
The Pnictogen Bond, Together with Other Non-Covalent Interactions, in the Rational Design of One-, Two- and Three-Dimensional Organic-Inorganic Hybrid Metal Halide Perovskite Semiconducting Materials, and Beyond




Abstract
:1. Introduction
2. Computational Details
3. Illustrative Crystal Systems
3.1. Diazanediium Dichloride
3.2. Methylammonium Lead Halide Perovskites
3.3. Methylammonium and Deutero-Methylammonium Halides
3.4. Ammonium Cyanate
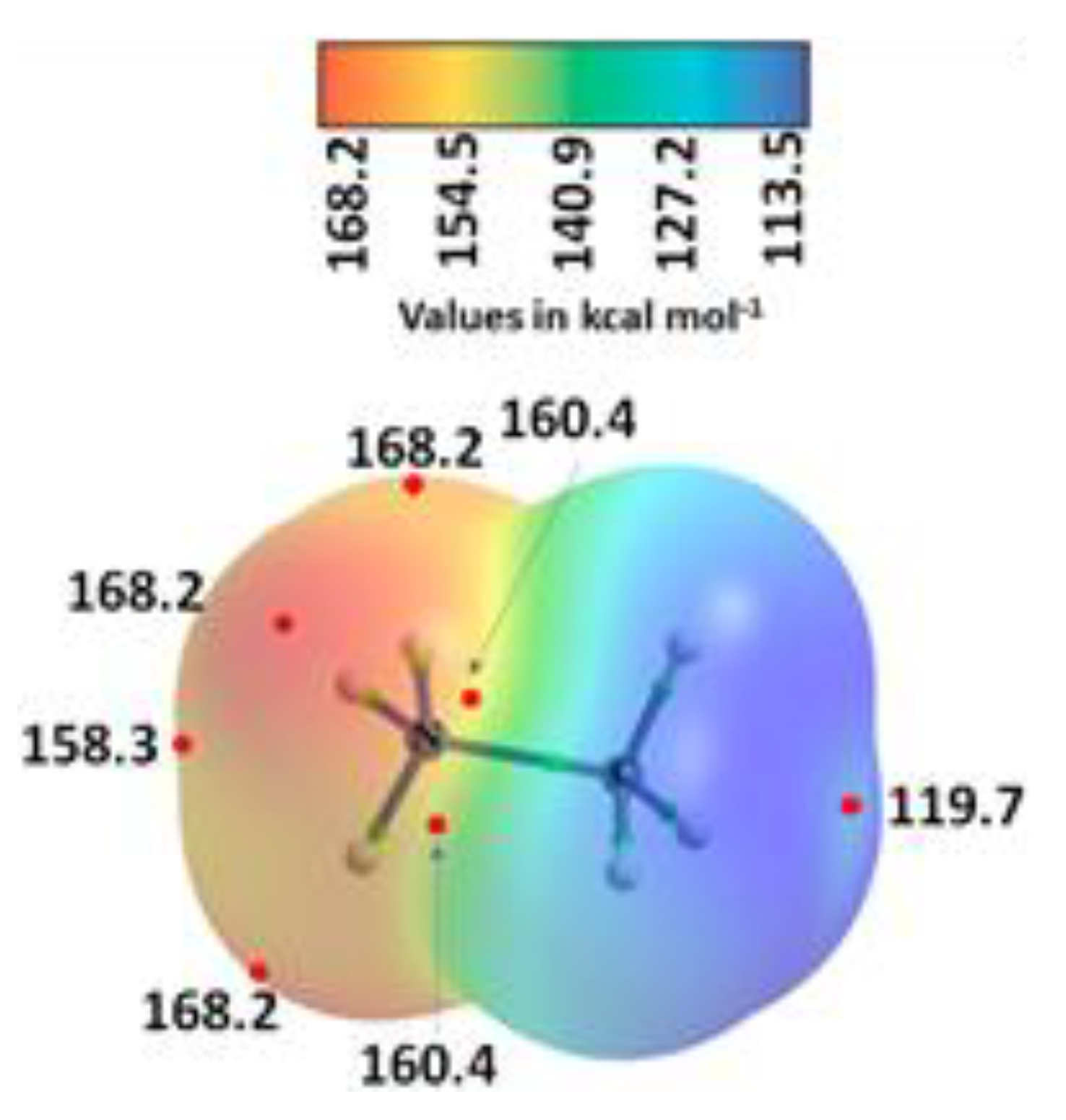
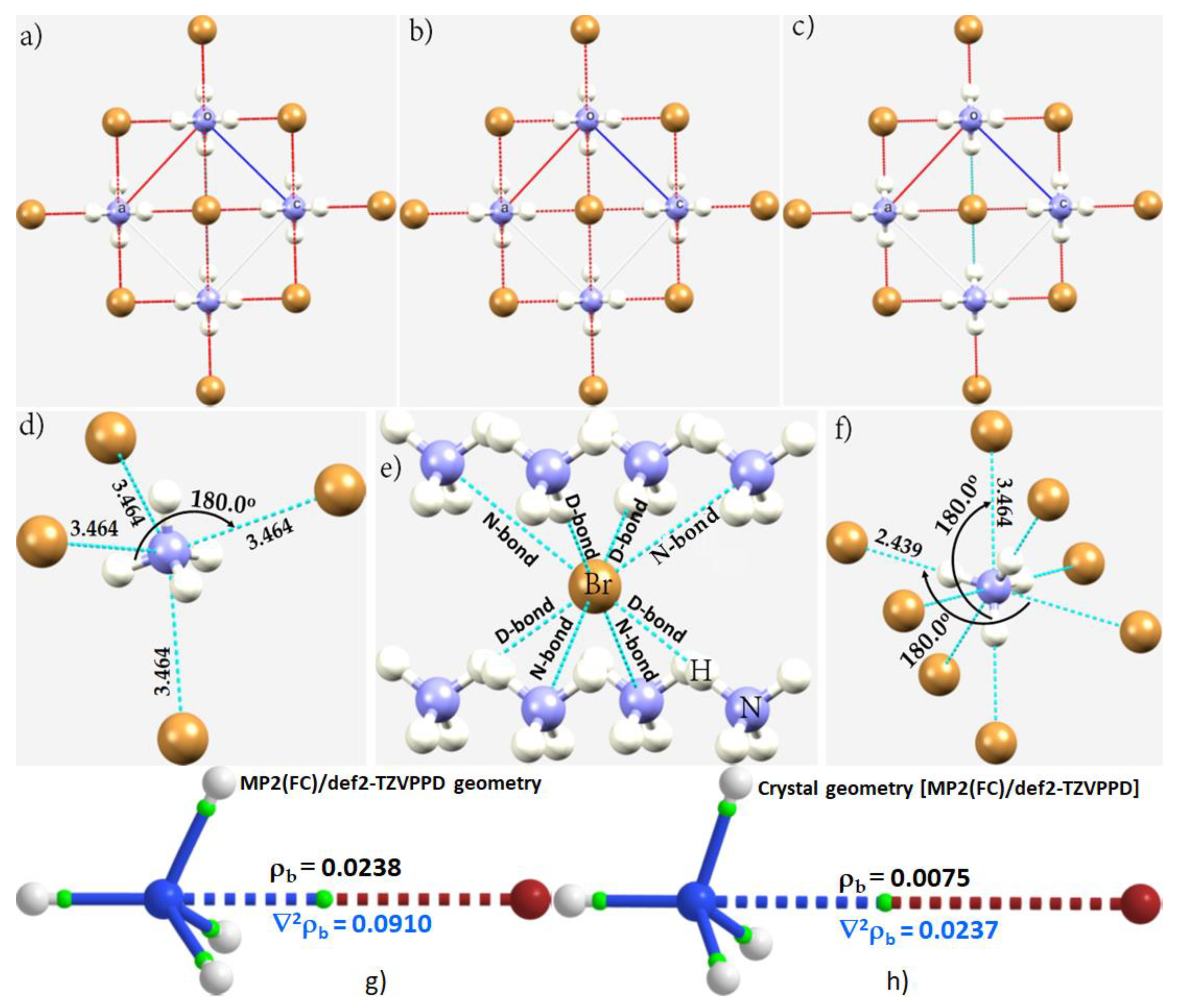
3.5. The Ammonium Halides
3.6. The Crystal Structure of ([I44Pb18][CH3NH3]
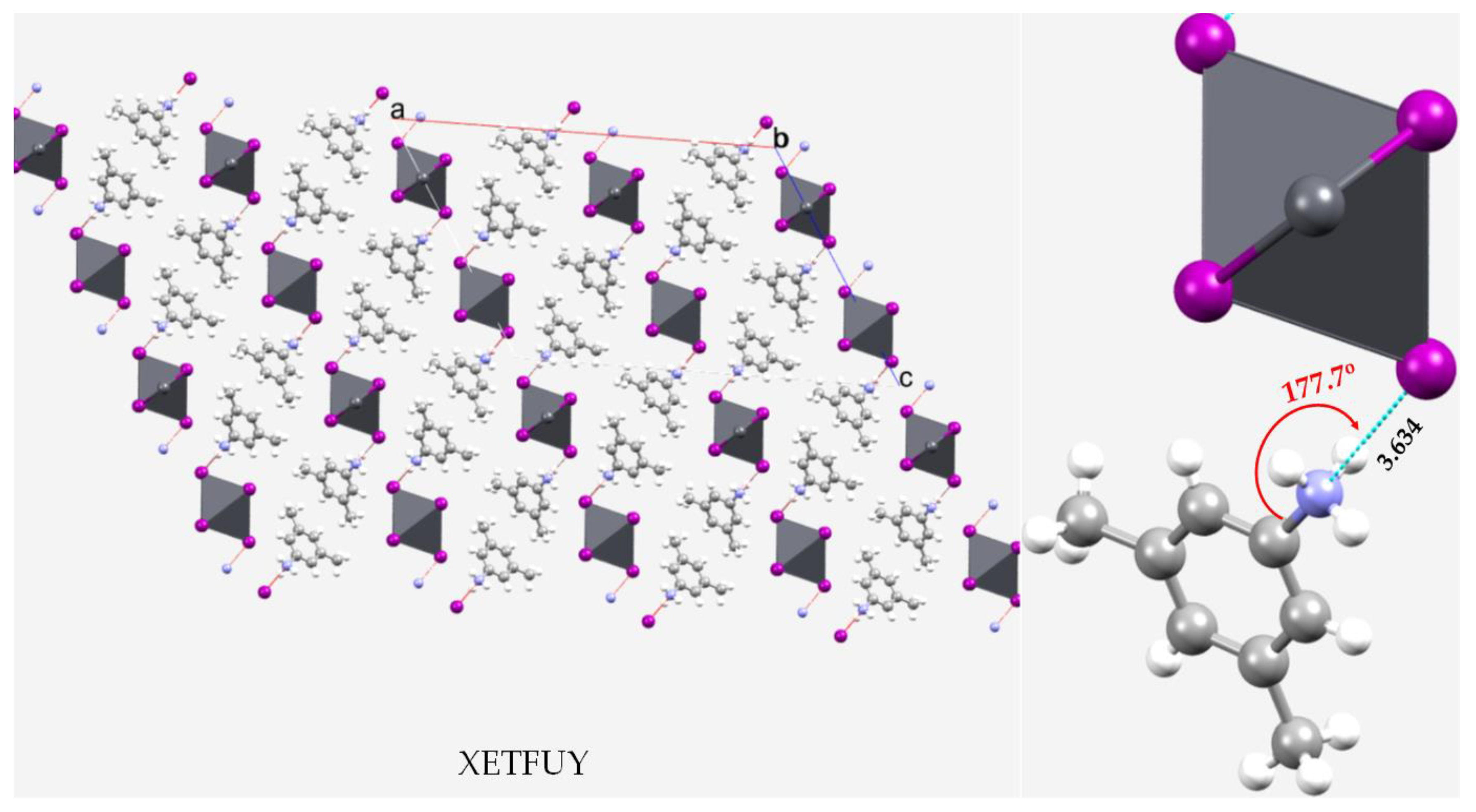
3.7. Pnictogen Bond in 2D Functional Crystals of Metal Halide Perovskites: Derivatives of Ammonium as Pnictogen Bond Donors
4. Pnictogen Bonding in 1D (One-Dimensional) Perovskite Systems
5. Pnictogen Bonding in Zero-Dimensional (0D) Crystal Systems
6. Statistical Analysis of N-Centered Pnictogen Bond Distances and Angles in Crystals
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Foster, S.L.; Bakovic, S.I.P.; Duda, R.D.; Maheshwari, S.; Milton, R.D.; Minteer, S.D.; Janik, M.J.; Renner, J.N.; Greenlee, L.F. Catalysts for nitrogen reduction to ammonia. Nat. Catal. 2018, 1, 490–500. [Google Scholar] [CrossRef]
- Uyanik, M.; Kato, T.; Sahara, N.; Katade, O.; Ishihara, K. High-Performance Ammonium Hypoiodite/Oxone Catalysis for Enantioselective Oxidative Dearomatization of Arenols. ACS Catal. 2019, 9, 11619–11626. [Google Scholar] [CrossRef]
- Hinokuma, S.; Sato, K. Ammonia Combustion Catalysts. Chem. Lett. 2021, 50, 752–759. [Google Scholar] [CrossRef]
- Lamb, K.E.; Dolan, M.D.; Kennedy, D.F. Ammonia for hydrogen storage; A review of catalytic ammonia decomposition and hydrogen separation and purification. Int. J. Hydrogen Energy 2019, 44, 3580–3593. [Google Scholar] [CrossRef]
- US Geological Survey. Mineral Commodity Summaries 2020; US Geological Survey: Reston, VA, USA, 2020. [CrossRef] [Green Version]
- MacFarlane, D.R.; Cherepanov, P.V.; Choi, J.; Suryanto, B.H.R.; Hodgetts, R.Y.; Bakker, J.M.; Ferrero Vallana, F.M.; Simonov, A.N. A Roadmap to the Ammonia Economy. Joule 2020, 4, 1186–1205. [Google Scholar] [CrossRef]
- Ogasawara, K.; Nakao, T.; Kishida, K.; Ye, T.-N.; Lu, Y.; Abe, H.; Niwa, Y.; Sasase, M.; Kitano, M.; Hosono, H. Ammonia Decomposition over CaNH-Supported Ni Catalysts via an NH2−-Vacancy-Mediated Mars–van Krevelen Mechanism. ACS Catal. 2021, 11, 11005–11015. [Google Scholar] [CrossRef]
- Mukherjee, S.; Devaguptapu, S.V.; Sviripa, A.; Lund, C.R.F.; Wu, G. Low-temperature ammonia decomposition catalysts for hydrogen generation. Appl. Catal. B: Environ. 2018, 226, 162–181. [Google Scholar] [CrossRef]
- Rouwenhorst, K.H.R.; Engelmann, Y.; van ‘t Veer, K.; Postma, R.S.; Bogaerts, A.; Lefferts, L. Plasma-driven catalysis: Green ammonia synthesis with intermittent electricity. Green Chem. 2020, 22, 6258–6287. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M.; Yamashita, K. The Nitrogen Bond, or The Nitrogen-centered Pnictogen Bond: The Covalently Bound Nitrogen Atom in Molecular Entities and Crystals as a Pnictogen Bond Donor. Compounds 2022, 2, 80–110. [Google Scholar] [CrossRef]
- Reiss, G.J. UJONUD, dep. no. 790189. CSD Commun. 2010. [Google Scholar] [CrossRef]
- Partridge, H.; Bauschlicher, C.W. Calculation of magnesium (1+)-ligand relative binding energies. J. Phys. Chem. 1992, 96, 8827–8832. [Google Scholar] [CrossRef]
- Kim, K.S.; Lee, S.; Mhin, B.J.; Cho, S.J.; Kim, J. Structures and energetics of (Zn (NH3)n2+ (n = 4–6). Coordination number of Zn2+ by ammine. Chem. Phys. Lett. 1993, 216, 309–312. [Google Scholar] [CrossRef]
- Dudev, T.; Cowan, J.A.; Lim, C. Competitive binding in magnesium coordination chemistry: Water versus ligands of biological interest. J. Am. Chem. Soc. 1999, 121, 7665–7673. [Google Scholar] [CrossRef]
- Bérces, A.; Nukada, T.; Margl, P.; Ziegler, T. Solvation of Cu2+ in Water and Ammonia. Insight from Static and Dynamical Density Functional Theory. J. Phys. Chem. A 1999, 103, 9693–9701. [Google Scholar] [CrossRef]
- Pavelka, M.; Burda, J.V. Theoretical description of copper Cu (I)/Cu (II) complexes in mixed ammine-aqua environment. DFT and ab initio quantum chemical study. Chem. Phys. 2005, 312, 193–204. [Google Scholar] [CrossRef]
- Hancock, R.D.; Bartolotti, L.J. Density Functional Theory-Based Prediction of the Formation Constants of Complexes of Ammonia in Aqueous Solution: Indications of the Role of Relativistic Effects in the Solution Chemistry of Gold(I). Inorg. Chem. 2005, 44, 7175–7183. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Cukrowski, I.; Marques, H.M. DFT-X3LYP Studies on the Coordination Chemistry of Ni2+. Part 1: Six Coordinate [Ni(NH3)n(H2O)6-n]2+ Complexes. J. Phys. Chem. A 2008, 112, 10657–10666. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Cukrowski, I.; Marques, H.M. Low-spin complexes of Ni2+ with six NH3 and H2O ligands: A DFT–RX3LYP study. J. Mol. Str. (THEOCHEM) 2009, 915, 20–32. [Google Scholar] [CrossRef] [Green Version]
