
Is There a Connection between the Metabolism of Copper, Sulfur, and Molybdenum in Alzheimer’s Disease? New Insights on Disease Etiology

 , , , and
, , , and
Abstract
:1. Introduction
2. Linking AD Predisposing Factors to Cu and S Metabolism
3. Linking Cu to AD Pathology or Multiple AD-Associated Factors
4. Linking S Metabolism to AD Pathology or Multiple AD-Associated Factors
5. Hypothetical Relationship between Mo and AD Pathology or Multiple AD-Associated Factors
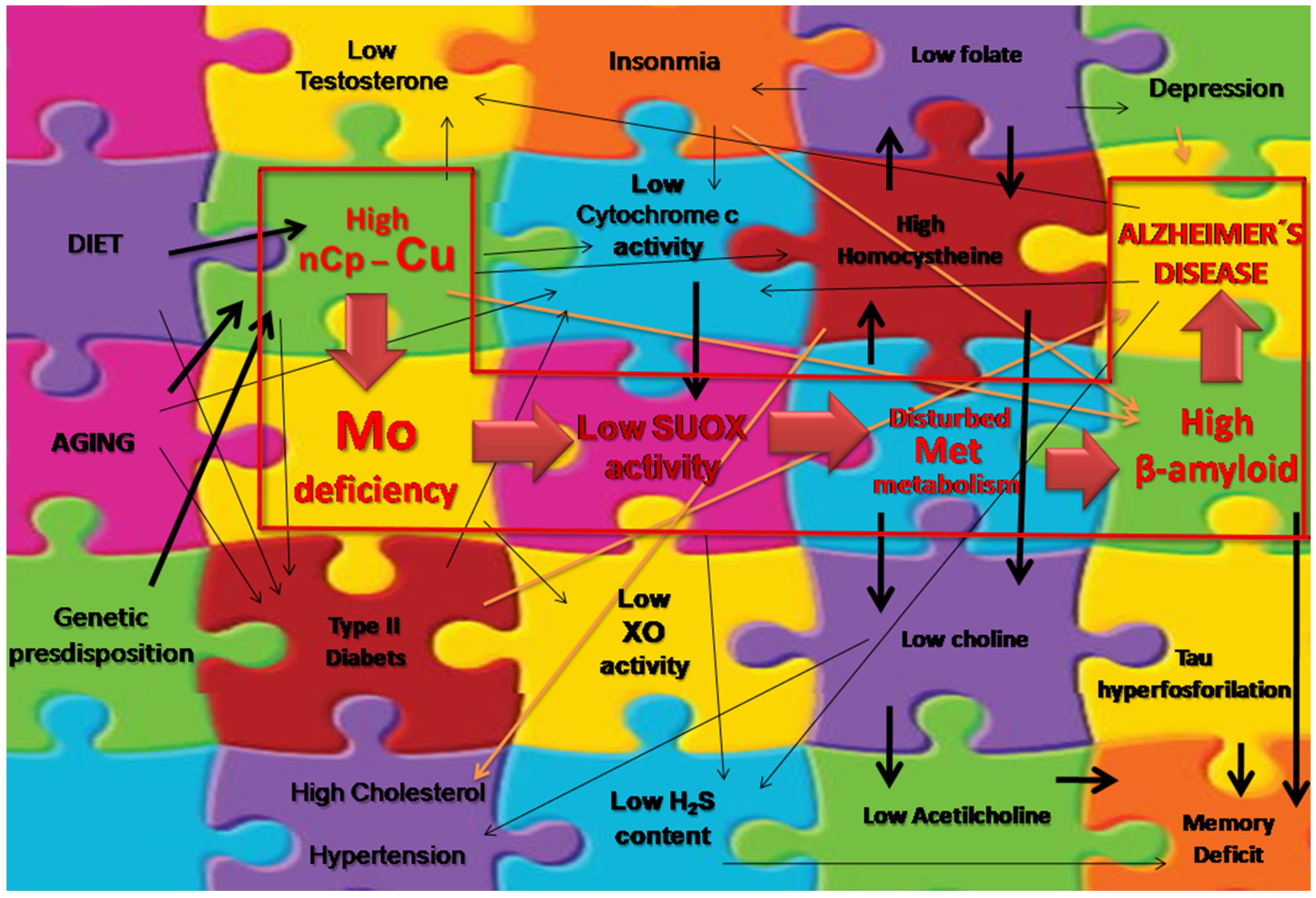
6. Linking Metabolic Interaction of Cu, Mo, and S to AD Pathology or Multiple AD-Associated Factors
7. Cu and Mo Increase in the Blood Vessels and Evidence Sustaining a Decreased Cu and Mo Transport in the Brain with Aging
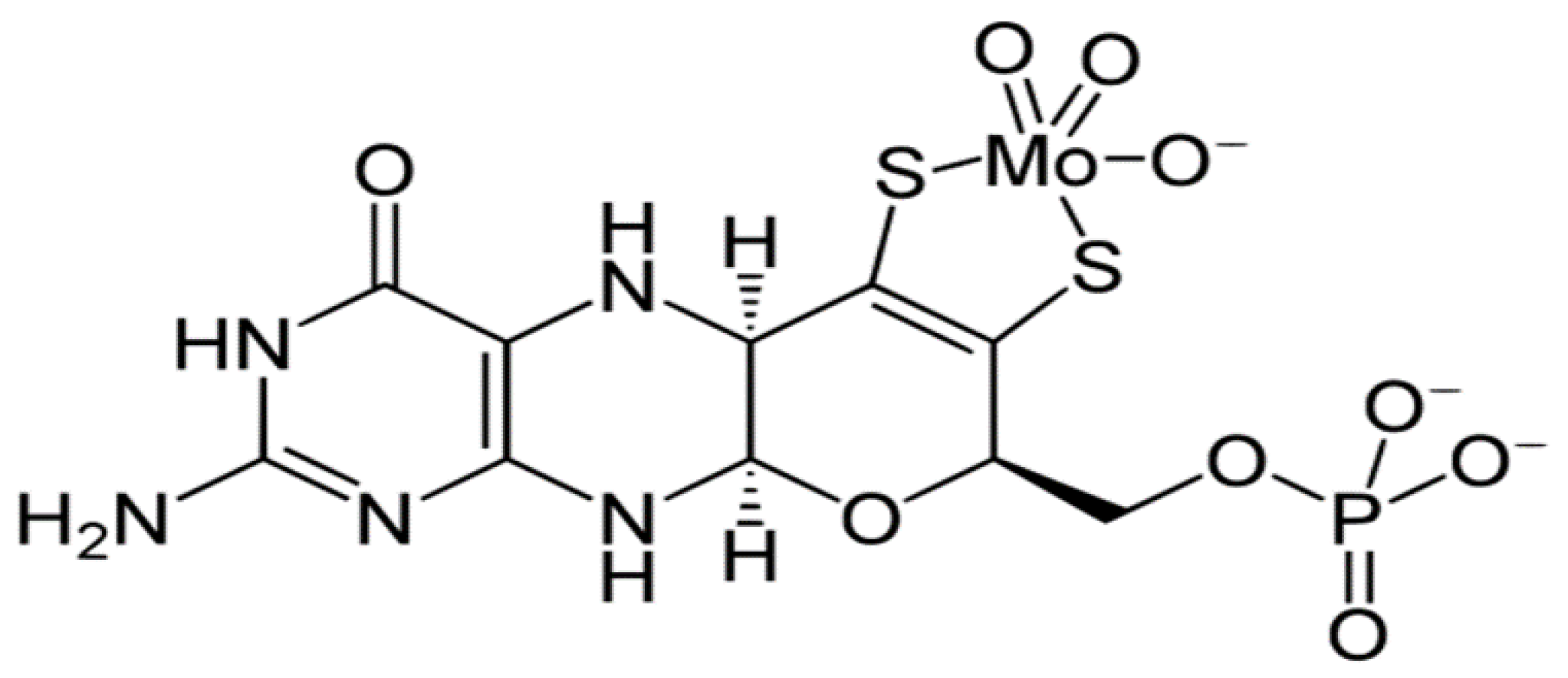
8. Mo Deficiency and Low SUOX and Cytochrome c Activities Are Possibly Linked to AD
9. Clinical Perspective for AD and the “Cu–Mo–S Circuitry”
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- WHO. Risk Reduction of Cognitive Decline and Dementia: WHO Guidelines; Licence: CC BY-NC-SA 3.0 IGO; World Health Organization: Geneva, Switzerland, 2019; ISBN 978-92-4-155054-3. [Google Scholar]
- Socha, K.; Klimiuk, K.; Naliwajko, S.K.; Soroczyńska, J.; Puścion-Jakubik, A.; Markiewicz-Żukowska, R.; Kochanowicz, J. Dietary Habits, Selenium, Copper, Zinc and Total Antioxidant Status in Serum in Relation to Cognitive Functions of Patients with Alzheimer’s Disease. Nutrients 2021, 13, 287. [Google Scholar] [CrossRef]
- Stocker, H.; Nabers, A.; Perna, L.; Möllers, T.; Rujescu, D.; Hartmann, A.; Holleczek, B.; Schöttker, B.; Stockmann, J.; Gerwert, K.; et al. Genetic predisposition, Aβ misfolding in blood plasma, and Alzheimer’s disease. Transl. Psychiatry 2021, 11, 261. [Google Scholar] [CrossRef]
