
Investigating the Role of GABA in Neural Development and Disease Using Mice Lacking GAD67 or VGAT Genes


{kind=link}
Abstract
:1. Preface
2. Importance of GABA in the Nervous System
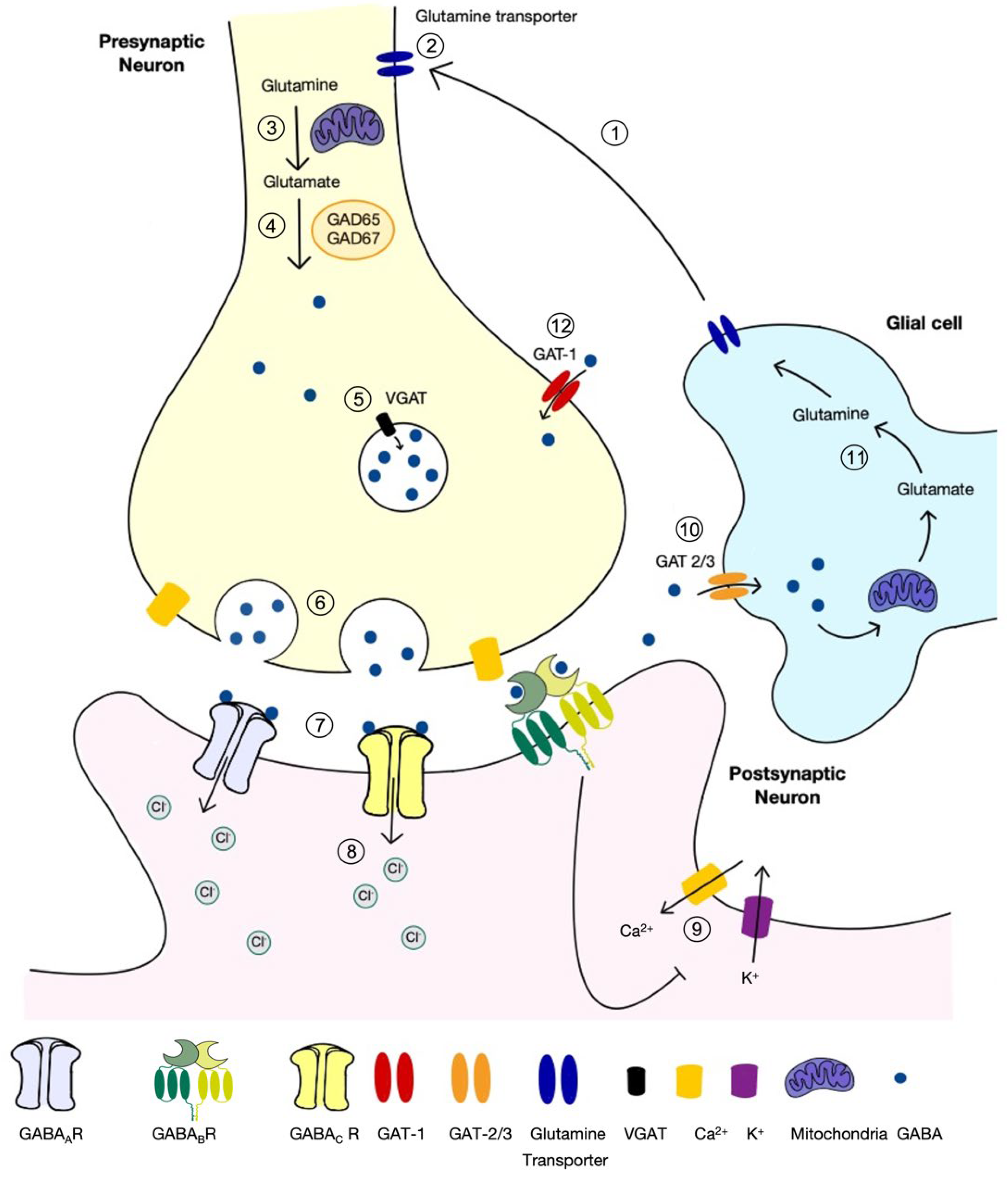
2.1. Inhibition in the Forebrain Is Mediated by GABA
2.2. The Role of GABA in the Mature Central Nervous System
2.3. The Role of VGAT in GABA and Glycine Signalling
2.4. GABA in Neurodevelopment
2.5. Excitatory–Inhibitory Balance Is Crucial in Normal Neurodevelopment
3. Disorders of Synaptic Inhibition
3.1. Synaptic Inhibition Dysfunction in Epilepsy
3.2. Synaptic Inhibition Dysfunction in Schizophrenia
3.3. Autism Spectrum Disorders and GABAergic Dysfunction
4. GABA Deficient Mouse Models
4.1. The GAD67+/− Mouse
4.2. The VGAT Deficient Mouse
5. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Asada, H.; Kawamura, Y.; Maruyama, K.; Kume, H.; Ding, R.G.; Kanbara, N.; Obata, K. Cleft palate and decreased brain γ-aminobutyric acid in mice lacking the 67-kDa isoform of glutamic ac-id decarboxylase. Proc. Natl. Acad. Sci. USA 1997, 94, 6496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojcik, S.M.; Katsurabayashi, S.; Guillemin, I.; Friauf, E.; Rosenmund, C.; Brose, N.; Rhee, J.-S. A Shared Vesicular Carrier Allows Synaptic Corelease of GABA and Glycine. Neuron 2006, 50, 575–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, H.-Y.; Cline, H.T. What Is Excitation/Inhibition and How Is It Regulated? A Case of the Elephant and the Wisemen. J. Exp. Neurosci. 2019, 13, 1179069519859371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamlin, C.R.; Yu, W.-Q.; Wong, R.O.L.; Hoon, M. Assembly and maintenance of GABAergic and Glycinergic circuits in the mammalian nervous system. Neural Dev. 2018, 13, 12. [Google Scholar] [CrossRef] [Green Version]
- Jewett, B.E.; Sharma, S. Physiology, GABA. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Kilb, W.; Kirischuk, S.; Luhmann, H.J. Role of tonic GABAergic currents during pre- and early postnatal rodent development. Front. Neural Circuits 2013, 7, 139. [Google Scholar] [CrossRef] [Green Version]
- Cellot, G.; Cherubini, E. Functional role of ambient GABA in refining neuronal circuits early in postnatal development. Front. Neural Circuits 2013, 7, 136. [Google Scholar] [CrossRef] [Green Version]
- Manent, J.-B.; Demarque, M.; Jorquera, I.; Pellegrino, C.; Ben-Ari, Y.; Aniksztejn, L.; Represa, A. A Noncanonical Release of GABA and Glutamate Modulates Neuronal Migration. J. Neurosci. 2005, 25, 4755–4765. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Z.; Huguenard, J.R.; Prince, D.A. GABAA receptor-mediated currents in interneurons and pyramidal cells of rat visual cortex. J. Physiol. 1998, 506, 715–730. [Google Scholar] [CrossRef]
- Huang, Z.J.; Di Cristo, G.; Ango, F. Development of GABA innervation in the cerebral and cerebellar cortices. Nat. Rev. Neurosci. 2007, 8, 673–686. [Google Scholar] [CrossRef]
- Decavel, C.; Pol, A.N.V.D. GABA: A dominant neurotransmitter in the hypothalamus. J. Comp. Neurol. 1990, 302, 1019–1037. [Google Scholar] [CrossRef]
- Halassa, M.M.; Acsády, L. Thalamic Inhibition: Diverse Sources, Diverse Scales. Trends Neurosci. 2016, 39, 680–693. [Google Scholar] [CrossRef] [Green Version]
- Fogarty, M.J.; Kanjhan, R.; Bellingham, M.C.; Noakes, P.G. Glycinergic Neurotransmission: A Potent Regulator of Embryonic Motor Neuron Dendritic Morphology and Synaptic Plasticity. J. Neurosci. 2016, 36, 80–87. [Google Scholar] [CrossRef] [Green Version]
- Moriguchi, Y.; Hiraki, K. Prefrontal cortex and executive function in young children: A review of NIRS studies. Front. Hum. Neurosci. 2013, 7, 867. [Google Scholar] [CrossRef] [Green Version]
- Dumoulin, A.; Triller, A.; Dieudonné, S. IPSC Kinetics at Identified GABAergic and Mixed GABAergic and Glycinergic Synapses onto Cerebellar Golgi Cells. J. Neurosci. 2001, 21, 6045–6057. [Google Scholar] [CrossRef] [Green Version]
- Wässle, H.; Koulen, P.; Brandstätter, J.H.; Fletcher, E.; Becker, C.-M. Glycine and GABA receptors in the mammalian retina. Vis. Res. 1998, 38, 1411–1430. [Google Scholar] [CrossRef] [Green Version]
- Pol, A.N.V.D.; Görcs, T. Glycine and glycine receptor immunoreactivity in brain and spinal cord. J. Neurosci. 1988, 8, 472–492. [Google Scholar] [CrossRef] [Green Version]
- Sanes, D.H.; Geary, W.A.; Wooten, G.F.; Rubel, E.W. Quantitative distribution of the glycine receptor in the auditory brain stem of the gerbil. J. Neurosci. 1987, 7, 3793–3802. [Google Scholar] [CrossRef] [Green Version]
- Trombley, P.Q.; Shepherd, G.M. Glycine exerts potent inhibitory actions on mammalian olfactory bulb neurons. J. Neurophysiol. 1994, 71, 761–767. [Google Scholar] [CrossRef]
- Song, W.; Chattipakorn, S.C.; McMahon, L.L. Glycine-Gated Chloride Channels Depress Synaptic Transmission in Rat Hippo-campus. J. Neurophysiol. 2006, 95, 2366–2379. [Google Scholar] [CrossRef] [Green Version]
- Diamond, J.S. Inhibitory Interneurons in the Retina: Types, Circuitry, and Function. Annu. Rev. Vis. Sci. 2017, 3, 1–24. [Google Scholar] [CrossRef]
- Jonas, P.; Bischofberger, J.; Sandkuühler, J. Corelease of Two Fast Neurotransmitters at a Central Synapse. Science 1998, 281, 419–424. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, J.A.; Berger, A.J. Cotransmission of GABA and Glycine to Brain Stem Motoneurons. J. Neurophysiol. 1999, 82, 1638–1641. [Google Scholar] [CrossRef] [Green Version]