- Varadwaj, P.R.; Marques, H.M. The physical chemistry of coordinated aqua-, ammine-, and mixed-ligand Co2+ complexes: DFT studies on the structure, energetics, and topological properties of the electron density. Phys. Chem. Chem. Phys. 2010, 12, 2126–2138. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Varadwaj, A.; Jin, B.Y. Ligand(s)-to-metal charge transfer as a factor controlling the equilibrium constants of late first-row transition metal complexes: Revealing the Irving-Williams thermodynamical series. Phys. Chem. Chem. Phys. 2015, 17, 805–811. [Google Scholar] [CrossRef]
- Saparov, B.; Mitzi, D.B. Organic–Inorganic Perovskites: Structural Versatility for Functional Materials Design. Chem. Rev. 2016, 116, 4558–4596. [Google Scholar] [CrossRef] [PubMed]
- CSD 5.43; Cambridge Crystallographic Data Centre (CCDC): Cambridge, UK, 2022.
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Varadwaj, A.; Varadwaj, P.R.; Marques, H.M.; Yamashita, K. Halogen in Materials Design: Revealing the Nature of Hydrogen Bonding and Other Non-Covalent Interactions in the Polymorphic Transformations of Methylammonium Lead Tribromide Perovskite. Mater. Chem. Today 2018, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M.; Yamashita, K. Significance of hydrogen bonding and other noncovalent interactions in determining octahedral tilting in the CH3NH3PbI3 hybrid organic-inorganic halide perovskite solar cell semiconductor. Sci. Rep. 2019, 9, 50. [Google Scholar] [CrossRef] [Green Version]
- Varadwaj, P.R.; Varadwaj, A.; Jin, B.Y. Significant evidence of C···O and C···C long-range contacts in several heterodimeric complexes of CO with CH3-X, should one refer to them as carbon and dicarbon bonds! Phys. Chem. Chem. Phys. 2014, 16, 17238–17252. [Google Scholar] [CrossRef] [PubMed]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. Carbon−carbon bonding between nitrogen heterocyclic carbenes and CO2. J. Phys. Chem. A 2017, 121, 8136–8146. [Google Scholar] [CrossRef] [PubMed]
- Marín-Luna, M.; Alkorta, I.; Elguero, J. Cooperativity in Tetrel Bonds. J. Phys Chem. A 2016, 120, 648–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellenbrandt, M. The Inorganic Crystal Structure Database (ICSD)—Present and Future. Crystallogr. Rev. 2004, 10, 17–22. [Google Scholar] [CrossRef]
- Belsky, A.; Hellenbrandt, M.; Karen, V.L.; Luksch, P. New developments in the Inorganic Crystal Structure Database (ICSD): Accessibility in support of materials research and design. Acta Crystallogr. B 2002, 58, 364–369. [Google Scholar] [CrossRef] [Green Version]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The σ-hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Janjić, G.V.; Zarić, S.D. σ-Hole interactions of covalently-bonded nitrogen, phosphorus and arsenic: A survey of crystal structures. Crystals 2014, 4, 12–31. [Google Scholar] [CrossRef] [Green Version]
- Varadwaj, A.; Varadwaj, P.R.; Marques, H.M.; Yamashita, K. The Pnictogen Bond: The Covalently Bound Arsenic Atom in Molecular Entities in Crystals as a Pnictogen Bond Donor. Molecules 2022, 27, 3421. [Google Scholar] [CrossRef] [PubMed]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M.; Yamashita, K. The Phosphorous Bond, or the Phosphorous-Centered Pnictogen Bond: The Covalently Bound Phosphorous Atom in Molecular Entities and Crystals as a Pnictogen Bond Donor. Molecules 2022, 27, 1487. [Google Scholar] [CrossRef] [PubMed]
- Varadwaj, A.; Varadwaj, P.R.; Marques, H.M.; Yamashita, K. The Stibium Bond or the Antimony-Centered Pnictogen Bond: The Covalently Bound Antimony Atom in Molecular Entities in Crystal Lattices as a Pnictogen Bond Donor. Int. J. Mol. Sci. 2022, 23, 4674. [Google Scholar] [CrossRef]
- Mokrai, R.; Barrett, J.; Apperley, D.C.; Batsanov, A.S.; Benkő, Z.; Heift, D. Weak Pnictogen Bond with Bismuth: Experimental Evidence Based on Bi−P Through-Space Coupling. Chem. Eur. J. 2019, 25, 4017–4024. [Google Scholar] [CrossRef] [Green Version]
- Politzer, P.; Murray, J.S. The use and misuse of van der Waals radii. Struct. Chem. 2021, 32, 623–629. [Google Scholar] [CrossRef]
- Schiemenz, G.P. The sum of van der Waals radii—A pitfall in the search for bonding. Z. Naturforsch. B 2007, 62, 235–243. [Google Scholar] [CrossRef] [Green Version]
- Dean, P.A.W. Facets of van der Waals Radii That Are Not Commonly Included in Undergraduate Textbooks. J. Chem. Ed. 2014, 91, 154–157. [Google Scholar] [CrossRef]
- Alvarez, S. A cartography of the van der Waals territories. Dalton Trans. 2013, 42, 8617–8636. [Google Scholar] [CrossRef] [Green Version]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M. Halogen Bonding: A Halogen-Centered Noncovalent Interaction Yet to Be Understood. Inorganics 2019, 7, 40. [Google Scholar] [CrossRef] [Green Version]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefebvre, C.; Rubez, G.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Hénon, E. Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density. Phys. Chem. Chem. Phys. 2017, 19, 17928–17936. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, C.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Piquemal, J.-P.; Hénon, E. The Independent Gradient Model: A New Approach for Probing Strong and Weak Interactions in Molecules from Wave Function Calculations. ChemPhysChem 2018, 19, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Head-Gordon, M.; Pople, J.A. A direct MP2 gradient method. Chem. Phys. Lett. 1990, 166, 275–280. [Google Scholar] [CrossRef]