- Lalioti, V.; Muruais, G.; Tsuchiya, Y.; Pulido, D.; Sandoval, I.V. Molecular mechanisms of copper homeostasis. Front. Biosci. 2009, 14, 4878–4903. [Google Scholar] [CrossRef] [Green Version]
- Palego, L.; Betti, L.; Giannaccini, G. Sulfur Metabolism and Sulfur-Containing Amino Acids: I-Molecular Effectors. Biochem. Pharmacol. 2015, 4, 1. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Trace Elements in Human Nutrition and Health; Prepared in Collaboration with the FAO of the UN and the IAEA; World Health Organization: Geneva, Switzerland, 1996. [Google Scholar]
- Heafield, M.T.; Fearn, S.; Steventon, G.B.; Waring, R.H.; Williams, A.C. Plasma cysteine and sulphate levels in patients with motor neurone, Parkinson’s and Alzheimer’s disease. Neurosci. Lett. 1990, 110, 216–220. [Google Scholar] [CrossRef]
- Squitti, R.; Faller, P.; Hureau, C.; Granzotto, A.; White, A.R.; Kepp, K.P. Copper Imbalance in Alzheimer’s Disease and Its Link with the Amyloid Hypothesis: Towards a Combined Clinical, Chemical, and Genetic Etiology. J. Alzheimers Dis. 2021, 83, 23–41. [Google Scholar] [CrossRef]
- Irreverre, F.; Mudd, H.; Heizer, W.D.; Laster, L. Sulfite oxidase deficiency: Studies of a patient with mental retardation, dislocated ocular lenses, and abnormal urinary excretion of S-sulfo-l-cysteine, sulfite, and thiosulfate. Biochem. Med. 1967, 1, 187–217. [Google Scholar] [CrossRef]
- Brown, O.K.; Scholem, R.D.; Croll, H.B.; Wraith, J.E.; McGill, J.J. Sulfite oxidase deficiency. Clinical, neuroradiologic, and biochemical features in two new patients. Neurology 1989, 39, 252. [Google Scholar] [CrossRef]
- Bianchi, V.E. Impact of Testosterone on Alzheimer’s Disease. World J. Mens Health 2022, 40, 243–256. [Google Scholar] [CrossRef]
- Ingenbleek, Y. Implications of protein malnutrition and inflammatory disorders in the pathophysiology of Alzheimer’s disease. Asia Pac. J. Clin. Nutr. 2020, 29, 450–461. [Google Scholar] [CrossRef]
