
Hypoxia Aggravates Inhibition of Alveolar Epithelial Na-Transport by Lipopolysaccharide-Stimulation of Alveolar Macrophages


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
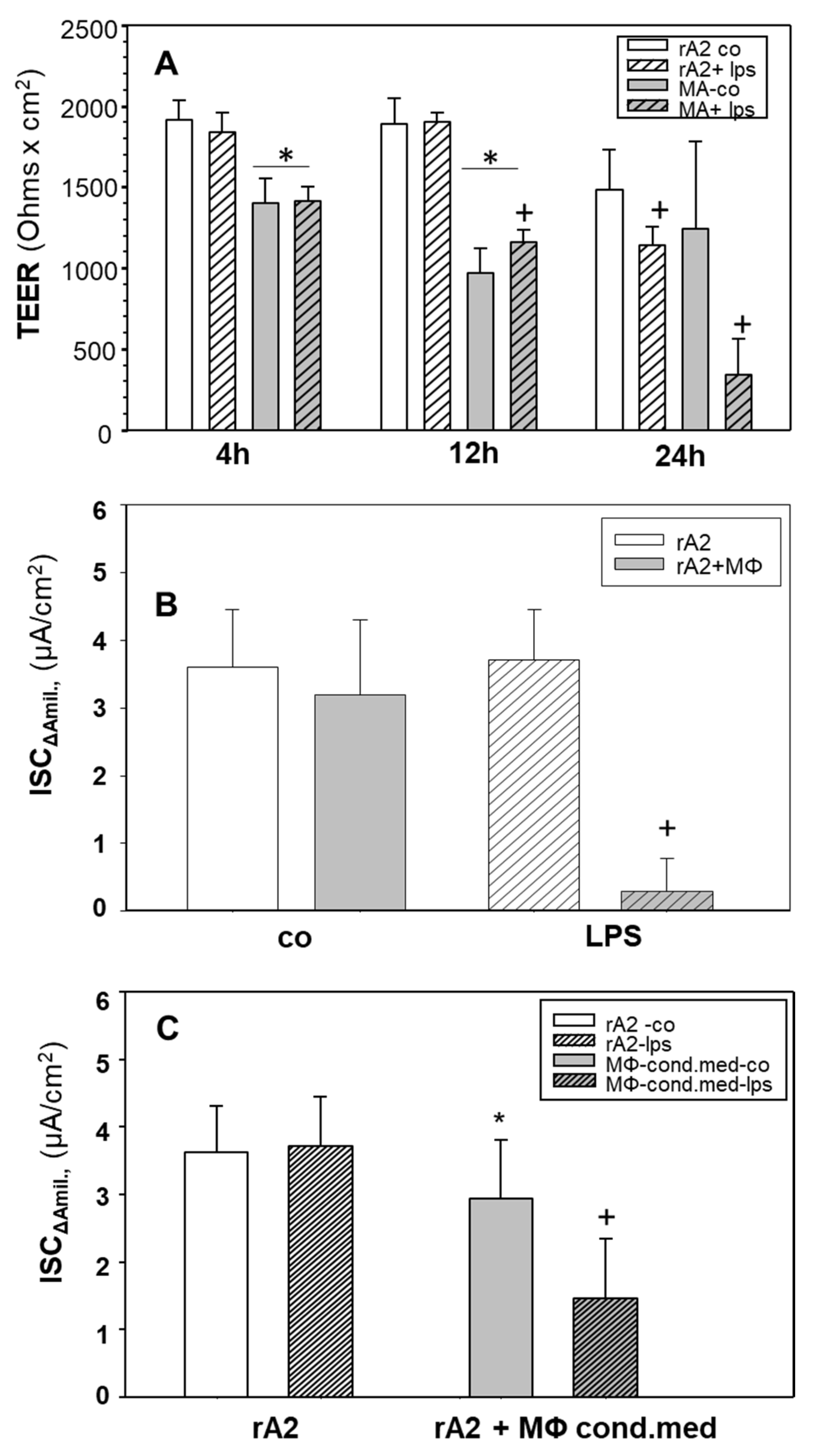
2.1. Effects of LPS Stimulated Macrophages on Ion-Transport of rA2 Cells
2.2. Cytokines, iNOS, NO/Nitrite
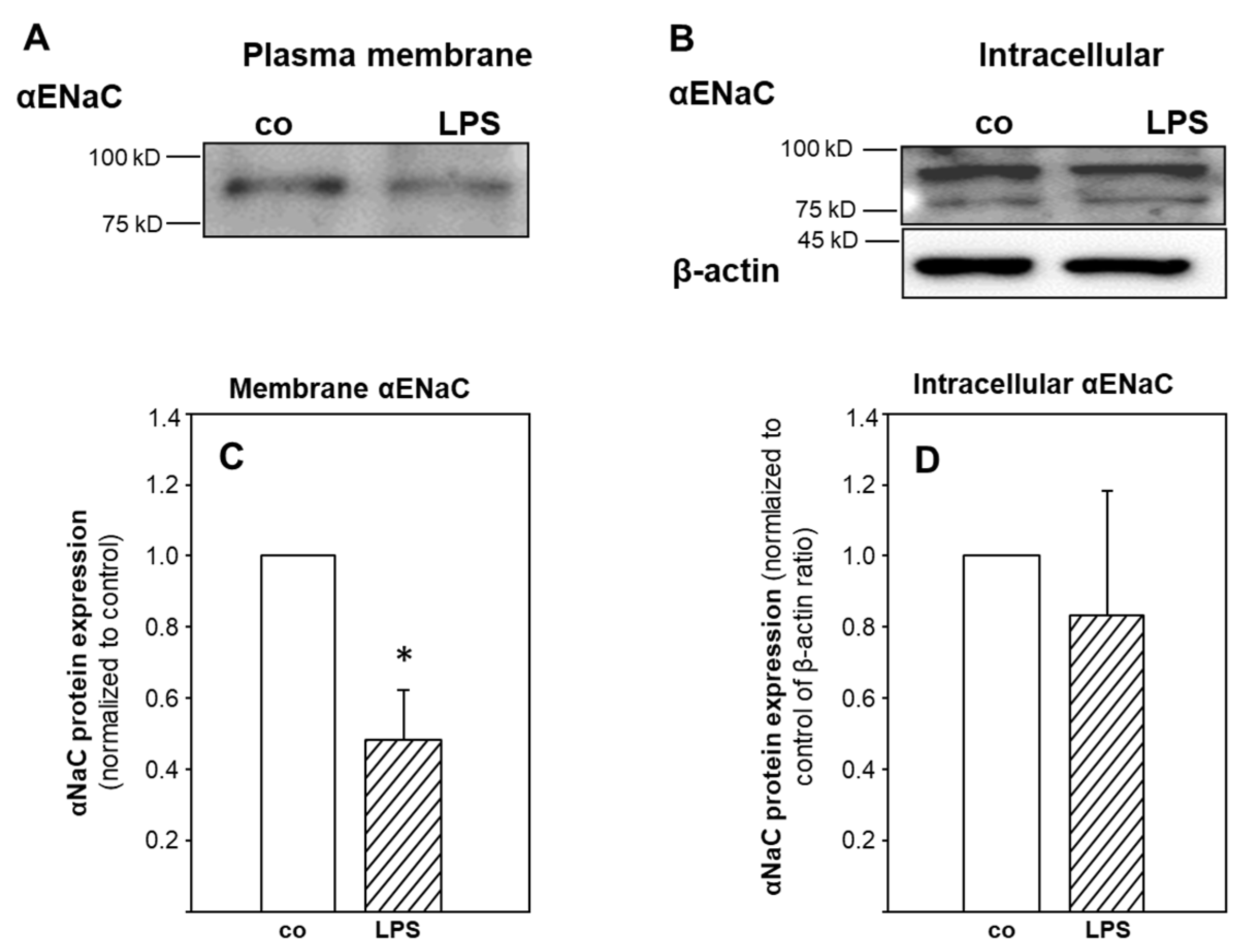
2.3. ENaC Surface Abundance
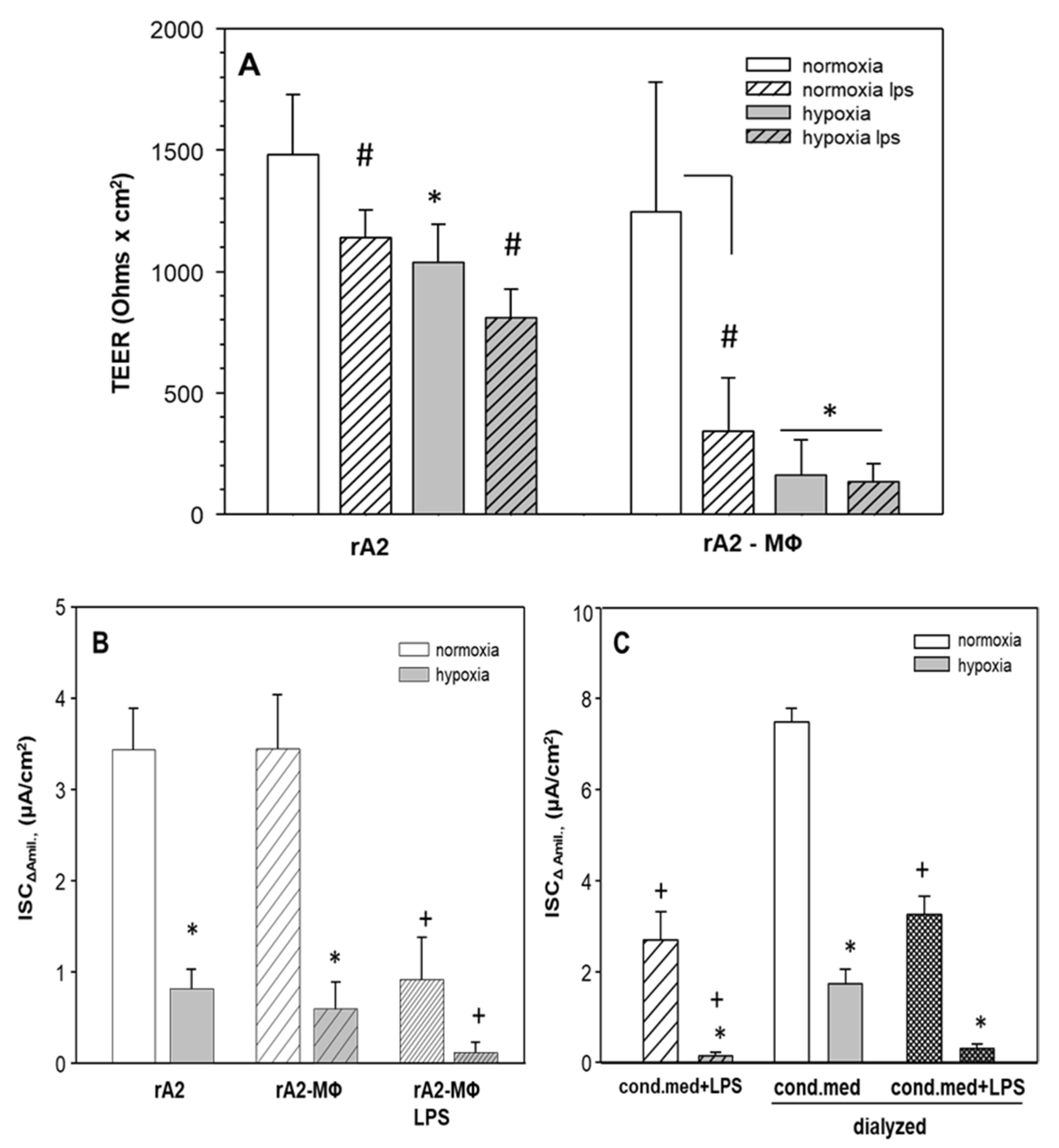
2.4. Hypoxia
3. Discussion
3.1. Inflammatory Signaling and ENaC-Activity
3.2. Mechanisms of Na-Transport Inhibition
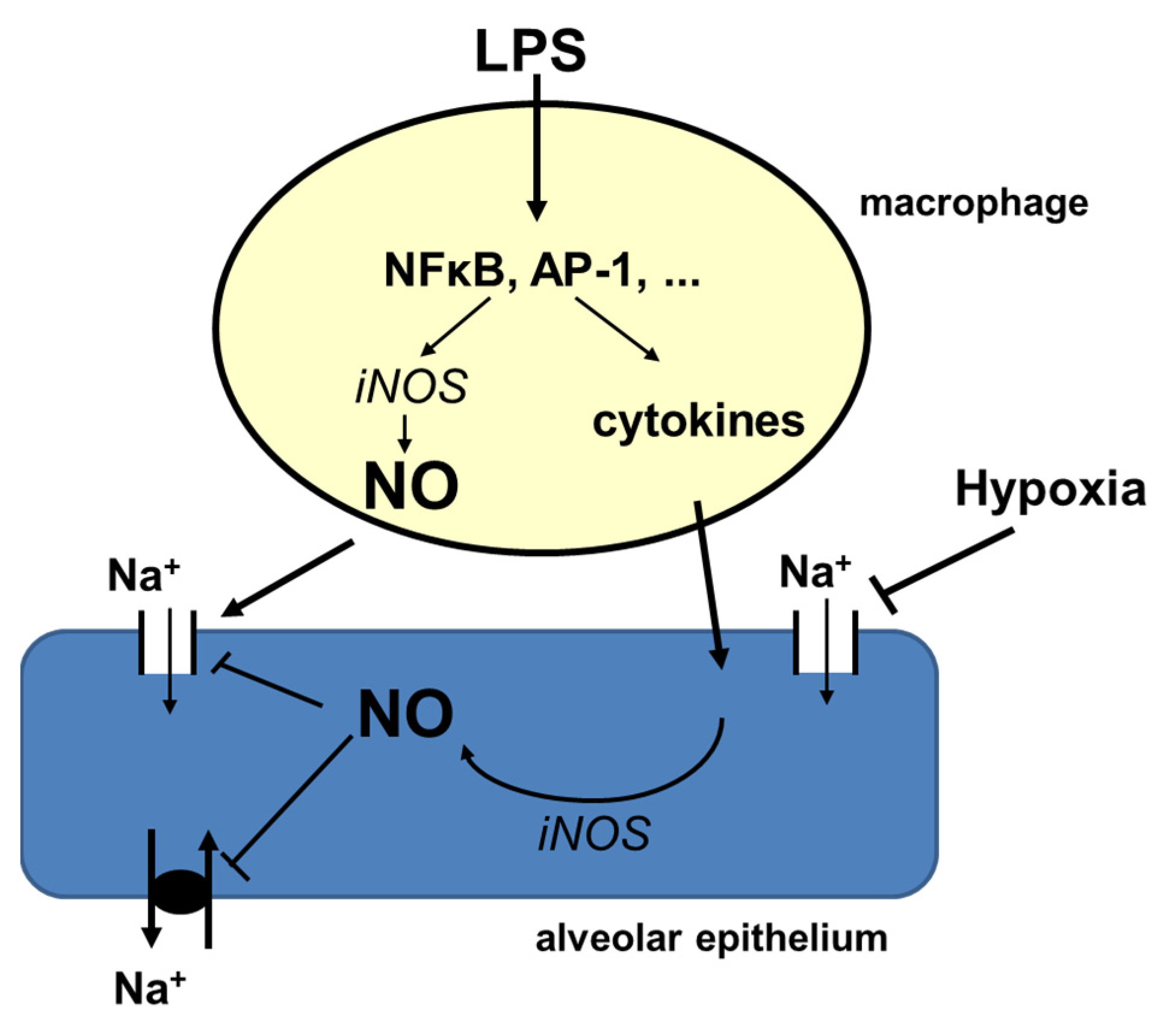
3.3. Hypoxia and Inflammation
3.4. Conclusions
4. Materials and Methods
4.1. Preparation of Rat Lung Primary Alveolar Epithelial Cells (rA2)
4.2. NR8383 Rat Alveolar Macrophage Cell Line (NR8383)
4.3. Co-Culture of rA2 with NR8383
4.4. Preparation of Macrophage-Conditioned Media and Dialysis
4.5. Treatments
4.6. Electrophysiological Measurements
4.7. RNA Isolation and Quantitative RT-PCR
4.8. Cytokine Measurements
4.9. Cell Surface Biotinylation and Western Blotting
4.10. Other Measurements
4.11. Statistical Evaluation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Delclaux, C.; Azoulay, E. Inflammatory response to infectious pulmonary injury. Eur. Respir. J. 2003, 22, 10s–14s. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bode, J.G.; Ehlting, C.; Haussinger, D. The macrophage response towards LPS and its control through the p38(MAPK)-STAT3 axis. Cell. Signal. 2012, 24, 1185–1194. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.T.; Li, S.L.; Cai, E.Q.; Wu, W.L.; Jin, J.S.; Zhu, B. LPS induces pulmonary intravascular macrophages producing inflammatory mediators via activating NF-kappaB. J. Cell. Biochem. 2003, 89, 1206–1214. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Ye, L.; Ye, L.; Li, B.; Gao, B.; Zeng, Y.; Kong, L.; Fang, X.; Zheng, H.; Wu, Z.; et al. Up-regulation of IL-6 and TNF-alpha induced by SARS-coronavirus spike protein in murine macrophages via NF-kappaB pathway. Virus Res. 2007, 128, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Haddad, I.Y.; Zhu, S.; Crow, J.; Barefield, E.; Gadilhe, T.; Matalon, S. Inhibition of alveolar type II cell ATP and surfactant synthesis by nitric oxide. Am. J. Physiol. 1996, 14, L898–L906. [Google Scholar] [CrossRef]