- Head-Gordon, M.; Head-Gordon, T. Analytic MP2 frequencies without fifth-order storage. Theory and application to bifurcated hydrogen bonds in the water hexamer. Chem. Phys. Lett. 1994, 220, 122–128. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Wilson, A.K.; Woon, D.E.; Peterson, K.A.; Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. IX. The atoms gallium through krypton. J. Chem. Phys. 1999, 110, 7667–7676. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Politzer, P.; Murray, J.S.; Clark, T.; Resnati, G. The s-hole revisited. Phys. Chem. Chem. Phys. 2017, 19, 32166–32178. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. The π-hole revisited. Phys. Chem. Chem. Phys. 2021, 23, 16458–16468. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding: An electrostatically-driven highly directional noncovalent interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Clark, T.; Riley, K.E.; Politzer, P. σ-Holes, π-holes and electrostatically-driven interactions. J. Mol. Model. 2012, 18, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S. σ-Hole Interactions: Perspectives and Misconceptions. Crystals 2017, 7, 212. [Google Scholar] [CrossRef] [Green Version]
- Varadwaj, P.R.; Varadwaj, A.; Jin, B.-Y. Unusual bonding modes of perfluorobenzene in its polymeric (dimeric, trimeric and tetrameric) forms: Entirely negative fluorine interacting cooperatively with entirely negative fluorine. Phys. Chem. Chem. Phys. 2015, 17, 31624–31645. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M.; Yamashita, K. Can Combined Electrostatic and Polarization Effects Alone Explain the F···F Negative-Negative Bonding in Simple Fluoro-Substituted Benzene Derivatives? A First-Principles Perspective. Computation 2018, 6, 51. [Google Scholar] [CrossRef] [Green Version]
- Tschakert, J.; Zhong, Q.; Martin-Jimenez, D.; Carracedo-Cosme, J.; Romero-Muñiz, C.; Henkel, P.; Schlöder, T.; Ahles, S.; Mollenhauer, D.; Wegner, H.A.; et al. Surface-controlled reversal of the selectivity of halogen bonds. Nat. Commun. 2020, 11, 5630. [Google Scholar] [CrossRef]
- Riley, K.E.; Murray, J.S.; Fanfrlík, J.; Řezáč, J.; Solá, R.J.; Concha, M.C.; Ramos, F.M.; Politzer, P. Halogen bond tunability I: The effects of aromatic fluorine substitution on the strengths of halogen-bonding interactions involving chlorine, bromine, and iodine. J. Mol. Modeling 2011, 17, 3309–3318. [Google Scholar] [CrossRef] [PubMed]
- Varadwaj, A.; Varadwaj, P.R.; Jin, B.-Y. Can an entirely negative fluorine in a molecule, viz. perfluorobenzene, interact attractively with the entirely negative site (s) on another molecule (s)? Like liking like! RSC Adv. 2016, 6, 19098–19110. [Google Scholar] [CrossRef]
- Varadwaj, A.; Varadwaj, P.R.; Marques, H.M.; Yamashita, K.; Pradeep, R.; Varadwaj, H.M.M.; Koichi, Y. Comment on “Extended Halogen Bonding between Fully Fluorinated Aromatic Molecules: Kawai et al., ACS Nano, 2015, 9, 2574”. arXiv 2017, arXiv:1802.09995. [Google Scholar]
- Varadwaj, A.; Marques, H.M.; Varadwaj, P.R. Is the Fluorine in Molecules Dispersive? Is Molecular Electrostatic Potential a Valid Property to Explore Fluorine-Centered Non-Covalent Interactions? Molecules 2019, 24, 379. [Google Scholar] [CrossRef] [Green Version]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Popelier, P.L.A. Atoms in Molecules: An Introduction; Pearson Education: Harlow, UK, 2000. [Google Scholar]
- Matta, C.F.; Boyd, R.J.T. The Quantum Theory of Atoms in Molecules; Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- Love, I. An AIM analysis of S,O bonds. J. Phys. Chem. A 2009, 113, 2640–2646. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally ob-served electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Kuznetsov, M.L. Relationships between Interaction Energy and Electron Density Properties for Homo Halogen Bonds of the [(A)nY–X···X–Z(B)m] Type (X = Cl, Br, I). Molecules 2019, 24, 2733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mata, I.; Alkorta, I.; Espinosa, E.; Molins, E. Relationships between interaction energy, intermolecular distance and electron density properties in hydrogen bonded complexes under external electric fields. Chem. Phys. Lett. 2011, 507, 185–189. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinhold, F. Natural localized molecular orbitals. J. Chem. Phys. 1985, 83, 1736–1740. [Google Scholar] [CrossRef]
- Reed, A.R.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Weinhold, F.; Carpenter, J.E. The Structure of Small Molecules; Naaman, R., Vager, Z., Eds.; Plenum: New York, NY, USA, 1988; pp. 227–236. [Google Scholar]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Molec. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Keith, T.A. AIMAll (V. 19.10.12); TK Gristmill Software. Overland Park, KS, USA, 2019. Available online: http://aim.tkgristmill.com (accessed on 21 March 2022).