- Kelsom, C.; Lu, W. Development and specification of GABAergic cortical interneurons. Cell Biosci. 2013, 3, 19. [Google Scholar] [CrossRef] [Green Version]
- Sigel, E.; Steinmann, M.E. Structure, Function, and Modulation of GABA(A) Receptors. J. Biol. Chem. 2012, 287, 40224–40231. [Google Scholar] [CrossRef] [Green Version]
- Olsen, R.W.; DeLorey, T.M. GABA Receptor Physiology and Pharmacology. In Basic Neurochemistry: Molecular, Cellular and Medical Aspects, 6th ed.; Siegel, G.J., Agranoff, B.W., Albers, R.W., Eds.; Lippincott-Raven: Philadelphia, PA, USA, 1999. [Google Scholar]
- DeLorey, T.M.; Sahbaie, P.; Hashemi, E.; Homanics, G.E.; Clark, J.D. Gabrb3 gene deficient mice exhibit impaired social and exploratory behaviors, deficits in non-selective attention and hypoplasia of cerebellar vermal lobules: A potential model of autism spectrum disorder. Behav. Brain Res. 2008, 187, 207–220. [Google Scholar] [CrossRef] [Green Version]
- Zafar, S.; Jabeen, I. Structure, Function, and Modulation of γ-Aminobutyric Acid Transporter 1 (GAT1) in Neurological Disorders: A Pharmacoinformatic Prospective. Front. Chem. 2018, 11, 397. [Google Scholar] [CrossRef] [PubMed]
- Varoqueaux, F.; Jamain, S.; Brose, N. Neuroligin 2 is exclusively localized to inhibitory synapses. Eur. J. Cell Biol. 2004, 83, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, T.J.; Craig, A.M. Synaptic organizing complexes. Curr. Opin. Neurobiol. 2011, 21, 132–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, C.I.; Chen, L. Postsynaptic assembly induced by neurexin-neuroligin interaction and neurotransmitter. Proc. Natl. Acad. Sci. USA 2005, 102, 6137–6142. [Google Scholar] [CrossRef] [Green Version]
- Scheiffele, P.; Fan, J.; Choih, J.; Fetter, R.; Serafini, T. Neuroligin Expressed in Nonneuronal Cells Triggers Presynaptic Development in Contacting Axons. Cell 2000, 101, 657–669. [Google Scholar] [CrossRef] [Green Version]
- Modi, J.; Prentice, H.; Wu, J.-Y. Regulation of GABA Neurotransmission by Glutamic Acid Decarboxylase (GAD). Curr. Pharm. Des. 2015, 21, 4939–4942. [Google Scholar] [CrossRef]
- Erlander, M.G.; Tillakaratne, N.J.; Feldblum, S.; Patel, N.; Tobin, A.J. Two genes encode distinct glutamate decarboxylases. Neuron 1991, 7, 91–100. [Google Scholar] [CrossRef]
- Gaetz, W.; Bloy, L.; Wang, D.; Port, R.; Blaskey, L.; Levy, S.; Roberts, T. GABA estimation in the brains of children on the autism spectrum: Measurement precision and regional cortical variation. NeuroImage 2013, 86, 1. [Google Scholar] [CrossRef] [Green Version]
- Kuwana, S.; Okada, Y.; Sugawara, Y.; Tsunekawa, N.; Obata, K. Disturbance of neural respiratory control in neonatal mice lacking gaba synthesizing enzyme 67-kda iso-form of glutamic acid decarboxylase. Neuroscience 2003, 120, 861–870. [Google Scholar] [CrossRef]
- Stork, O.; Ji, F.-Y.; Kaneko, K.; Stork, S.; Yoshinobu, Y.; Moriya, T.; Shibata, S.; Obata, K. Postnatal development of a GABA deficit and disturbance of neural functions in mice lacking GAD65. Brain Res. 2000, 865, 45–58. [Google Scholar] [CrossRef]
- Sandhu, K.V.; Lang, D.; Muller, B.; Nullmeier, S.; Yanagawa, Y.; Schwegler, H.; Stork, O. Glutamic acid decarboxylase 67 haplodeficiency impairs social behavior in mice. Genes Brain Behav. 2014, 13, 439–450. [Google Scholar] [CrossRef] [Green Version]
- Sagné, C.; El Mestikawy, S.; Isambert, M.-F.; Hamon, M.; Henry, J.-P.; Giros, B.; Gasnier, B. Cloning of a functional vesicular GABA and glycine transporter by screening of genome databases. FEBS Lett. 1997, 417, 177–183. [Google Scholar] [CrossRef] [Green Version]
- Dumoulin, A.; Rostaing, P.; Bedet, C.; Lévi, S.; Isambert, M.F.; Henry, J.P.; Gasnier, B. Presence of the vesicular inhibitory amino acid transporter in GABAergic and glycinergic synaptic ter-minal boutons. J. Cell Sci. 1999, 112, 811–823. [Google Scholar] [CrossRef]
- Chaudhry, F.A.; Reimer, R.J.; Bellocchio, E.E.; Danbolt, N.C.; Osen, K.K.; Edwards, R.H.; Storm-Mathisen, J. The Vesicular GABA Transporter, VGAT, Localizes to Synaptic Vesicles in Sets of Glycinergic as Well as GABAergic Neurons. J. Neurosci. 1998, 18, 9733–9750. [Google Scholar] [CrossRef] [Green Version]
- Bruining, H.; Hardstone, R.; Juarez-Martinez, E.L.; Sprengers, J.; Avramiea, A.-E.; Simpraga, S.; Houtman, S.J.; Poil, S.-S.; Dallares, E.; Palva, S.; et al. Measurement of excitation-inhibition ratio in autism spectrum disorder using critical brain dynamics. Sci. Rep. 2020, 10, 9195. [Google Scholar] [CrossRef]
- Ito, S. GABA and glycine in the developing brain. J. Physiol. Sci. 2016, 66, 375–379. [Google Scholar] [CrossRef]
- Wang, W.; Xu, T.L. Chloride homeostasis differentially affects GABA(A) receptor- and glycine receptor-mediated effects on spontaneous circuit activity in hippocampal cell culture. Neurosci. Lett. 2006, 406, 11–16. [Google Scholar] [CrossRef]
- Blaesse, P.; Airaksinen, M.S.; Rivera, C.; Kaila, K. Cation-Chloride Cotransporters and Neuronal Function. Neuron 2009, 61, 820–838. [Google Scholar] [CrossRef] [Green Version]