- Trapnell, B.C.; Whitsett, J.A. Gm-CSF regulates pulmonary surfactant homeostasis and alveolar macrophage-mediated innate host defense. Annu. Rev. Physiol. 2002, 64, 775–802. [Google Scholar] [CrossRef]
- Hamacher, J.; Hadizamani, Y.; Borgmann, M.; Mohaupt, M.; Mannel, D.N.; Moehrlen, U.; Lucas, R.; Stammberger, U. Cytokine-Ion Channel Interactions in Pulmonary Inflammation. Front. Immunol. 2017, 8, 1644. [Google Scholar] [CrossRef] [Green Version]
- Mairbäurl, H.; Mayer, K.; Kim, K.J.; Borok, Z.; Bärtsch, P.; Crandall, E.D. Hypoxia decreases active Na transport across primary rat alveolar epithelial cell monolayers. Am. J. Physiol. 2002, 282, L659–L665. [Google Scholar] [CrossRef]
- Mairbäurl, H. Role of alveolar epithelial sodium transport in high altitude pulmonary edema (HAPE). Respir. Physiol. Neurobiol. 2006, 151, 178–191. [Google Scholar] [CrossRef]
- Clerici, C. Sodium transport in alveolar epithelial cells: Modulation by O2 tension. Kidney Int. 1998, 65, S79–S83. [Google Scholar]
- Ogawa, S.; Koga, S.; Kuwabara, K.; Brett, J.; Morrow, B.; Morris, S.A.; Bilezikian, J.P.; Silverstein, S.C.; Stern, D. Hypoxia-induced increased permeability of endothelial monolayers occurs through lowering of cellular cAMP levels. Am. J. Physiol. 1992, 262, C546–C554. [Google Scholar] [CrossRef]
- Dehler, M.; Zessin, E.; Bärtsch, P.; Mairbäurl, H. Hypoxia causes permeability edema in the constant-pressure perfused rat lung. Eur. Respir. J. 2006, 27, 600–606. [Google Scholar] [CrossRef] [Green Version]
- Chao, J.; Wood, J.G.; Gonzalez, N.C. Alveolar hypoxia, alveolar macrophages, and systemic inflammation. Respir. Res. 2009, 10, 54. [Google Scholar] [CrossRef] [Green Version]
- Matthay, M.A.; Clerici, C.; Saumon, G. Active fluid clearance from distal airspaces. J. Appl. Physiol. 2002, 93, 1533–1541. [Google Scholar] [CrossRef] [Green Version]