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comp. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Varadwaj, P.R. Methylammonium Lead Trihalide Perovskite Solar Cell Semiconductors Are Not Organometallic: A Perspective. Helv. Chim. Acta 2017, 100, e1700090. [Google Scholar] [CrossRef]
- Bolte, M. AWEJEW, dep. no. 2090637. CSD Commun. 2021. [Google Scholar] [CrossRef]
- Qiu, H.; Li, F.; Jin, C.; Lu, J.; Yang, Z.; Pan, S.; Mutailipu, M. (N2H6)[HPO3F]2: Maximizing the optical anisotropy of deep-ultraviolet fluorophosphates. Chem. Commun. 2022, 58, 5594–5597. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Zuo, C.; Niu, M.; Zhou, C.; Bradley, S.J.; Hall, C.R.; Xu, W.; Wen, X.; Hao, X.; Gao, M.; et al. Revealing the Role of Methylammonium Chloride for Improving the Performance of 2D Perovskite Solar Cells. ACS Appl. Mater. Interfaces 2020, 12, 25980–25990. [Google Scholar] [CrossRef] [PubMed]
- Varadwaj, A.; Varadwaj, P.R.; Yamashita, K. Revealing the Chemistry between Bandgap and Binding Energy for Pb/Sn-based Trihalide Perovskite Solar Cell Semiconductors. ChemSusChem 2018, 11, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Varadwaj, A.; Varadwaj, P.R.; Yamashita, K. Revealing the Cooperative Chemistry of the Organic Cation in the Methylammonium Lead Triiodide Perovskite Semiconductor System. ChemistrySelect 2018, 3, 7269–7282. [Google Scholar] [CrossRef]
- Chi, L.; Swainson, I.; Cranswick, L.; Her, J.-H.; Stephens, P.; Knop, O. The ordered phase of methylammonium lead chloride CH3ND3PbCl3. J. Solid State Chem. 2005, 178, 1376–1385. [Google Scholar] [CrossRef]
- Swainson, I.P.; Hammond, R.P.; Soullière, C.; Knop, O.; Massa, W. Phase transitions in the perovskite methylammonium lead bromide, CH3ND3PbBr3. J. Solid State Chem. 2003, 176, 97–104. [Google Scholar] [CrossRef]
- Whitfield, P.S.; Herron, N.; Guise, W.E.; Page, K.; Cheng, Y.Q.; Milas, I.; Crawford, M.K. Structures, Phase Transitions and Tricritical Behavior of the Hybrid Perovskite Methyl Ammonium Lead Iodide. Sci. Rep. 2016, 6, 35685. [Google Scholar] [CrossRef] [Green Version]
- Weller, M.T.; Weber, O.J.; Henry, P.F.; Di Pumpo, A.M.; Hansen, T.C. Complete structure and cation orientation in the perovskite photovoltaic methylammonium lead iodide between 100 and 352 K. Chem. Commun. 2015, 51, 4180–4183. [Google Scholar]
- Yamamuro, O.; Matsuo, T.; Suga, H.; David, W.I.F.; Ibberson, R.M.; Leadbetter, A.J. Neutron diffraction and calorimetric studies of methylammonium iodide. Acta Cryst. B 1992, 48, 329–336. [Google Scholar] [CrossRef]
- Wang, R.T.; Xu, A.F.; Chen, J.Y.; Yang, L.W.; Xu, G.; Jarvis, V.; Britten, J.F. KUBNOK02, dep. no. 1946189. CSD Commun. 2019. [Google Scholar] [CrossRef]
- Li, W.; Huang, D.; Lv, Y. Theoretical study on the mechanism and stereochemistry of the cinchona–thiourea organocatalytic hydrophosphonylation of an α-ketoester. Org. Biomol. Chem. 2013, 11, 7497–7506. [Google Scholar] [CrossRef]
- Sen, S.; Patwari, G.N. Electrostatics and Dispersion in X–H···Y (X = C, N, O; Y = N, O) Hydrogen Bonds and Their Role in X–H Vibrational Frequency Shifts. ACS Omega 2018, 3, 18518–18527. [Google Scholar] [CrossRef]
- Varadwaj, A.; Varadwaj, P.R.; Yamashita, K. Hybrid organic–inorganic CH3NH3PbI3 perovskite building blocks: Revealing ultra-strong hydrogen bonding and mulliken inner complexes and their implications in materials design. J. Comp. Chem. 2017, 38, 2802–2818. [Google Scholar] [CrossRef] [PubMed]
- Parker, T.M.; Burns, L.A.; Parrish, R.M.; Ryno, A.G.; Sherrill, C.D. Levels of symmetry adapted perturbation theory (SAPT). I. Efficiency and performance for interaction energies. J. Chem. Phys. 2014, 140, 094106. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.E.; Weinhold, F. Natural bond orbital analysis of near-Hartree–Fock water dimer. J. Chem. Phys. 1983, 78, 4066–4073. [Google Scholar] [CrossRef]
- MacLean, E.J.; Harris, K.D.M.; Kariuki, B.M.; Kitchin, S.J.; Tykwinski, R.R.; Swainson, I.P.; Dunitz, J.D. Ammonium Cyanate Shows N−H···N Hydrogen Bonding, Not N−H···O. J. Am. Chem. Soc. 2003, 125, 14449–14451. [Google Scholar] [CrossRef] [PubMed]
- Nelyubina, Y.V.; Antipin, M.Y.; Lyssenko, K.A. Are Halide···Halide Contacts a Feature of Rock-Salts Only? J. Phys. Chem. A 2007, 111, 1091–1095. [Google Scholar] [CrossRef]
- Nelyubina, Y.V.; Korlyukov, A.A.; Lyssenko, K.A. Experimental Charge Density Evidence for Pnicogen Bonding in a Crystal of Ammonium Chloride. ChemPhysChem 2015, 16, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Levy, H.A.; Peterson, S.W. Neutron Diffraction Determination of the Crystal Structure of Ammonium Bromide in Four Phases1. J. Am. Chem. Soc. 1953, 75, 1536–1542. [Google Scholar] [CrossRef]
- Kolomiichuk, V.N.; Dvoryankin, V.F. An electron diffraction determination of the positions of hydrogen atoms in NH4Br. Kristallografiya 1964, 9, 50–56. [Google Scholar]
- Kolomiichuk, V.N. An electron-diffraction study of a low-temperature ammonium bromide phase. Kristallografiya 1965, 10, 565–567. [Google Scholar]