- Virtanen, M.A.; Uvarov, P.; Hübner, C.A.; Kaila, K. NKCC1, an Elusive Molecular Target in Brain Development: Making Sense of the Existing Data. Cells 2020, 9, 2607. [Google Scholar] [CrossRef]
- Gatto, C.L.; Broadie, K. Genetic controls balancing excitatory and inhibitory synaptogenesis in neurodevelopmental dis-order models. Front. Synaptic Neurosci. 2010, 2, 4. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Jaenisch, R.; Sur, M. The role of GABAergic signalling in neurodevelopmental disorders. Nat. Rev. Neurosci. 2021, 22, 290–307. [Google Scholar] [CrossRef]
- Ben-Ari, Y. Excitatory actions of gaba during development: The nature of the nurture. Nat. Rev. Neurosci. 2002, 3, 728–739. [Google Scholar] [CrossRef]
- Schmidt, M.J.; Mirnics, K. Neurodevelopment, GABA System Dysfunction, and Schizophrenia. Neuropsychopharmacology 2014, 40, 190–206. [Google Scholar] [CrossRef] [Green Version]
- Murata, Y.; Colonnese, M.T. GABAergic interneurons excite neonatal hippocampus in vivo. Sci. Adv. 2020, 6, eaba1430. [Google Scholar] [CrossRef]
- Payne, J.A.; Rivera, C.; Voipio, J.; Kaila, K. Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 2003, 26, 199–206. [Google Scholar] [CrossRef]
- Kirmse, K.; Hübner, C.A.; Isbrandt, D.; Witte, O.W.; Holthoff, K. GABAergic Transmission during Brain Development: Multiple Effects at Multiple Stages. Neuroscientist 2017, 24, 36–53. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Fukuda, A. Development and regulation of chloride homeostasis in the central nervous system. Front. Cell. Neurosci. 2015, 9, 371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kendler, A.; Golden, J.A. Progenitor Cell Proliferation Outside the Ventricular and Subventricular Zones during Human Brain Development. J. Neuropathol. Exp. Neurol. 1996, 55, 1253–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owens, D.F.; Kriegstein, A.R. Is there more to GABA than synaptic inhibition? Nat. Rev. Neurosci. 2002, 3, 715–727. [Google Scholar] [CrossRef]
- Haydar, T.F.; Wang, F.; Schwartz, M.L.; Rakic, P. Differential modulation of proliferation in the neocortical ventricular and subventricular zones. J. Neurosci. 2000, 20, 5764–5774. [Google Scholar] [CrossRef] [Green Version]
- Bortone, D.; Polleux, F. KCC2 Expression Promotes the Termination of Cortical Interneuron Migration in a Voltage-Sensitive Calcium-Dependent Manner. Neuron 2009, 62, 53–71. [Google Scholar] [CrossRef] [Green Version]
- LoTurco, J.J.; Owens, D.F.; Heath, M.J.; Davis, M.B.; Kriegstein, A.R. GABA and glutamate depolarize cortical progenitor cells and inhibit DNA synthesis. Neuron 1995, 15, 1287–1298. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Kakizaki, T.; Sakagami, H.; Saito, K.; Ebihara, S.; Kato, M.; Hirabayashi, M.; Saito, Y.; Furuya, N.; Yanagawa, Y. Fluorescent labeling of both GABAergic and glycinergic neurons in vesicular GABA transporter (VGAT)–Venus transgenic mouse. Neuroscience 2009, 164, 1031–1043. [Google Scholar] [CrossRef]
- Marty, S.; Berninger, B.; Carroll, P.; Thoenen, H. GABAergic Stimulation Regulates the Phenotype of Hippocampal Interneurons through the Regulation of Brain-Derived Neurotrophic Factor. Neuron 1996, 16, 565–570. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Sun, D. GABA receptors in brain development, function, and injury. Metab. Brain Dis. 2015, 30, 367–379. [Google Scholar] [CrossRef] [Green Version]
- Ganguly, K.; Schinder, A.F.; Wong, S.T.; Poo, M.-M. GABA Itself Promotes the Developmental Switch of Neuronal GABAergic Responses from Excitation to Inhibition. Cell 2001, 105, 521–532. [Google Scholar] [CrossRef] [Green Version]
- Carment, L.; Dupin, L.; Guedj, L.; Térémetz, M.; Cuenca, M.; Krebs, M.-O.; Amado, I.; Maier, M.A.; Lindberg, P.G. Neural noise and cortical inhibition in schizophrenia. Brain Stimul. 2020, 13, 1298–1304. [Google Scholar] [CrossRef]
- Alwis, D.S.; Rajan, R. Environmental enrichment causes a global potentiation of neuronal responses across stimulus com-plexity and lamina of sensory cortex. Front. Cell. Neurosci. 2013, 7, 124. [Google Scholar] [CrossRef] [Green Version]
- Villa, K.L.; Nedivi, E. Dendrites; Springer: Tokyo, Japan, 2016; pp. 467–487. [Google Scholar]
- Fogarty, M.J.; Kanjhan, R.; Yanagawa, Y.; Noakes, P.G.; Bellingham, M. Alterations in hypoglossal motor neurons due to GAD67 and VGAT deficiency in mice. Exp. Neurol. 2017, 289, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Forrest, M.P.; Hill, M.J.; Kavanagh, D.H.; Tansey, K.E.; Waite, A.J.; Blake, D.J. The Psychiatric Risk Gene Transcription Factor 4 (TCF4) Regulates Neurodevelopmental Pathways Asso-ciated With Schizophrenia, Autism, and Intellectual Disability. Schizophr Bull. 2018, 44, 1100–1110. [Google Scholar] [CrossRef] [Green Version]
- Penzes, P.; Woolfrey, K.M.; Srivastava, D.P. Epac2-mediated dendritic spine remodeling: Implications for disease. Mol. Cell. Neurosci. 2011, 46, 368–380. [Google Scholar] [CrossRef] [Green Version]
- Klenowski, P.M.; Shariff, M.; Belmer, A.; Fogarty, M.J.; Mu, E.W.H.; Bellingham, M.; Bartlett, S.E. Prolonged Consumption of Sucrose in a Binge-Like Manner, Alters the Morphology of Medium Spiny Neurons in the Nucleus Accumbens Shell. Front. Behav. Neurosci. 2016, 10, 54. [Google Scholar] [CrossRef] [Green Version]