- Guney, S.; Schuler, A.; Ott, A.; Höschele, S.; Baloglu, E.; Bärtsch, P.; Mairbäurl, H. Dexamethasone prevents transport inhibition by hypoxia in rat lung and alveolar epithelial cells by stimulating activity and expression of Na+/K+-ATPase and epithelial Na+ channels. Am. J. Physiol. 2007, 293, L1332–L1338. [Google Scholar] [CrossRef] [Green Version]
- Baloglu, E.; Reingruber, T.; Bärtsch, P.; Mairbäurl, H. beta2-Adrenergics in hypoxia desensitize receptors but blunt inhibition of reabsorption in rat lungs. Am. J. Respir. Cell Mol. Biol. 2011, 45, 1059–1068. [Google Scholar] [CrossRef]
- Planes, C.; Escoubet, B.; BlotChabaud, M.; Friedlander, G.; Farman, N.; Clerici, C. Hypoxia downregulates expression and activity of epithelial sodium channels in rat alveolar epithelial cells. Am. J. Respir. Cell Mol. Biol. 1997, 17, 508–518. [Google Scholar] [CrossRef] [Green Version]
- Vivona, M.L.; Matthay, M.A.; Chabaud, M.B.; Friedlander, G.; Clerici, C. Hypoxia reduces alveolar epithelial sodium and fluid transport in rats: Reversal by beta-adrenergic agonist treatment. Am. J. Respir. Cell Mol. Biol. 2001, 25, 554–561. [Google Scholar] [CrossRef]
- Price, L.C.; McAuley, D.F.; Marino, P.S.; Finney, S.J.; Griffiths, M.J.; Wort, S.J. Pathophysiology of pulmonary hypertension in acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L803–L815. [Google Scholar] [CrossRef] [Green Version]
- Ware, L.B.; Matthay, M.A. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2001, 163, 1376–1383. [Google Scholar] [CrossRef]
- Matthay, M.A. Alveolar fluid clearance in patients with ARDS: Does it make a difference? Chest 2002, 122, 340S–343S. [Google Scholar] [CrossRef]
- Thorley, A.J.; Ford, P.A.; Giembycz, M.A.; Goldstraw, P.; Young, A.; Tetley, T.D. Differential regulation of cytokine release and leukocyte migration by lipopolysaccharide-stimulated primary human lung alveolar type II epithelial cells and macrophages. J. Immunol. 2007, 178, 463–473. [Google Scholar] [CrossRef]
- Whitsett, J.A.; Alenghat, T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat. Immunol. 2015, 16, 27–35. [Google Scholar] [CrossRef] [Green Version]