- Balagurov, A.M.; Kozlenko, D.P.; Savenko, B.N.; Glazkov, V.P.; Somenkov, V.A.; Hull, S. Neutron diffraction study of structural changes in ammonium halides under high pressure. Phys. B Cond. Matter 1999, 265, 92–96. [Google Scholar] [CrossRef]
- Pistorius, C.W.F.T. Phase relations and structures of solids at high pressures. Prog. Solid State Chem. 1976, 11, 1–151. [Google Scholar] [CrossRef]
- Huang, Y.; Huang, X.; Wang, L.; Wu, G.; Duan, D.; Bao, K.; Zhou, Q.; Liu, B.; Cui, T. Structural properties of ammonium iodide under high pressure. RSC Adv. 2015, 5, 40336–40340. [Google Scholar] [CrossRef]
- Fateev, S.A.; Petrov, A.A.; Khrustalev, V.N.; Dorovatovskii, P.V.; Zubavichus, Y.V.; Goodilin, E.A.; Tarasov, A.B. Solution Processing of Methylammonium Lead Iodide Perovskite from γ-Butyrolactone: Crystallization Mediated by Solvation Equilibrium. Chem. Mater. 2018, 30, 5237–5244. [Google Scholar] [CrossRef]
- Krautscheid, H.; Vielsack, F. [Pb18I44]8−—An Iodoplumbate with an Unusual Structure. Angew. Chem. Int. Ed. Engl. 1995, 34, 2035–2037. [Google Scholar] [CrossRef]
- Billing, D.G.; Lemmerer, A. Synthesis, characterization and phase transitions of the inorganic–organic layered perovskite-type hybrids [(CnH2n+1NH3)2PbI4] (n = 12, 14, 16 and 18). New J. Chem. 2008, 32, 1736–1746. [Google Scholar] [CrossRef]
- Yang, W.; Xiao, X.; He, H.; Tong, G.; Hu, J.; Xiao, X.; Chen, J.; Li, M.; He, Y. Intermolecular Hydrogen-Bonding Correlated Structure Distortion and Broadband White-Light Emission in 5-Ammonium Valeric Acid Templated Lead Chloride Perovskites. Cryst. Growth Des. 2021, 21, 5731–5739. [Google Scholar] [CrossRef]
- Lemmerer, A.; Billing, D.G. Lead halide inorganic–organic hybrids incorporating diammonium cations. CrystEngComm 2012, 14, 1954–1966. [Google Scholar] [CrossRef]
- Lemmerer, A.; Billing, D.G. Synthesis, characterization and phase transitions of the inorganic–organic layered perovskite-type hybrids [(CnH2n+1NH3)2PbI4], n = 7, 8, 9 and 10. Dalton Trans. 2012, 41, 1146–1157. [Google Scholar] [CrossRef]
- Lee, J.-H.; Bristowe, N.C.; Lee, J.H.; Lee, S.-H.; Bristowe, P.D.; Cheetham, A.K.; Jang, H.M. Resolving the Physical Origin of Octahedral Tilting in Halide Perovskites. Chem. Mater. 2016, 28, 4259–4266. [Google Scholar] [CrossRef] [Green Version]
- Pradeesh, K.; Yadav, G.S.; Singh, M.; Vijaya Prakash, G. Synthesis, structure and optical studies of inorganic–organic hybrid semiconductor, NH3(CH2)12NH3PbI4. Mater. Chem. Phys. 2010, 124, 44–47. [Google Scholar] [CrossRef]
- Harchani, A.; Carpenter-Warren, C.L.; Slawin, A.M.Z.; Haddad, A. Structure and properties evolution with inorganic and organic acids of a new organo-chlorocadmate compound (C6H20N3)2[Cd2Cl10]: Theoretical approach. J. Mol. Str. 2019, 1192, 49–58. [Google Scholar] [CrossRef]
- Kefi, R.; Maher, E.G.; Zeller, M.; Lefebvre, F.; Ben Nasr, C. CCDC 851463: Experimental Crystal Structure Determination; University of Texas Arlington: Austin, TX, USA, 2012. [Google Scholar] [CrossRef]
- Song, N.; Chen, S.-P.; Fan, X.-W.; Tan, Y.-H.; Wei, W.-J.; Tang, Y.-Z. Regulating Reversible Phase Transition Behaviors by Poly-H/F Substitution in Hybrid Perovskite-Like 2[CH2FCH2NH3]·[CdCl4]. ACS Omega 2020, 5, 6773–6780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lartigue-Bourdeau, C.; Chanh, N.B.; Duplessix, R.; Gallois, B. Thermal study and crystal structure of a perovskite-type unsaturated molecular composite: Propargylamine and cadmium chloride complex salt. J. Phys. Chem. Solids 1993, 54, 349–356. [Google Scholar] [CrossRef]
- Bonamartini Corradi, A.; Cramarossa, M.R.; Manfredini, T.; Giusti, J.; Battaglia, L.P.; Saccani, A.; Sandrolini, F. Structural, Thermal, and Electrical Characterization of Bis(ethylethylene)diammonium Dichloride Tetrachlorocadmate(II) with Perovskite-like Structure. Chem. Mater. 1994, 6, 1499–1503. [Google Scholar] [CrossRef]
- Kind, R. Structural phase transitions in perovskite layer structures. Ferroelectrics 1980, 24, 81–88. [Google Scholar] [CrossRef]
- De Jongh, L.J.; Miedema, A.R. Experiments on simple magnetic model systems. Adv. Phys. 2001, 50, 947–1170. [Google Scholar] [CrossRef]
- Kind, R.; Plesko, S.; Arend, H.; Blinc, R.; Zeks, B.; Seliger, J.; Lozar, B.; Slak, J.; Levstik, A.; Filipic, C.; et al. Dynamics of the n-decylammonium chains in the perovskite-type layer structure compound (C10H21NH3)2CdCl4. J. Chem. Phys. 1979, 71, 2118–2130. [Google Scholar] [CrossRef]
- Depmeier, W. The uniqueness of the propyl compound in the series (CnH2n+1NH3)2MnCl4 with n = 1–10. J. Solid State Chem. 1979, 29, 15–26. [Google Scholar] [CrossRef]
- Mokhlisse, R.; Couzi, M.; Chanh, N.B.; Haget, Y.; Hauw, C.; Meresse, A. Raman scattering and X-ray diffraction study of structural phase transitions in the perovskite-type layer compound (C3H7NH3)2CdCl4. J. Phys. Chem. Solids 1985, 46, 187–195. [Google Scholar] [CrossRef]
- Chanh, N.B.; Hauw, C.; Meresse, A.; Rey-Lafon, M.; Ricard, L. X-ray ditffraction, differential scanning calorimetric and spectroscopic studies of phase transition in the bidimensional compound (C12H25NH3)2CdCl4. J. Phys. Chem. Solids 1985, 46, 1413–1420. [Google Scholar] [CrossRef]