- Li, S.; Sun, W.; Zhang, D. Association of Zinc, Iron, Copper, and Selenium Intakes with Low Cognitive Performance in Older Adults: A Cross-Sectional Study from National Health and Nutrition Examination Survey (NHANES). J. Alzheimers Dis. 2019, 72, 1145–1157. [Google Scholar] [CrossRef]
- Gouaref, I.; Bellahsene, Z.; Zekri, S.; Alamir, B.; Koceir, E.A. The link between trace elements and metabolic syndrome/oxidative stress in essential hypertension with or without type 2 diabetes. Ann. Biol. Clin. 2016, 74, 233–243. [Google Scholar] [CrossRef]
- Squitti, R.; Mendez, A.J.; Simonelli, I.; Ricordi, C. Diabetes and Alzheimer’s Disease: Can Elevated Free Copper Predict the Risk of the Disease? J. Alzheimers Dis. 2017, 56, 1055–1064. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.H.; Lu, F.; Chen, S.D.; Zhou, Z.H.; Han, Y.Z.; Hu, J.Y. Effect of electroacupuncture on serum copper, zinc, calcium and magnesium levels in the depression rats. J. Tradit. Chin. Med. 2011, 31, 112–114. [Google Scholar] [CrossRef] [Green Version]
- Salustri, C.; Squitti, R.; Zappasodi, F.; Ventriglia, M.; Bevacqua, M.G.; Fontana, M.; Tecchio, F. Oxidative stress and brain glutamate-mediated excitability in depressed patients. J. Affect. Disord. 2010, 127, 321–325. [Google Scholar] [CrossRef]
- Ni, M.; You, Y.; Chen, J.; Zhang, L. Copper in depressive disorder: A systematic review and meta-analysis of observational studies. Psychiatry Res. 2018, 267, 506–515. [Google Scholar] [CrossRef]
- Xu, J.; He, K.; Zhang, K.; Yang, C.; Nie, L.; Dan, D.; Liu, J.; Zhang, C.; Yang, X. Low-Dose Copper Exposure Exacerbates Depression-Like Behavior in ApoE4 Transgenic Mice. Oxidative Med. Cell. Longev. 2021, 25, 6634181. [Google Scholar] [CrossRef]