- Shehata, M.F. The Epithelial Sodium Channel alpha subunit (alpha ENaC) alternatively spliced form “b” in Dahl rats: What’s next? Int. Arch. Med. 2010, 3, 14. [Google Scholar] [CrossRef] [Green Version]
- Eaton, D.C.; Helms, M.N.; Koval, M.; Bao, H.F.; Jain, L. The contribution of epithelial sodium channels to alveolar function in health and disease. Annu. Rev. Physiol. 2009, 71, 403–423. [Google Scholar] [CrossRef]
- Boncoeur, E.; Tardif, V.; Tessier, M.C.; Morneau, F.; Lavoie, J.; Gendreau-Berthiaume, E.; Grygorczyk, R.; Dagenais, A.; Berthiaume, Y. Modulation of epithelial sodium channel activity by lipopolysaccharide in alveolar type II cells: Involvement of purinergic signaling. Am. J. Physiol. 2010, 298, L417–L426. [Google Scholar] [CrossRef]
- Haddad, J.J.; Land, S.C. Redox signaling-mediated regulation of lipopolysaccharide-induced proinflammatory cytokine biosynthesis in alveolar epithelial cells. Antioxid. Redox Signal. 2002, 4, 179–193. [Google Scholar] [CrossRef]
- Ward, P.A. Phagocytes and the lung. Ann. N. Y. Acad. Sci. 1997, 832, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Duvall, M.D.; Crow, J.P.; Matalon, S. Nitric oxide inhibits Na + absorption across alveolar type II monolayers. Am. J. Physiol. 1998, 274, L369–L377. [Google Scholar] [CrossRef] [PubMed]
- Jain, L.; Chen, X.J.; Brown, L.A.; Eaton, D.C. Nitric oxide inhibits lung sodium transport through a cGMP-mediated inhibition of epithelial cation channels. Am. J. Physiol. 1998, 18, L475–L484. [Google Scholar] [CrossRef] [PubMed]
- Helms, M.N.; Yu, L.; Malik, B.; Kleinhenz, D.J.; Hart, C.M.; Eaton, D.C. Role of SGK1 in nitric oxide inhibition of ENaC in Na+-transporting epithelia. Am. J. Physiol. Cell Physiol. 2005, 289, C717–C726. [Google Scholar] [CrossRef] [Green Version]
- Althaus, M.; Pichl, A.; Clauss, W.G.; Seeger, W.; Fronius, M.; Morty, R.E. Nitric oxide inhibits highly selective sodium channels and the Na+/K+-ATPase in H441 cells. Am. J. Respir. Cell Mol. Biol. 2011, 44, 53–65. [Google Scholar] [CrossRef]