- White, M.A. Energetics of long alkyl chains embedded in a crystalline matrix: (n-C18H37NH3)2CdCl4. J. Chem. Phys. 1984, 81, 6100–6105. [Google Scholar] [CrossRef] [Green Version]
- Couzi, M.; Chanh, N.B.; Meresse, A.; Negrier, P.; Papoular, R.J.; Millet, R. A neutron diffraction study of the high pressure structural phase transition in (CD3ND3)2MnCl4. Phase Trans. 1989, 14, 69–78. [Google Scholar] [CrossRef]
- Negrie, R.P.; Couzi, M.; Chanh, N.B.; Hauw, C.; Meresse, A. Structural phase transitions in the perovskite-type layer compound NH3(CH2)5NH3CdCl4. J. Phys. France 1989, 50, 405–430. [Google Scholar] [CrossRef]
- Chhor, K.; Abello, L.; Pommier, C.; Sourisseau, C. Reorientational motions in a perovskite-type layer compound [NH3(CH2)5NH3]MnCl4. A calorimetric study. J. Phys. Chem. Solids 1988, 49, 1079–1085. [Google Scholar] [CrossRef]
- Schenk, K.J.; Chapuis, G. Thermotropic phase transitions in bis(n-tetradecylammonium) tetrachlorocadmate(II) and some homologous compounds. J. Phys Chem. 1988, 92, 7141–7147. [Google Scholar] [CrossRef]
- Ishihara, T.; Takahashi, J.; Goto, T. Optical properties due to electronic transitions in two-dimensional semiconductors CnH2n+1NH3)2PbI4. Phys. Rev. B 1990, 42, 11099–11107. [Google Scholar] [CrossRef] [PubMed]
- Zangar, H.; Miane, J.L.; Courseille, C.; Chanh, N.B.; Couzi, M.; Mlik, Y. Structural phase transition in the perovskite-type layer compound (C3H7NH3)2PbCl4. Phys. Stat. Sol. 1989, 115, 107–118. [Google Scholar] [CrossRef]
- Robinson, S.W.; Mustoe, C.L.; White, N.G.; Brown, A.; Thompson, A.L.; Kennepohl, P.; Beer, P.D. Evidence for Halogen Bond Covalency in Acyclic and Interlocked Halogen-Bonding Receptor Anion Recognition. J. Am. Chem. Soc. 2015, 137, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Kellett, C.W.; Kennepohl, P.; Berlinguette, C.P. π covalency in the halogen bond. Nature Commun. 2020, 11, 3310. [Google Scholar] [CrossRef]
- Anyfanti, G.; Bauzá, A.; Gentiluomo, L.; Rodrigues, J.; Portalone, G.; Frontera, A.; Rissanen, K.; Puttreddy, R. Short X···N Halogen Bonds With Hexamethylenetetraamine as the Acceptor. Front. Chem. 2021, 9, 623595. [Google Scholar] [CrossRef]
- Wolters, L.P.; Smits, N.W.G.; Guerra, C.F. Covalency in resonance-assisted halogen bonds demonstrated with cooperativity in N-halo-guanine quartets. Phys. Chem. Chem. Phys. 2015, 17, 1585–1592. [Google Scholar] [CrossRef]
- Lemmerer, A.; Billing, D.G. Effect of heteroatoms in the inorganic–organic layered perovskite-type hybrids [(ZCnH2nNH3)2PbI4], n = 2, 3, 4, 5, 6; Z = OH, Br and I; and [(H3NC2H4S2C2H4NH3)PbI4]. CrystEngComm 2010, 12, 1290–1301. [Google Scholar] [CrossRef]
- Xi, J.; Spanopoulos, I.; Bang, K.; Xu, J.; Dong, H.; Yang, Y.; Malliakas, C.D.; Hoffman, J.M.; Kanatzidis, M.G.; Wu, Z. Alternative Organic Spacers for More Efficient Perovskite Solar Cells Containing Ruddlesden–Popper Phases. J. Am. Chem. Soc. 2020, 142, 19705–19714. [Google Scholar] [CrossRef]
- Jana, M.K.; Song, R.; Liu, H.; Khanal, D.R.; Janke, S.M.; Zhao, R.; Liu, C.; Vardeny, Z.V.; Blum, V.; Mitzi, D.B. CCDC 2015614: Experimental Crystal Structure Determination; University of Texas Arlington: Austin, TX, USA, 2020. [Google Scholar] [CrossRef]
- Liu, X.; Xu, Z.; Long, P.; Yao, Y.; Ji, C.; Li, L.; Sun, Z.; Hong, M.; Luo, J. A Multiaxial Layered Halide Double Perovskite Ferroelectric with Multiple Ferroic Orders. Chem. Mater. 2020, 32, 8965–8970. [Google Scholar] [CrossRef]
- Sourisseau, S.; Louvain, N.; Bi, W.; Mercier, N.; Rondeau, D.; Boucher, F.; Buzaré, J.-Y.; Legein, C. Reduced Band Gap Hybrid Perovskites Resulting from Combined Hydrogen and Halogen Bonding at the Organic−Inorganic Interface. Chem. Mater. 2007, 19, 600–607. [Google Scholar] [CrossRef]
- Connor, B.A.; Leppert, L.; Smith, M.D.; Neaton, J.B.; Karunadasa, H.I. Layered Halide Double Perovskites: Dimensional Reduction of Cs2AgBiBr6. J. Am. Chem. Soc. 2018, 140, 5235–5240. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wu, Z.; Guan, T.; Jiang, H.; Long, P.; Li, X.; Ji, C.; Chen, S.; Sun, Z.; Luo, J. Giant room temperature electrocaloric effect in a layered hybrid perovskite ferroelectric: [(CH3)2CHCH2NH3]2PbCl4. Nat. Commun. 2021, 12, 5502. [Google Scholar] [CrossRef] [PubMed]
- McClure, E.T.; McCormick, A.P.; Woodward, P.M. Four Lead-free Layered Double Perovskites with the n = 1 Ruddlesden–Popper Structure. Inorg. Chem. 2020, 59, 6010–6017. [Google Scholar] [CrossRef]
- Mei, A.; Li, X.; Liu, L.; Ku, Z.; Liu, T.; Rong, Y.; Xu, M.; Hu, M.; Chen, J.; Yang, Y.; et al. A hole-conductor-free, fully printable mesoscopic perovskite solar cell with high stability. Science 2014, 345, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Krummer, M.; Zimmermann, B.; Klingenberg, P.; Daub, M.; Hillebrecht, H. Perovskite-Related 2D Compounds in the System 5-Amino Valerian Acid Cation/MA/Pb/X (X = Cl, Br)—Synthesis, Crystal Structures, and Optical Properties. Eur. J. Inorg. Chem. 2020, 2020, 4581–4592. [Google Scholar] [CrossRef]