- Chatopadhyay, A.; Sarkar, M.; Biswas, N.M. Dose-dependent effect of copper chloride on malereproductive function in immature rats. Kathmandu Univ. Med. J. 2005, 3, 392–400. [Google Scholar]
- Chang, C.S.; Choi, J.B.; Kim, H.J.; Park, S.B. Correlation between serum testosterone level and concentrations of copper and zinc in hair tissue. Biol. Trace Elem. Res. 2011, 144, 264–271. [Google Scholar] [CrossRef]
- Rosario, E.R.; Stanczyk, F.Z.; Pike, C.J. Age-Related Testosterone Depletion and the Development of Alzheimer Disease. JAMA J. Am. Med. Assoc. 2004, 292, 1431–1432. [Google Scholar] [CrossRef]
- Lewis, R.C.; Meeker, J.D. Biomarkers of Exposure to Molybdenum and Other Metals in Relation to Testosterone among Men from NHANES 2011–2012. Fertil. Steril. 2015, 103, 172–178. [Google Scholar] [CrossRef] [Green Version]
- Khosravi, M.; Sotoudeh, G.; Amini, M.; Raisi, F.; Mansoori, A.; Hosseinzadeh, M. The relationship between dietary patterns and depression mediated by serum levels of Folate and vitamin B12. Psychiatry 2020, 20, 63. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, P.; Singh, N. Homocysteine excess: Delineating the possible mechanism of neurotoxicity and depression. Fundam. Clin. Pharmacol. 2015, 29, 522–528. [Google Scholar] [CrossRef]
- Namekawa, Y.; Baba, H.; Maeshima, H.; Nakano, Y.; Satomur, E.; Takebayashi, N.; Nomoto, H.; Suzuki, T.; Arai, H. Heterogeneity of elderly depression: Increased risk of Alzheimer’s disease and Aβ protein metabolism. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2013, 43, 203–208. [Google Scholar] [CrossRef]
- WHO. Evaluations of the Joint FAO/WHO Expert Committee on Food Additives (JECFA). Copper. CAS Number: 7440-50-8. Tox Monograph: FAS 17-JECFA 26/265. 1982. Available online: http://www.inchem.org/documents/jecfa/jecmono/v17je31.htm (accessed on 12 January 2020).