- Zsengeller, Z.K.; Ross, G.F.; Trapnell, B.C.; Szabo, C.; Whitsett, J.A. Adenovirus infection increases iNOS and peroxynitrite production in the lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 280, L503–L511. [Google Scholar] [CrossRef]
- Farley, K.S.; Wang, L.F.; Law, C.; Mehta, S. Alveolar macrophage inducible nitric oxide synthase-dependent pulmonary microvascular endothelial cell septic barrier dysfunction. Microvasc. Res. 2008, 76, 208–216. [Google Scholar] [CrossRef]
- Farley, K.S.; Wang, L.; Mehta, S. Septic pulmonary microvascular endothelial cell injury: Role of alveolar macrophage NADPH oxidase. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L480–L488. [Google Scholar] [CrossRef] [Green Version]
- Hua, Y.; Han, A.; Yu, T.; Hou, Y.; Ding, Y.; Nie, H. Small Extracellular Vesicles Containing miR-34c Derived from Bone Marrow Mesenchymal Stem Cells Regulates Epithelial Sodium Channel via Targeting MARCKS. Int. J. Mol. Sci. 2022, 23, 5196. [Google Scholar] [CrossRef]
- Iles, K.E.; Song, W.; Miller, D.W.; Dickinson, D.A.; Matalon, S. Reactive species and pulmonary edema. Expert Rev. Respir. Med. 2009, 3, 487–496. [Google Scholar] [CrossRef] [Green Version]
- Willis, B.C.; Kim, K.J.; Li, X.; Liebler, J.; Crandall, E.D.; Borok, Z. Modulation of ion conductance and active transport by TGF- beta 1 in alveolar epithelial cell monolayers. Am. J. Physiol. 2003, 285, L1192–L1200. [Google Scholar]
- Dagenais, A.; Frechette, R.; Yamagata, K.; Yamagata, T.; Carmel, J.F.; Clermont, M.E.; Brochiero, E.; Masse, C.; Berthiaume, Y. Downregulation of ENaC activity and expression by TNF-à in alveolar epithelial cells. Am. J. Physiol. 2004, 286, L301–L311. [Google Scholar] [CrossRef]
- Frank, J.; Roux, J.; Kawakatsu, H.; Su, G.; Dagenais, A.; Berthiaume, Y.; Howard, M.; Canessa, C.M.; Fang, X.; Sheppard, D.; et al. Transforming growth factor-beta1 decreases expression of the epithelial sodium channel alphaENaC and alveolar epithelial vectorial sodium and fluid transport via an ERK1/2-dependent mechanism. J. Biol. Chem. 2003, 278, 43939–43950. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, V.G.; Baird, M.S.; Chen, L.; Matalon, S. DETANONOate, a nitric oxide donor, decreases amiloride- sensitive alveolar fluid clearance in rabbits. Am. J. Respir. Crit. Care Med. 2000, 161, 1154–1160. [Google Scholar] [CrossRef]