- Moy, S.S.; Nadler, J.J.; Young, N.B.; Perez, A.; Holloway, L.P.; Barbaro, R.P.; Barbaro, J.R.; Wilson, L.M.; Threadgill, D.W.; Lauder, J.M.; et al. Mouse behavioral tasks relevant to autism: Phenotypes of 10 inbred strains. Behav. Brain Res. 2007, 176, 4–20. [Google Scholar] [CrossRef] [Green Version]
- Purves, D.; Augustine, G.J.; Fitzpatrick, D.; Katz, L.C.; LaMantia, A.S.; McNamara, J.O.; Williams, S.M. GABA and Glycine. In Neuroscience, 2nd ed.; Sinauer Associates: Sunderland, MA, USA, 2001. [Google Scholar]
- Fujihara, K.; Miwa, H.; Kakizaki, T.; Kaneko, R.; Mikuni, M.; Tanahira, C.; Yanagawa, Y. Glutamate Decarboxylase 67 Deficiency in a Subset of GABAergic Neurons Induces Schizophrenia-Related Pheno-types. Neuropsychopharmacology 2015, 40, 2475–2486. [Google Scholar] [CrossRef] [Green Version]
- Thompson, M.; Weickert, C.S.; Wyatt, E.; Webster, M.J. Decreased glutamic acid decarboxylase67 mRNA expression in multiple brain areas of patients with schizophrenia and mood disorders. J. Psychiatr. Res. 2009, 43, 970–977. [Google Scholar] [CrossRef]
- Whitney, E.R.; Kemper, T.L.; Bauman, M.L.; Rosene, D.; Blatt, G.J. Cerebellar Purkinje Cells are Reduced in a Subpopulation of Autistic Brains: A Stereological Experiment Using Calbindin-D28k. Cerebellum 2008, 7, 406–416. [Google Scholar] [CrossRef]
- Fatemi, S.H.; Reutiman, T.J.; Folsom, T.D.; Thuras, P.D. GABA(A) receptor downregulation in brains of subjects with autism. J. Autism. Dev. Disord. 2009, 39, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Maglóczky, Z.; Freund, T.F. Impaired and repaired inhibitory circuits in the epileptic human hippocampus. Trends Neurosci. 2005, 28, 334–340. [Google Scholar] [CrossRef]
- Lin, C.C.; Huang, T.L. Chapter 3—Epigenetic biomarkers in neuropsychiatric disorders. In Neuropsychiatric Disorders and Epi-genetics; Yasui, D.H., Peedicayil, J., Grayson, D.R., Eds.; Academic Press: Boston, MA, USA, 2017; pp. 35–66. [Google Scholar]
- Thijs, R.D.; Surges, R.; O’Brien, T.J.; Sander, J.W. Epilepsy in adults. Lancet 2019, 393, 689–701. [Google Scholar] [CrossRef]
- Singh, A.; Trevick, S. The Epidemiology of Global Epilepsy. Neurol. Clin. 2016, 34, 837–847. [Google Scholar] [CrossRef]
- Pitkänen, A.; Engel, J., Jr. Past and Present Definitions of Epileptogenesis and Its Biomarkers. Neurotherapeutics 2014, 11, 231–241. [Google Scholar] [CrossRef] [Green Version]
- Fisher, R.S.; van Emde Boas, W.; Blume, W.; Elger, C.; Genton, P.; Lee, P.; Engel, J., Jr. Epileptic Seizures and Epilepsy: Definitions Proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia 2005, 46, 470–472. [Google Scholar] [CrossRef]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshe, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Termi-nology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [Green Version]
- Yuen, A.W.; Keezer, M.R.; Sander, J.W. Epilepsy is a neurological and a systemic disorder. Epilepsy Behav. 2018, 78, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Scharfman, H.E. The neurobiology of epilepsy. Curr. Neurol. Neurosci. Rep. 2007, 7, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Aroniadou-Anderjaska, V.; Fritsch, B.; Qashu, F.; Braga, M.F. Pathology and pathophysiology of the amygdala in epileptogenesis and epilepsy. Epilepsy Res. 2008, 78, 102–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avanzini, G.; Franceschetti, S. Cellular biology of epileptogenesis. Lancet Neurol. 2003, 2, 33–42. [Google Scholar] [CrossRef]
- Coan, A.C.; Cendes, F. Epilepsy as progressive disorders: What is the evidence that can guide our clinical decisions and how can neuroimaging help? Epilepsy Behav. 2013, 26, 313–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malmgren, K.; Thom, M. Hippocampal sclerosis-Origins and imaging. Epilepsia 2012, 53, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Engel, J. The Temporal Lobe and Limbic System. J. Clin. Neurophysiol. 1998, 15, 277. [Google Scholar] [CrossRef]
- De Lanerolle, N.; Kim, J.; Robbins, R.; Spencer, D. Hippocampal interneuron loss and plasticity in human temporal lobe epilepsy. Brain Res. 1989, 495, 387–395. [Google Scholar] [CrossRef]
- Robbins, R.J.; Brines, M.L.; Kim, J.H.; Adrian, T.; De Lanerolle, N.; Welsh, S.; Spencer, D.D. A selective loss of somatostatin in the hippocampus of patients with temporal lobe epilepsy. Ann. Neurol. 1991, 29, 325–332. [Google Scholar] [CrossRef]
- Loescher, W. Critical review of current animal models of seizures and epilepsy used in the discovery and development of new antiepileptic drugs. Seizure 2011, 20, 359–368. [Google Scholar] [CrossRef] [Green Version]
- Kandratavicius, L.; Balista, P.A.; Lopes-Aguiar, C.; Ruggiero, R.N.; Umeoka, E.H.; Garcia-Cairasco, N.; Bueno-Junior, L.S.; Leite, J.P. Animal models of epilepsy: Use and limitations. Neuropsychiatr. Dis. Treat. 2014, 10, 1693–1705. [Google Scholar] [CrossRef] [Green Version]
- Escayg, A.; Goldin, A.L. Sodium channel SCN1A and epilepsy: Mutations and mechanisms. Epilepsia 2010, 51, 1650–1658. [Google Scholar] [CrossRef] [Green Version]
- Ogiwara, I.; Miyamotor, H.; Morita, N.; Atapour, N.; Mazaki, E.; Inoue, I.; Takeuchi, T.; Itohara, S.; Yanagawa, Y.; Obata, K.; et al. Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: A circuit basis for epileptic sei-zures in mice carrying an Scn1a gene mutation. J. Neurosci. 2007, 27, 5903–5914. [Google Scholar] [CrossRef] [Green Version]