- Pohanka, M. Copper and copper nanoparticles toxicity and their impact on basic functions in the body. Bratisl. Med. J. 2019, 120, 397–409. [Google Scholar] [CrossRef] [Green Version]
- Di Donato, M.; Sarkar, B. Copper transport and its alterations in Menkes and Wilson diseases. Biochim. Biophys. Acta 1997, 1360, 3–16. [Google Scholar] [CrossRef] [Green Version]
- Flores, C.R.; Puga, M.P.; Wrobel, K.; Sevilla, M.E.G.; Wrobel, K. Trace elements status in diabetes mellitus type 2: Possible role of the interaction between molybdenum and copper in the progress of typical complications. Diabetes Res. Clin. Pract. 2011, 91, 333–341. [Google Scholar] [CrossRef]
- Institute of Medicine—US. Panel on Micronutrients. Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc; National Academy Press: Washington, DC, USA, 2002; 773p. [Google Scholar]
- Massie, H.R.; Aiello, V.R.; Iodice, A.A. Changes with age in copper and superoxide dismutase levels in brains of C57BL/6J mice. Mech. Ageing Dev. 1979, 10, 93–99. [Google Scholar] [CrossRef]
- Zatta, P.; Drago, D.; Zambenedetti, P.; Bolognin, S.; Nogara, E.; Peruffo, A.; Cozzi, B. Accumulation of copper and other metal ions, and metallothionein I/II expression in the bovine brain as a function of aging. J. Chem. Neuroanat. 2008, 36, 1–5. [Google Scholar] [CrossRef]
- Vasudevaraju, P.; Bharathi; Jyothsna, T.; Shamasundar, N.M.; Rao, K.S.; Balaraj, B.M.; Rao, K.S.J.; Sathyanarayana, R.T.S. New evidence on iron, copper accumulation and zinc depletion and its correlation with DNA integrity in aging human brain regions. Indian J. Psychiatry 2010, 52, 140–144. [Google Scholar] [CrossRef]
- Kepp, K.P.; Squitti, R. Copper imbalance in Alzheimer’s disease: Convergence of the chemistry and the clinic. Coord. Chem. Rev. 2019, 397, 168–187. [Google Scholar] [CrossRef]
- Squitti, R.; Ventriglia, M.; Simonelli, I.; Bonvicini, C.; Costa, A.; Perini, G.; Binetti, G.; Benussi, L.; Ghidoni, R.; Koch, G.; et al. Copper Imbalance in Alzheimer’s Disease: Meta-Analysis of Serum, Plasma, and Brain Specimens, and Replication Study Evaluating ATP7B Gene Variants. Biomolecules 2021, 11, 960. [Google Scholar] [CrossRef]
- Coelho, F.C.; Squitti, R.; Ventriglia, M.; Cerchiaro, G.; Daher, J.P.; Rocha, J.G.; Rongioletti, M.C.A.; Moonen, A.C. Agricultural Use of Copper and Its Link to Alzheimer’s Disease. Biomolecules 2020, 10, 897. [Google Scholar] [CrossRef]