- Boncoeur, E.; Bouvet, G.F.; Migneault, F.; Tardif, V.; Ferraro, P.; Radzioch, D.; de Sanctis, J.B.; Eidelman, D.; Govindaraju, K.; Dagenais, A.; et al. Induction of nitric oxide synthase expression by lipopolysaccharide is mediated by calcium-dependent PKCalpha-beta1 in alveolar epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L175–L184. [Google Scholar] [CrossRef] [Green Version]
- Matalon, S.; Hu, P.; Ischiropoulos, H.; Beckman, J.S. Peroxynitrite Inhibition of Oxygen Consumption and Ion Transport in Alveolar Type II Pneumocytes. Chest 1994, 105, S74. [Google Scholar] [CrossRef]
- Roux, J.; Kawakatsu, H.; Gartland, B.; Pespeni, M.; Sheppard, D.; Matthay, M.A.; Canessa, C.M.; Pittet, J.F. Interleukin-1 beta decreases expression of the epithelial sodium channel alpha-subunit in alveolar epithelial cells via a p3 8 MAPK-dependent signaling pathway. J. Biol. Chem. 2005, 280, 18579–18589. [Google Scholar] [CrossRef] [Green Version]
- Elia, N.; Tapponnier, M.; Matthay, M.A.; Hamacher, J.; Pache, J.C.; Brundler, M.A.; Totsch, M.; De Baetselier, P.; Fransen, L.; Fukuda, N.; et al. Functional identification of the alveolar edema reabsorption activity of murine tumor necrosis factor-alpha. Am. J. Respir. Crit. Care Med. 2003, 168, 1043–1050. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Hamacher, J.; Gorshkov, B.; White, R.; Sridhar, S.; Verin, A.; Chakraborty, T.; Lucas, R. The Dual Role of TNF in Pulmonary Edema. J. Cardiovasc. Dis. Res. 2010, 1, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Xu, H.R.; Xiang, S.Y.; Zhang, C.; Ye, Y.; Shen, C.X.; Mei, H.X.; Zhang, P.H.; Ma, H.Y.; Zheng, S.X.; et al. Resolvin Conjugates in Tissue Regeneration 1 Promote Alveolar Fluid Clearance by Activating Alveolar Epithelial Sodium Channels and Na, K-ATPase in Lipopolysaccharide-Induced Acute Lung Injury. J. Pharmacol. Exp. Ther. 2021, 379, 156–165. [Google Scholar] [CrossRef]