- Escayg, A.; Macdonald, B.T.; Meisler, M.H.; Baulac, S.; Huberfeld, G.; An-Gourfinkel, I.; Brice, A.; LeGuern, E.; Moulard, B.; Chaigne, D.; et al. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat. Genet. 2000, 24, 343–345. [Google Scholar] [CrossRef]
- Steinlein, O.K. Mechanisms underlying epilepsies associated with sodium channel mutations. Prog. Brain Res. 2014, 213, 97–111. [Google Scholar] [CrossRef]
- Macdonald, R.L.; Kang, J.Q.; Gallagher, M.J. Mutations in GABAA receptor subunits associated with genetic epilepsies. J. Physiol. 2010, 588 Pt 11, 1861–1869. [Google Scholar] [CrossRef]
- Matsumoto, H.; Marsan, C.A. Cellular Mechanisms in Experimental Epileptic Seizures. Science 1964, 144, 193–194. [Google Scholar] [CrossRef]
- Prince, D.A.; Wilder, B.J. Control mechanisms in cortical epileptogenic foci. “Surround” inhibition. Arch. Neurol. 1967, 16, 194–202. [Google Scholar] [CrossRef]
- Walther, H.; Lambert, J.D.C.; Jones, R.S.G.; Heinemann, U.; Hamon, B. Epileptiform activity in combined slices of the hippocampus, subiculum and entorhinal cortex during per-fusion with low magnesium medium. Neurosci. Lett. 1986, 69, 156–161. [Google Scholar] [CrossRef]
- Prince, D.A. The depolarization shift in “epileptic” neurons. Exp. Neurol. 1968, 21, 467–485. [Google Scholar] [CrossRef]
- Dichter, M.; Spencer, W.A. Penicillin-induced interictal discharges from the cat hippocampus. I. Characteristics and topographical features. J. Neurophysiol. 1969, 32, 649–662. [Google Scholar] [CrossRef]
- Khazipov, R.; Holmes, G.L. Synchronization of kainate-induced epileptic activity via GABAergic inhibition in the super-fused rat hippocampus in vivo. J. Neurosci. 2003, 23, 5337–5341. [Google Scholar] [CrossRef] [Green Version]
- Gutnick, M.; Connors, B.; Prince, D.A. Mechanisms of neocortical epileptogenesis in vitro. J. Neurophysiol. 1982, 48, 1321–1335. [Google Scholar] [CrossRef]
- Miles, R.; Wong, R.K.S. Single neurones can initiate synchronized population discharge in the hippocampus. Nature 1983, 306, 371–373. [Google Scholar] [CrossRef]
- Wiechert, P.; Herbst, A. Provocation of cerebral seizures by derangement of the natural balance between glutamic acid and gamma-aminobutyric acid. J. Neurochem. 1966, 13, 59–64. [Google Scholar] [CrossRef]
- Croucher, M.J.; Collins, J.F.; Meldrum, B.S. Anticonvulsant Action of Excitatory Amino Acid Antagonists. Science 1982, 216, 899–901. [Google Scholar] [CrossRef]
- Calcagnotto, M.E.; Paredes, M.F.; Tihan, T.; Barbaro, N.M.; Baraban, S.C. Dysfunction of Synaptic Inhibition in Epilepsy Associated with Focal Cortical Dysplasia. J. Neurosci. 2005, 25, 9649–9657. [Google Scholar] [CrossRef] [Green Version]
- Zhong, S.; Zhao, Z.; Xie, W.; Cai, Y.; Zhang, Y.; Ding, J.; Wang, X. GABAergic Interneuron and Neurotransmission Are mTOR-Dependently Disturbed in Experimental Focal Cortical Dysplasia. Mol. Neurobiol. 2021, 58, 156–169. [Google Scholar] [CrossRef]
- Braff, D.L.; Geyer, M.A.; Swerdlow, N.R. Human studies of prepulse inhibition of startle: Normal subjects, patient groups, and pharmacological studies. Psychopharmacology 2001, 156, 234–258. [Google Scholar] [CrossRef]
- Couture, S.M.; Penn, D.L.; Roberts, D.L. The Functional Significance of Social Cognition in Schizophrenia: A Review. Schizophr. Bull. 2006, 32 (Suppl. 1), S44–S63. [Google Scholar] [CrossRef] [Green Version]
- Mueser, K.T.; Jeste, D.V. Clinical Handbook of Schizophrenia; Guilford Press: New York, NY, USA, 2008. [Google Scholar]
- Siever, L.J.; Davis, K.L. The Pathophysiology of Schizophrenia Disorders: Perspectives from the Spectrum. Am. J. Psychiatry 2004, 161, 398–413. [Google Scholar] [CrossRef]
- Dipiro, J.T.; Talbert, R.L.; Yee, G.C.; Matzke, G.R.; Wells, B.G.; Posey, L.M. Pharmacotherapy: A Pathophysiologic Approach, 9th ed.; McGraw-Hill Education Medical: New York, NY, USA, 2014. [Google Scholar]
- Insel, T.R. Rethinking schizophrenia. Nature 2010, 468, 187–193. [Google Scholar] [CrossRef] [Green Version]
- McDonald, C.; Murphy, K.C. The new genetics of schizophrenia. Psychiatr. Clin. North Am. 2003, 26, 41–63. [Google Scholar] [CrossRef]
- Guidotti, A.; Auta, J.; Davis, J.M.; Di-Giorgi-Gerevini, V.D.; Dwivedi, Y.; Grayson, D.R.; Impagnatiello, F.; Pandey, G.; Presold, C.; Sharma, R.; et al. Decrease in Reelin and Glutamic Acid Decarboxylase67 (GAD67) Expression in Schizophrenia and Bipolar Disorder: A Postmortem Brain Study. Arch. Gen. Psychiatry 2000, 57, 1061–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veldic, M.; Guidotti, A.; Maloku, E.; Davis, J.M.; Costa, E. In psychosis, cortical interneurons overexpress DNA-methyltransferase 1. Proc. Natl. Acad. Sci. USA 2005, 102, 2152–2157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, T.U.; Walsh, J.P.; Benes, F.M. Density of glutamic acid decarboxylase 67 messenger RNA-containing neurons that express the N-methyl-D-aspartate receptor subunit NR2A in the anterior cingulate cortex in schizophrenia and bipolar disorder. Arch. Gen. Psychiatry 2004, 61, 649–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatemi, S.H.; Stary, J.M.; Earle, J.A.; Araghi-Niknam, M.; Eagan, E. GABAergic dysfunction in schizophrenia and mood disorders as reflected by decreased levels of glutamic acid decarboxylase 65 and 67 kDa and Reelin proteins in cerebellum. Schizophr. Res. 2005, 72, 109–122. [Google Scholar] [CrossRef]
