neocortical thickness. Behav. Neurosci. 1985, 99, 691–706. [Google Scholar] [CrossRef]
- Carter, P.A.; Swallow, J.G.; Davis, S.J.; Garland, T., Jr. Nesting behavior of house mice (Mus domesticus) selected for increased wheel-running activity. Behav. Genet. 2000, 30, 85–94. [Google Scholar] [CrossRef]
- Filali, M.; Lalonde, R. Age-related cognitive decline and nesting behavior in an APPswe/PS1 bigenic model of Alzheimer’s disease. Brain Res. 2009, 1292, 93–99. [Google Scholar] [CrossRef]
- Villemagne, V.L.; Pike, K.E.; Chételat, G.; Ellis, K.A.; Mulligan, R.S.; Bourgeat, P.; Ackermann, U.; Jones, G.; Szoeke, C.; Salvado, O.; et al. Longitudinal assessment of Aβ and cognition in aging and Alzheimer disease. Ann. Neurol. 2011, 69, 181–192. [Google Scholar] [CrossRef]
- Aizenstein, H.J.; Nebes, R.D.; Saxton, J.A.; Price, J.C.; Mathis, C.A.; Tsopelas, N.D.; Ziolko, S.K.; James, J.A.; Snitz, B.E.; Houck, P.R.; et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch. Neurol. 2008, 65, 1509–1517. [Google Scholar] [CrossRef]
- Sturchio, A.; Dwivedi, A.K.; Young, C.B.; Malm, T.; Marsili, L.; Sharma, J.S.; Mahajan, A.; Hill, E.J.; Andaloussi, S.E.L.; Poston, K.L.; et al. High cerebrospinal amyloid-β; 42 is associated with normal cognition in individuals with brain amyloidosis. eClinicalMedicine 2021, 38, 100988. [Google Scholar] [CrossRef]
- Hamprecht, B.; Löffler, F. Primary glial cultures as a model for studying hormone action. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1985; Volume 109, pp. 341–345. [Google Scholar]
- Gingras, B.A.; Suprunchuk, T.; Bayley, C.H. The Preparation of Some Thiosemicarbazones and Their Copper Complexes: Part III. Can. J. Chem. 1962, 40, 1053–1059. [Google Scholar] [CrossRef]
- Baker, E.N.; Hall, D.; Waters, T.N. Conformational influences in copper co-ordination compounds. Part IV. Crystal structure of the chloroform adduct of NN′-ethylenebis(salicylideneiminato)copper(II). J. Chem. Soc. A Inorg. Phys. Theor. 1970, 406–409. [Google Scholar] [CrossRef]
- Nayak, M.; Koner, R.; Lin, H.-H.; Flörke, U.; Wei, H.-H.; Mohanta, S. Syntheses, Structures, and Magnetic Properties of Mononuclear CuII and Tetranuclear CuII3MII (M = Cu, Co, or Mn) Compounds Derived from N,N′-Ethylenebis(3-ethoxysalicylaldimine): Cocrystallization Due to Potential Encapsulation of Water. Inorg. Chem. 2006, 45, 10764–10773. [Google Scholar] [CrossRef]
- Paterson, B.M.; Karas, J.A.; Scanlon, D.B.; White, J.M.; Donnelly, P.S. Versatile New Bis(thiosemicarbazone) Bifunctional Chelators: Synthesis, Conjugation to Bombesin(7−14)-NH2, and Copper-64 Radiolabeling. Inorg. Chem. 2010, 49, 1884–1893. [Google Scholar] [CrossRef]
- Grubman, A.; Lidgerwood, G.E.; Duncan, C.; Bica, L.; Tan, J.-L.; Parker, S.J.; Caragounis, A.; Meyerowitz, J.; Volitakis, I.; Moujalled, D.; et al. Deregulation of subcellular biometal homeostasis through loss of the metal transporter, Zip7, in a childhood neurodegenerative disorder. Acta Neuropathol. Commun. 2014, 2, 25. [Google Scholar] [CrossRef]
- Deacon, R.M. Assessing nest building in mice. Nat. Protoc. 2006, 1, 1117–1119. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choo, X.Y.; McInnes, L.E.; Grubman, A.; Wasielewska, J.M.; Belaya, I.; Burrows, E.; Quek, H.; Martín, J.C.; Loppi, S.; Sorvari, A.; et al. Novel Anti-Neuroinflammatory Properties of a Thiosemicarbazone–Pyridylhydrazone Copper(II) Complex. Int. J. Mol. Sci. 2022, 23, 10722. https://doi.org/10.3390/ijms231810722
Choo XY, McInnes LE, Grubman A, Wasielewska JM, Belaya I, Burrows E, Quek H, Martín JC, Loppi S, Sorvari A, et al. Novel Anti-Neuroinflammatory Properties of a Thiosemicarbazone–Pyridylhydrazone Copper(II) Complex. International Journal of Molecular Sciences. 2022; 23(18):10722. https://doi.org/10.3390/ijms231810722
Chicago/Turabian StyleChoo, Xin Yi, Lachlan E. McInnes, Alexandra Grubman, Joanna M. Wasielewska, Irina Belaya, Emma Burrows, Hazel Quek, Jorge Cañas Martín, Sanna Loppi, Annika Sorvari, and et al. 2022. "Novel Anti-Neuroinflammatory Properties of a Thiosemicarbazone–Pyridylhydrazone Copper(II) Complex" International Journal of Molecular Sciences 23, no. 18: 10722. https://doi.org/10.3390/ijms231810722