- Deng, W.; Qi, D.; Tang, X.M.; Deng, X.Y.; He, J.; Wang, D.X. The WNK4/SPAK Pathway Stimulates Alveolar Fluid Clearance By Up-Regulation of Epithelial Sodium Channel In Mice with Lipopolysaccharide-Induced Acute Respiratory Distress Syndrome. Shock, 2022; online ahead of print. [Google Scholar] [CrossRef]
- Zhou, Z.; Hua, Y.; Ding, Y.; Hou, Y.; Yu, T.; Cui, Y.; Nie, H. Conditioned Medium of Bone Marrow Mesenchymal Stem Cells Involved in Acute Lung Injury by Regulating Epithelial Sodium Channels via miR-34c. Front. Bioeng. Biotechnol. 2021, 9, 640116. [Google Scholar] [CrossRef]
- Baloglu, E.; Nonnenmacher, G.; Seleninova, A.; Berg, L.; Velineni, K.; Ermis-Kaya, E.; Mairbäurl, H. The role of hypoxia-induced modulation of alveolar epithelial Na(+)- transport in hypoxemia at high altitude. Pulm. Circ. 2020, 10, 50–58. [Google Scholar] [CrossRef]
- Planes, C.; BlotChabaud, M.; Matthay, M.A.; Couette, S.; Uchida, T.; Clerici, C. Hypoxia and beta(2)-agonists regulate cell surface expression of the epithelial sodium channel in native alveolar epithelial cells. J. Biol. Chem. 2002, 277, 47318–47324. [Google Scholar] [CrossRef] [Green Version]
- Dada, L.; Bertorello, A.; Pedemonte, C.; Chandel, N.; Sznajder, J.I. Hypoxia inhibits Na,K-ATPase function by endocytosis of its à1 subunit in alveolar epithelial cells. Am. J. Respir. Crit. Care Med. 2001, 163, A572. [Google Scholar]
- Scherrer, U.; Vollenweider, L.; Delabays, A.; Savcic, M.; Eichenberger, U.; Kleger, G.R.; Fikrle, A.; Ballmer, P.E.; Nicod, P.; Bärtsch, P. Inhaled nitric oxide for high-altitude pulmonary edema. N. Engl. J. Med. 1996, 334, 624–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ignarro, L.J. Inhaled NO and COVID-19. Br. J. Pharmacol. 2020, 177, 3848–3849. [Google Scholar] [CrossRef] [PubMed]
- Jorens, P.G.; Vermeire, P.A.; Herman, A.G. L-arginine-dependent nitric oxide synthase: A new metabolic pathway in the lung and airways. Eur. Respir. J. 1993, 6, 258–266. [Google Scholar]
- Haddad, I.Y.; Pataki, G.; Hu, P.; Galliani, C.; Beckman, J.S.; Matalon, S. Quantitation of nitrotyrosine levels in lung sections of patients and animals with acute lung injury. J. Clin. Investig. 1994, 94, 2407–2413. [Google Scholar] [CrossRef]