- Hashimoto, T.; Volk, D.W.; Eggan, S.M.; Mirnics, K.; Pierri, J.N.; Sun, Z.; Sampson, A.R.; Lewis, D.A. Gene Expression Deficits in a Subclass of GABA Neurons in the Prefrontal Cortex of Subjects with Schizophrenia. J. Neurosci. 2003, 23, 6315–6326. [Google Scholar] [CrossRef]
- Rocco, B.R.; Lewis, D.A.; Fish, K.N. Markedly Lower Glutamic Acid Decarboxylase 67 Protein Levels in a Subset of Bou-tons in Schizophrenia. Biol. Psychiatry 2016, 79, 1006–1015. [Google Scholar] [CrossRef] [Green Version]
- Lewis, D.A.; Hashimoto, T.; Volk, D.W. Cortical inhibitory neurons and schizophrenia. Nat. Rev. Neurosci. 2005, 6, 312–324. [Google Scholar] [CrossRef]
- Hashimoto, T.; Bazmi, H.H.; Mirnics, K.; Wu, Q.; Sampson, A.R.; Lewis, D.A. Conserved Regional Patterns of GABA-Related Transcript Expression in the Neocortex of Subjects with Schizophrenia. Am. J. Psychiatry 2008, 165, 479–489. [Google Scholar] [CrossRef] [Green Version]
- Guastella, J.; Nelson, N.; Nelson, H.; Czyzyk, L.; Keynan, S.; Miedel, M.C.; Davidson, N.; Lester, H.A.; Kanner, B.I. Cloning and Expression of a Rat Brain GABA Transporter. Science 1990, 249, 1303–1306. [Google Scholar] [CrossRef]
- Borden, L.A. GABA transporter heterogeneity: Pharmacology and cellular localization. Neurochem. Int. 1996, 29, 335–356. [Google Scholar] [CrossRef]
- Gong, N.; Li, Y.; Cai, G.-Q.; Niu, R.-F.; Fang, Q.; Wu, K.; Chen, Z.; Lin, L.-N.; Xu, L.; Fei, J.; et al. GABA Transporter-1 Activity Modulates Hippocampal Theta Oscillation and Theta Burst Stimulation-Induced Long-Term Potentiation. J. Neurosci. 2009, 29, 15836–15845. [Google Scholar] [CrossRef] [Green Version]
- Dalby, N.O. GABA-level increasing and anticonvulsant effects of three different GABA uptake inhibitors. Neuropharmacology 2000, 39, 2399–2407. [Google Scholar] [CrossRef]
- Semyanov, A.; Walker, M.; Kullmann, D. GABA uptake regulates cortical excitability via cell type–specific tonic inhibition. Nat. Neurosci. 2003, 6, 484–490. [Google Scholar] [CrossRef]
- Gur, R.E.; Cowell, P.E.; Latshaw, A.; Turetsky, B.I.; Grossman, R.I.; Arnold, S.E.; Gur, R.C. Reduced dorsal and orbital prefrontal gray matter volumes in schizophrenia. Arch. Gen. Psychiatry 2000, 57, 761–768. [Google Scholar] [CrossRef]
- Carpenter, W.T.; Koenig, J.I. The Evolution of Drug Development in Schizophrenia: Past Issues and Future Opportunities. Neuropsychopharmacology 2007, 33, 2061–2079. [Google Scholar] [CrossRef]
- Lapiz, M.D.S.; Fulford, A.; Muchimapura, S.; Mason, R.; Parker, T.; Marsden, C.A. Influence of Postweaning Social Isolation in the Rat on Brain Development, Conditioned Behavior, and Neurotransmission. Neurosci. Behav. Physiol. 2003, 33, 13–29. [Google Scholar] [CrossRef]
- Featherstone, R.E.; Rizos, Z.; Kapur, S.; Fletcher, P.J. A sensitizing regimen of amphetamine that disrupts attentional set-shifting does not disrupt work-ing or long-term memory. Behav. Brain Res. 2008, 189, 170–179. [Google Scholar] [CrossRef]
- Jentsch, J.D.; Roth, R.H. The neuropsychopharmacology of phencyclidine: From NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology 1999, 20, 201–225. [Google Scholar] [CrossRef] [Green Version]
- Neill, J.C.; Barnes, S.; Cook, S.; Grayson, B.; Idris, N.F.; McLean, S.L.; Snigdha, S.; Rajagopal, L.; Harte, M.K. Animal models of cognitive dysfunction and negative symptoms of schizophrenia: Focus on NMDA receptor antagonism. Pharmacol. Ther. 2010, 128, 419–432. [Google Scholar] [CrossRef] [Green Version]
- Lodge, D.J.; Behrens, M.M.; Grace, A. A Loss of Parvalbumin-Containing Interneurons Is Associated with Diminished Oscillatory Activity in an Animal Model of Schizophrenia. J. Neurosci. 2009, 29, 2344–2354. [Google Scholar] [CrossRef]
- Powell, C.M.; Miyakawa, T. Schizophrenia-Relevant Behavioral Testing in Rodent Models: A Uniquely Human Disorder? Biol. Psychiatry 2006, 59, 1198–1207. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Fang, Q.; Xiao, X.; Wang, Y.-Z.; Cai, Y.-Q.; Cao, H.; Hu, G.; Chen, Z.; Fei, J.; Gong, N.; et al. GABA Transporter-1 Deficiency Confers Schizophrenia-Like Behavioral Phenotypes. PLoS ONE 2013, 8, e69883. [Google Scholar] [CrossRef] [Green Version]
- Tamamaki, N.; Yanagawa, Y.; Tomioka, R.; Miyazaki, J.-I.; Obata, K.; Kaneko, T. Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the GAD67-GFP knock-in mouse. J. Comp. Neurol. 2003, 467, 60–79. [Google Scholar] [CrossRef] [PubMed]