- Soe, C.M.M.; Nagabhushana, G.P.; Shivaramaiah, R.; Tsai, H.; Nie, W.; Blancon, J.-C.; Melkonyan, F.; Cao, D.H.; Traoré, B.; Pedesseau, L.; et al. Structural and thermodynamic limits of layer thickness in 2D halide perovskites. Proc. Nail. Acad. Sci. USA 2019, 116, 58–66. [Google Scholar] [CrossRef] [Green Version]
- Li, L. CCDC 1939731: Experimental Crystal Structure Determination; University of Texas Arlington: Austin, TX, USA, 2020. [Google Scholar] [CrossRef]
- Hillebrecht, H. CCDC 1999300: Experimental Crystal Structure Determination; University of Texas Arlington: Austin, TX, USA, 2020. [Google Scholar] [CrossRef]
- Feng, L.-J.; Zhao, Y.-Y.; Song, R.-Y.; Lei, X.-W. Three homologous 1D lead halide perovskites with broadband white-light emissions. Inorg. Chem. Commun. 2022, 136, 109146. [Google Scholar] [CrossRef]
- Hoffman, J.M.; Che, X.; Sidhik, S.; Li, X.; Hadar, I.; Blancon, J.-C.; Yamaguchi, H.; Kepenekian, M.; Katan, C.; Even, J.; et al. From 2D to 1D Electronic Dimensionality in Halide Perovskites with Stepped and Flat Layers Using Propylammonium as a Spacer. J. Am. Chem. Soc. 2019, 141, 10661–10676. [Google Scholar] [CrossRef]
- Pipitone, C.; Boldrini, S.; Ferrario, A.; Garcìa-Espejo, G.; Guagliardi, A.; Masciocchi, N.; Martorana, A.; Giannici, F. Ultralow thermal conductivity in 1D and 2D imidazolium-based lead halide perovskites. Appl. Phys. Lett. 2021, 119, 101104. [Google Scholar] [CrossRef]
- Zheng, Y.; Xu, J.; Bu, X.-H. 1D Chiral Lead Halide Perovskites with Superior Second-Order Optical Nonlinearity. Adv. Opt. Mat. 2022, 10, 2101545. [Google Scholar] [CrossRef]
- Yuan, Z.; Zhou, C.; Tian, Y.; Shu, Y.; Messier, J.; Wang, J.C.; van de Burgt, L.J.; Kountouriotis, K.; Xin, Y.; Holt, E.; et al. One-dimensional organic lead halide perovskites with efficient bluish white-light emission. Nature Commun. 2017, 8, 14051. [Google Scholar] [CrossRef] [PubMed]
- Essalhi, R.; Abdelbaky, M.S.M.; Elleuch, S.; Zouari, F.; García-Granda, S. Crystal structure, Hirschfield surface analysis, thermal and DFT investigation accomplished with photoluminescence study of bis(N, N-diethylethylendiammonium)decabromodiantimoinate(III). J. Mol. Str. 2020, 1221, 128828. [Google Scholar] [CrossRef]
- Luo, J.; Zhang, H.-J.; Quasie, O.; Shan, S.-M.; Zhang, Y.-M.; Kong, L.-Y. Further C-15-acyl phragmalin derivatives from Chukrasia tabularis A. Juss. Phytochemistry 2015, 117, 410–416. [Google Scholar] [CrossRef]
- Mawhinney, T.P.; Li, Y.; Chance, D.L.; Kelley, S.P.; Mossine, V.V. Crystal structure of (R,S)-2-hydroxy-4-(methylsulfanyl)butanoic acid. Acta Crystallogr. E 2020, 76, 562–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senior, L.; Linden, A. Halobismuth(III) salts with substituted aminopyridinium cations. Acta Crystallogr. C 2020, 76, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Hao, F.; Stoumpos, C.C.; Liu, Z.; Chang, R.P.H.; Kanatzidis, M.G. Controllable Perovskite Crystallization at a Gas–Solid Interface for Hole Conductor-Free Solar Cells with Steady Power Conversion Efficiency over 10%. J. Am. Chem. Soc. 2014, 136, 16411–16419. [Google Scholar] [CrossRef] [PubMed]
- Lemmerer, A.; Billing, D.G. Two packing motifs based upon chains of edge-sharing PbI6 octahedra. Acta Crystallogr. C 2006, 62, m597–m601. [Google Scholar] [CrossRef] [PubMed]
- Guo, N.; Lin, Y.-H.; Zeng, G.-F.; Xi, S.-Q. Structure of 1,3-propanediammonium tetrachlorocobaltate(II). Acta Crystallogr. C 1992, 48, 542–543. [Google Scholar] [CrossRef] [Green Version]
- Wen, H.; Miller, S.E.; House, D.A.; McKee, V.; Robinson, W.T. The crystal structures of some chloromercury(II) anions with Co(III) complexes or protonated polyamines as cations. Inorg. Chim. Acta 1992, 193, 77–85. [Google Scholar] [CrossRef]
- Spengler, R.; Zouari, R.; Ben Salah, A.; Zimmermann, H.; Burzlaff, H. Redetermination of Bis(1,2-ethanediammonium) Dichloride Tetrachloromercurate(II). Acta Crystallogr. C 1998, 54, IUC9800034. [Google Scholar] [CrossRef]
- Smith, H.W.; Stratton, W.J. Preparation, properties, and crystal and molecular structure of ethylenediammonium tetrachlorocobaltate(II) chloride, (NH3CH2CH2NH3)2(CoCl4)Cl2. Inorg. Chem. 1977, 16, 1640–1645. [Google Scholar] [CrossRef]
- Kumar, M.; Verma, S.K.; Singh, B.; Thakur, A.; Kumar, A.; Jasrotia, D. 2D Interwoven Metal-Organic Framework in Tetrachloromercurate(II) based Hybrid Material. Chem. Sci. Trans. 2015, 4, 629–637. [Google Scholar]
- Ouerghi, Z.; Gornitzka, H.; Temel, E.; Dridi, I.; Kefi, R. A new non-centrosymmetric Chlorobismuthate(III) hybrid material: Crystal structure, optical properties and antibacterial study. J. Mol. Str. 2019, 1181, 338–347. [Google Scholar] [CrossRef]
- Mao, W.; Wang, J.; Hu, X.; Zhou, B.; Zheng, G.; Mo, S.; Li, S.; Long, F.; Zou, Z. Synthesis, crystal structure, photoluminescence properties of organic-inorganic hybrid materials based on ethylenediamine bromide. J. Saudi Chem. Soc. 2020, 24, 52–60. [Google Scholar] [CrossRef]
- Bourne, S.A.; Mangombo, Z. Phenylamines as building blocks to layered inorganic–organic structures. CrystEngComm 2004, 6, 438–442. [Google Scholar] [CrossRef]
- Dobrzycki, L.; Woźniak, K. 1D vs 2D crystal architecture of hybrid inorganic–organic structures with benzidine dication. J. Mol. Str. 2009, 921, 18–33. [Google Scholar] [CrossRef]