- Sittipunt, C.; Steinberg, K.P.; Ruzinski, J.T.; Myles, C.; Zhu, S.; Goodman, R.B.; Hudson, L.D.; Matalon, S.; Martin, T.R. Nitric oxide and nitrotyrosine in the lungs of patients with acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2001, 163, 503–510. [Google Scholar] [CrossRef]
- Dobbs, L.G. Isolation and culture of alveolar type II cells. Am. J. Physiol. 1990, 258, L134–L147. [Google Scholar] [CrossRef]
- Rao, X.; Huang, X.; Zhou, Z.; Lin, X. An improvement of the 2−ΔΔCT) method for quantitative real-time polymerase chain reaction data analysis. Biostat. Bioinforma. Biomath. 2013, 3, 71–85. [Google Scholar]
- Gottardi, C.J.; Dunbar, L.A.; Caplan, M.J. Biotinylation and assessment of membrane polarity: Caveats and methodological concerns. Am. J. Physiol. 1995, 268, F285–F295. [Google Scholar] [CrossRef]
- Peters, D.M.; Vadasz, I.; Wujak, L.; Wygrecka, M.; Olschewski, A.; Becker, C.; Herold, S.; Papp, R.; Mayer, K.; Rummel, S.; et al. TGF-beta directs trafficking of the epithelial sodium channel ENaC which has implications for ion and fluid transport in acute lung injury. Proc. Natl. Acad. Sci. USA 2014, 111, E374–E383. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baloglu, E.; Velineni, K.; Ermis-Kaya, E.; Mairbäurl, H. Hypoxia Aggravates Inhibition of Alveolar Epithelial Na-Transport by Lipopolysaccharide-Stimulation of Alveolar Macrophages. Int. J. Mol. Sci. 2022, 23, 8315. https://doi.org/10.3390/ijms23158315
Baloglu E, Velineni K, Ermis-Kaya E, Mairbäurl H. Hypoxia Aggravates Inhibition of Alveolar Epithelial Na-Transport by Lipopolysaccharide-Stimulation of Alveolar Macrophages. International Journal of Molecular Sciences. 2022; 23(15):8315. https://doi.org/10.3390/ijms23158315
Chicago/Turabian StyleBaloglu, Emel, Kalpana Velineni, Ezgi Ermis-Kaya, and Heimo Mairbäurl. 2022. "Hypoxia Aggravates Inhibition of Alveolar Epithelial Na-Transport by Lipopolysaccharide-Stimulation of Alveolar Macrophages" International Journal of Molecular Sciences 23, no. 15: 8315. https://doi.org/10.3390/ijms23158315