- Yin, D.-M.; Chen, Y.-J.; Lu, Y.-S.; Bean, J.C.; Sathyamurthy, A.; Shen, C.; Liu, X.; Lin, T.W.; Smith, C.A.; Xiong, W.-C.; et al. Reversal of Behavioral Deficits and Synaptic Dysfunction in Mice Overexpressing Neuregulin 1. Neuron 2013, 78, 644–657. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.Y.; Wu, Z.; Forsyth, C.T.; Hu, Y.; Yee, S.P.; Chen, G. GABAergic deficits and schizophrenia-like behaviors in a mouse model carrying patient-derived neurolig-in-2 R215H mutation. Mol. Brain 2018, 11, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefansson, H.; Petursson, H.; Sigurdsson, E.; Steinthorsdottir, V.; Bjornsdottir, S.; Sigmundsson, T.; Ghosh, S.; Brynjolfsson, J.; Gunnarsdottir, S.; Ivarsson, O.; et al. Neuregulin 1 and Susceptibility to Schizophrenia. Am. J. Hum. Genet. 2002, 71, 877–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, S.B.; Valakh, V. Excitatory/Inhibitory Balance and Circuit Homeostasis in Autism Spectrum Disorders. Neuron 2015, 87, 684–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: DSM-IV, 4th ed.; American Psychiatric Association: Washington, DC, USA, 1994. [Google Scholar]
- Centers for Disease Control and Prevention. Autism Spectrum Disorder (ASD). 2016. Available online: http://www.cdc.gov/ncbddd/autism/data.html (accessed on 19 October 2016).
- DeLorey, T.M.; Sahbaie, P.; Hashemi, E.; Li, W.W.; Salehi, A.; Clark, D.J. Somatosensory and sensorimotor consequences associated with the heterozygous disruption of the au-tism candidate gene, Gabrb3. Behav. Brain Res. 2011, 216, 36–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, H.-T.; Chen, H.; Samaco, R.; Xue, M.; Chahrour, M.; Yoo, J.; Neul, J.; Gong, S.; Lu, H.-C.; Heintz, N.; et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 2010, 468, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Werling, D. The role of sex-differential biology in risk for autism spectrum disorder. Biol. Sex Differ. 2016, 7, 58. [Google Scholar] [CrossRef] [Green Version]
- Gogolla, N.; LeBlanc, J.J.; Quast, K.B.; Südhof, T.C.; Fagiolini, M.; Hensch, T.K. Common circuit defect of excitatory-inhibitory balance in mouse models of autism. J. Neurodev. Disord. 2009, 1, 172–181. [Google Scholar] [CrossRef] [Green Version]
- Ackerman, S. Discovering the Brain. In The Development and Shaping of the Brain; National Academies Press (US): Washington, DC, USA, 1992; Volume 6. [Google Scholar]
- Riccomagno, M.M.; Kolodkin, A.L. Sculpting Neural Circuits by Axon and Dendrite Pruning. Annu. Rev. Cell Dev. Biol. 2015, 31, 779–805. [Google Scholar] [CrossRef] [Green Version]
- Markham, J.A.; Greenough, W.T. Experience-driven brain plasticity: Beyond the synapse. Neuron Glia Biol. 2004, 1, 351–363. [Google Scholar] [CrossRef] [Green Version]
- Coghlan, S.; Horder, J.; Inkster, B.; Mendez, M.A.; Murphy, D.G.; Nutt, D.J. GABA system dysfunction in autism and related disorders: From synapse to symptoms. Neurosci. Biobehav. Rev. 2012, 36, 2044–2055. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.D.; Kriegstein, A.R. Defining the role of GABA in cortical development. J. Physiol. 2009, 587, 1873–1879. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, Z.; Bai, Y.T.; Wu, G.Y.; Chen, G. Gad67 haploinsufficiency reduces amyloid pathology and rescues olfactory memory deficits in a mouse mod-el of Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 73. [Google Scholar] [CrossRef] [Green Version]
- Ono, M.; Yanagawa, Y.; Koyano, K. GABAergic neurons in inferior colliculus of the GAD67-GFP knock-in mouse: Elec-trophysiological and morphological properties. Neurosci. Res. 2005, 51, 475–492. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.E.; McKenna, J.T.; Winston, S.; Basheer, R.; Yanagawa, Y.; Thakkar, M.M.; McCarley, R.W. Characterization of GABAergic neurons in rapid-eye-movement sleep controlling regions of the brainstem reticular formation in GAD67-green fluorescent protein knock-in mice. Eur. J. Neurosci. 2008, 27, 352–363. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, H.; Yanagawa, Y.; Obata, K. Development of stellate and basket cells and their apoptosis in mouse cerebellar cortex. Neurosci. Res. 2004, 50, 13–22. [Google Scholar] [CrossRef]
- Berghuis, P.; Rajnicek, A.M.; Morozov, Y.M.; Ross, R.A.; Mulder, J.; Urbaán, G.M.; Monory, K.; Marsicano, G.; Matteoli, M.; Canty, A.; et al. Hardwiring the Brain: Endocannabinoids Shape Neuronal Connectivity. Science 2007, 316, 1212–1216. [Google Scholar] [CrossRef] [Green Version]
- Lau, C.G.; Murthy, V.N. Activity-Dependent Regulation of Inhibition via GAD67. J. Neurosci. 2012, 32, 8521–8531. [Google Scholar] [CrossRef] [Green Version]
- Lazarus, M.S.; Krishnan, K.; Huang, Z.J. GAD67 Deficiency in Parvalbumin Interneurons Produces Deficits in Inhibitory Transmission and Network Disinhibition in Mouse Prefrontal Cortex. Cereb. Cortex 2013, 25, 1290–1296. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, H.; Hashimoto, K.; Miyazaki, T.; Yanagawa, Y.; Obata, K.; Watanabe, M.; Kano, M. Strength of GABAergic transmission influences climbing fiber synapse elimination during cerebellar development. Neurosci. Res. 2007, 58, S52. [Google Scholar] [CrossRef]
- Chattopadhyaya, B.; Di Cristo, G.; Wu, C.Z.; Knott, G.; Kuhlman, S.; Fu, Y.; Palmiter, R.D.; Huang, Z.J. GAD67-Mediated GABA Synthesis and Signaling Regulate Inhibitory Synaptic Innervation in the Visual Cortex. Neuron 2007, 54, 889–903. [Google Scholar] [CrossRef] [Green Version]
- Novotny, M.; Harvey, S.; Jemiolo, B.; Alberts, J. Synthetic pheromones that promote inter-male aggression in mice. Proc. Natl. Acad. Sci. USA 1985, 82, 2059–2061. [Google Scholar] [CrossRef] [Green Version]
- Guillot, P.-V.; Chapouthier, G. Intermale aggression and dark/light preference in ten inbred mouse strains. Behav. Brain Res. 1996, 77, 211–213. [Google Scholar] [CrossRef]