- Su, B.; Song, G.; Molokeev, M.S.; Lin, Z.; Xia, Z. Synthesis, Crystal Structure and Green Luminescence in Zero-Dimensional Tin Halide (C8H14N2)2SnBr6. Inorg. Chem. 2020, 59, 9962–9968. [Google Scholar] [CrossRef]
- Ishiharaa, H.; Horiuchib, K.; Douc, S.-q.; Gesingc, T.M.; Buhlc, J.-C.; Paulus, H.; Fuessd, H. Isolated versus Condensed Anion Structure IV: An NQR Study and X-ray Structure Analysis of [H3N(CH2)3NH3]CdI4·2H2O, [H3CNH2(CH2)3NH3]CdBr4, [(CH3)4N]2CdBr4, and [(CH3)3S]2CdBr4. Z. Naturforsch. A: Phys. Sci. 1998, 53, 717–724. [Google Scholar] [CrossRef]
- Makhinya, A.N.; Shusharina, E.A.; Baidina, I.A.; Il’in, M.A. Structure of the first nitrosoammine complexes of ruthenium with a coordinated sulfate ion: [Ru(NO)(NH3)4(SO4)](HSO4)·H2O and [Ru(NO)(NH3)3Cl(SO4)]·2H2O. J. Struct. Chem. 2011, 52, 946–953. [Google Scholar] [CrossRef]
- Willermann, M.; Mulcahy, C.; Sigel, R.K.O.; Cerdà, M.M.; Freisinger, E.; Sanz Miguel, P.J.; Roitzsch, M.; Lippert, B. Pyrazine as a Building Block for Molecular Architectures with PtII. Inorg. Chem. 2006, 45, 2093–2099. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.-N.; Zhao, R.-Y.; Xu, H.; Wang, Z.-H.; Liu, Q.-S.; Shahid, M.Z.; Miao, J.-L.; Chen, G.; Li, C. The structures, water stabilities and photoluminescence properties of two types of iodocuprate(i)-based hybrids. Dalton Trans. 2018, 47, 2306–2317. [Google Scholar] [CrossRef] [PubMed]
- Siebel, S.; Dammann, C.; Sanz Miguel, P.J.; Drewello, T.; Kampf, G.; Teubner, N.; Bednarski, P.J.; Freisinger, E.; Lippert, B. Analogues of Cis- and Transplatin with a Rich Solution Chemistry: Cis-[PtCl2(NH3)(1-MeC-N3)] and trans-[PtI2(NH3)(1-MeC-N3)]. Chem. Eur. J. 2015, 21, 17827–17843. [Google Scholar] [CrossRef] [Green Version]
- Spackman, M.A. How Reliable Are Intermolecular Interaction Energies Estimated from Topological Analysis of Experimental Electron Densities? Cryst. Growth Des. 2015, 15, 5624–5628. [Google Scholar] [CrossRef]































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | (ωB97X-D)crystal geometry | (ωB97X-D)optimized geometry | [MP2(FC)]crystal geometry | [MP2(FC)]optimized geometry | ||||
|---|---|---|---|---|---|---|---|---|
| Eint | Eint(BSSE) | Eint | Eint(BSSE) | Eint | Eint(BSSE) | Eint | Eint(BSSE) | |
| H3C-H3N···I | −94.25 | −94.24 | −102.1 | −102.08 | −95.68 | −94.42 | −105.31 | −103.16 |
| H3N-H3C···I | −70.21 | −70.19 | −76.1 | −76.07 | −71.16 | −70.26 | −78.77 | −76.91 |
| Geometry (Complex Type) | Eels | Eexch | Eind | Edis | Eint(SAPT0) | Eint(SCF)(BSSE) a |
|---|---|---|---|---|---|---|
| Crystal geometry (H3C-H3N···I) | −91.00 | 9.32 | −8.87 | −2.87 | −93.44 | −94.24 |
| Crystal geometry (H3N-H3C···I) | −67.97 | 4.47 | −4.4 | −2.01 | −69.92 | −70.19 |
| ωB97X-D (H3C-H3N···I) | −108.8 | 26.84 | −14.12 | −5.93 | −102.00 | −102.08 |
| ωB97X-D (H3N-H3C···I) | −82.91 | 19.92 | −8.51 | −5.25 | −76.76 | −76.07 |
| MP2(FC)(H3C-H3N···I) | −111.74 | 31.04 | −14.95 | −6.55 | −102.2 | −105.31 |
| MP2(FC)(H3N-H3C···I) | −85.94 | 24.51 | −9.46 | −6.03 | −76.93 | −78.77 |
| Interaction Type | Donor NBO | Acceptor NBO | E(2)[MP2(FC] Geometry] | E(2)[ωB97X-D Geometry] | |
|---|---|---|---|---|---|
| (C)N···I | LP(4) I | → | RY*(1)N | 2.86 | 2.66 |
| (C)N···I | LP(4) I | → | BD*(1)N-C | 4.51 | 3.99 |
| N-H···I | LP(2)I | → | BD*(1)N-H | 0.62 | 0.51 |
| N-H···I | LP(2)I | → | BD*(1)N-H | 0.62 | 0.51 |
| N-H···I | LP(3)I | → | BD*(1)N-H | 0.83 | 0.68 |
| N-H···I | LP(4) I | → | BD*(1)N-H | 1.14 | 0.94 |
| N-H···I | LP(4) I | → | BD*(1)N-H | 1.14 | 0.94 |
| N-H···I | LP(4) I | → | BD*(1)N-H | 1.14 | 0.94 |
| (N)C···I | LP(4)I | → | RY*(1)(C) | 2.58 | 2.26 |
| (N)C···I | LP(1)I | → | BD*(1)C-N | 0.56 | 0.41 |
| (N)C···I | LP(4)I | → | BD*(C-N) | 10.22 | 8.14 |
| N-H···I | LP(2)I | → | BD*(1)C-H | 0.12 | 0.08 |
| N-H···I | LP(2)I | → | BD*(1)C-H | 0.12 | 0.08 |
| N-H···I | LP(3)I | → | BD*(1)C-H | 0.12 | 0.11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varadwaj, A.; Varadwaj, P.R.; Marques, H.M.; Yamashita, K. The Pnictogen Bond, Together with Other Non-Covalent Interactions, in the Rational Design of One-, Two- and Three-Dimensional Organic-Inorganic Hybrid Metal Halide Perovskite Semiconducting Materials, and Beyond. Int. J. Mol. Sci. 2022, 23, 8816. https://doi.org/10.3390/ijms23158816
Varadwaj A, Varadwaj PR, Marques HM, Yamashita K. The Pnictogen Bond, Together with Other Non-Covalent Interactions, in the Rational Design of One-, Two- and Three-Dimensional Organic-Inorganic Hybrid Metal Halide Perovskite Semiconducting Materials, and Beyond. International Journal of Molecular Sciences. 2022; 23(15):8816. https://doi.org/10.3390/ijms23158816
Chicago/Turabian StyleVaradwaj, Arpita, Pradeep R. Varadwaj, Helder M. Marques, and Koichi Yamashita. 2022. "The Pnictogen Bond, Together with Other Non-Covalent Interactions, in the Rational Design of One-, Two- and Three-Dimensional Organic-Inorganic Hybrid Metal Halide Perovskite Semiconducting Materials, and Beyond" International Journal of Molecular Sciences 23, no. 15: 8816. https://doi.org/10.3390/ijms23158816