- Hendrickson, R.C.; Krauthamer, S.; Essenberg, J.M.; Holy, T.E. Inhibition Shapes Sex Selectivity in the Mouse Accessory Olfactory Bulb. J. Neurosci. 2008, 28, 12523–12534. [Google Scholar] [CrossRef]
- Malaspina, D.; Goetz, R.; Keller, A.; Messinger, J.W.; Bruder, G.; Goetz, D.; Opler, M.; Harlap, S.; Harkavy-Friedman, J.; Antonius, D. Olfactory processing, sex effects and heterogeneity in schizophrenia. Schizophr. Res. 2012, 135, 144–151. [Google Scholar] [CrossRef] [Green Version]
- Nullmeier, S.; Elmers, C.; D’Hanis, W.; Sandhu, K.V.K.; Stork, O.; Yanagawa, Y.; Panther, P.; Schwegler, H. Glutamic acid decarboxylase 67 haplodeficiency in mice: Consequences of postweaning social isolation on behavior and changes in brain neurochemical systems. Anat. Embryol. 2020, 225, 1719–1742. [Google Scholar] [CrossRef]
- Häfner, H.; Löffler, W.; Maurer, K.; Hambrecht, M.; Der Heiden, W.A. Depression, negative symptoms, social stagnation and social decline in the early course of schizophrenia. Acta Psychiatr. Scand. 1999, 100, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.M. Hyperactivity in mice lacking one allele of the glutamic acid decarboxylase 67 gene. ADHD Atten. Deficit Hyperact. Disord. 2018, 10, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Edden, R.A.E.; Crocetti, D.; Zhu, H.; Gilbert, D.; Mostofsky, S.H. Reduced GABA Concentration in Attention-Deficit/Hyperactivity Disorder. Arch. Gen. Psychiatry 2012, 69, 750–753. [Google Scholar] [CrossRef] [PubMed]
- Bruxel, E.M.; Akutagava-Martins, G.C.; Salatino-Oliveira, A.; Genro, J.P.; Zeni, C.P.; Polanczyk, G.V.; Hutz, M.H. GAD1 gene polymorphisms are associated with hyperactivity in Attention-Deficit/Hyperactivity Disorder. Am. J. Med. Genet. Neuropsychiatr. Genet. 2016, 171, 1099–1104. [Google Scholar] [CrossRef]
- Naaijen, J.; Bralten, J.; Poelmans, G.; Glennon, J.C.; Franke, B.; Buitelaar, J.K. Glutamatergic and GABAergic gene sets in attention-deficit/hyperactivity disorder: Association to overlap-ping traits in ADHD and autism. Transl. Psychiatry 2017, 7, e999. [Google Scholar] [CrossRef]
- Kinney, D.K.; Munir, K.M.; Crowley, D.J.; Miller, A.M. Prenatal stress and risk for autism. Neurosci. Biobehav. Rev. 2008, 32, 1519–1532. [Google Scholar] [CrossRef] [Green Version]
- Fine, R.; Zhang, J.; Stevens, H.E. Prenatal stress and inhibitory neuron systems: Implications for neuropsychiatric disorders. Mol. Psychiatry 2014, 19, 641–651. [Google Scholar] [CrossRef]
- Ronald, A.; Pennell, C.E.; Whitehouse, A.J.O. Prenatal Maternal Stress Associated with ADHD and Autistic Traits in early Childhood. Front. Psychol. 2011, 1, 223. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Burgos, G.; Fish, K.N.; Lewis, D.A. GABA Neuron Alterations, Cortical Circuit Dysfunction and Cognitive Def-icits in Schizophrenia. Neural Plast. 2011, 2011, 723184. [Google Scholar] [CrossRef]
- Uchida, T.; Oki, Y.; Yanagawa, Y.; Fukuda, A. A heterozygous deletion in the glutamate decarboxylase 67 gene enhances maternal and fetal stress vulnerability. Neurosci. Res. 2011, 69, 276–282. [Google Scholar] [CrossRef] [Green Version]
- Uchida, T.; Furukawa, T.; Iwata, S.; Yanagawa, Y.; Fukuda, A. Selective loss of parvalbumin-positive GABAergic interneurons in the cerebral cortex of maternally stressed Gad1-heterozygous mouse offspring. Transl. Psychiatry 2014, 4, e371. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, Y.; Lee, J.; Chun, H.; et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 2014, 20, 886–896. [Google Scholar] [CrossRef]
- Wu, Z.; Guo, Z.; Gearing, M.; Chen, G. Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzheimer’s disease model. Nat. Commun. 2014, 5, 4159. [Google Scholar] [CrossRef] [Green Version]
- Selkoe, D.J. The therapeutics of Alzheimer’s disease: Where we stand and where we are heading. Ann. Neurol. 2013, 74, 328–336. [Google Scholar] [CrossRef]
- McIntire, S.L.; Reimer, R.J.; Schuske, K.; Edwards, R.H.; Jorgensen, E.M. Identification and characterization of the vesicular GABA transporter. Nature 1997, 389, 870–876. [Google Scholar] [CrossRef]
- Saito, K.; Kakizaki, T.; Hayashi, R.; Nishimaru, H.; Furukawa, T.; Nakazato, Y.; Takamori, S.; Ebihara, S.; Uematsu, M.; Mishina, M.; et al. The physiological roles of vesicular GABA transporter during embryonic development: A study using knock-out mice. Mol. Brain 2010, 3, 40. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bolneo, E.; Chau, P.Y.S.; Noakes, P.G.; Bellingham, M.C. Investigating the Role of GABA in Neural Development and Disease Using Mice Lacking GAD67 or VGAT Genes. Int. J. Mol. Sci. 2022, 23, 7965. https://doi.org/10.3390/ijms23147965
Bolneo E, Chau PYS, Noakes PG, Bellingham MC. Investigating the Role of GABA in Neural Development and Disease Using Mice Lacking GAD67 or VGAT Genes. International Journal of Molecular Sciences. 2022; 23(14):7965. https://doi.org/10.3390/ijms23147965
Chicago/Turabian StyleBolneo, Erika, Pak Yan S. Chau, Peter G. Noakes, and Mark C. Bellingham. 2022. "Investigating the Role of GABA in Neural Development and Disease Using Mice Lacking GAD67 or VGAT Genes" International Journal of Molecular Sciences 23, no. 14: 7965. https://doi.org/10.3390/ijms23147965