- Masaldana, S.; Sharnel, A.S.; Clatworthya, C.G.; Smithd, Z.M.; Francis, P.S.; Denoyera, D.; Meggyesya, P.M.; Fontainea, S.L.; Cater, M.A. Copper accumulation in senescent cells: Interplay between copper transporters and impaired autophagy. Redox Biol. 2018, 16, 322–333. [Google Scholar] [CrossRef]
- Boilan, E.; Winant, V.; Dumortier, E.; Piret, J.P.; Bonfitto, F.; Osiewacz, H.D.; Debacq-Chainiaux, F.; Toussaint, O. Role of p38MAPK and oxidative stress in copper-induced senescence. Age 2013, 35, 2255–2271. [Google Scholar] [CrossRef] [Green Version]
- Gaetke, L.M.; Chow, C.K. Copper toxicity, oxidative, and antioxidant nutrients. Toxicology 2003, 189, 147–163. [Google Scholar] [CrossRef]
- Brewer, G.J. Copper-2 Hypothesis for Causation of the Current Alzheimer’s Disease Epidemic Together with Dietary Changes That Enhance the Epidemic. Chem. Res. Toxicol. 2017, 30, 763–768. [Google Scholar] [CrossRef]
- Squitti, R.; Ghidoni, R.; Simonelli, I.; Ivanova, I.D.; Colabufo, N.A.; Zuin, M.; Benussi, L.; Binetti, G.; Cassetta, E.; Rongioletti, M.; et al. Copper dyshomeostasis in Wilson disease and Alzheimer’s disease as shown by serum and urine copper indicators. J. Trace Elem. Med. Biol. 2018, 45, 181–188. [Google Scholar] [CrossRef]
- Hsu, H.; Rodriguez-Ortiz, C.J.; Lam Lim, S.; Zumkehr, J.; Kilian, J.G.; Vidal, J.; Kitazawa, M. Copper-Induced Upregulation of MicroRNAs Directs the Suppression of Endothelial LRP1 in Alzheimer’s Disease Model. Toxicol. Sci. 2019, 170, 144–156. [Google Scholar] [CrossRef]
- Esmieu, C.; Guettas, D.; Conte-Daban, A.; Sabater, L.; Faller, P.; Hureau, C. Copper-Targeting Approaches in Alzheimer’s Disease: How to Improve the Fallouts Obtained from In Vitro Studies. Inorg. Chem. 2019, 58, 13509–13527. [Google Scholar] [CrossRef] [Green Version]
- Hsu, H.-W.; Stephen, C.B.; Masashi, K. Environmental and Dietary Exposure to Copper and Its Cellular Mechanisms Linking to Alzheimer’s Disease. Toxicol. Sci. 2018, 163, 338–345. [Google Scholar] [CrossRef]
- James, S.A.; Volitakis, I.; Adlard, P.A.; Duce, J.A.; Masters, C.L.; Cherny, R.A.; Bush, A.I. Elevated labile Cu is associated with oxidative pathology in Alzheimer’s Disease. Free Radic. Biol. Med. 2012, 52, 298–302. [Google Scholar] [CrossRef]
- Pérez-Aguilar, F.; Burguera, J.A.; Benlloch, S.; Berenguer, M.; Rayón, J.M. Aceruloplasminemia in an asymptomatic patient with a new mutation. Diagn. Fam. Genet. Anal. 2005, 42, 947–949. [Google Scholar] [CrossRef]
- Squitti, R.; Siotto, M.; Arciello, M.; Rossi, L. Non-ceruloplasmin bound copper and ATP7B gene variants in Alzheimer’s disease. Metallomics 2016, 8, 863–873. [Google Scholar] [CrossRef]
- Bagheri, S.; Squitti, R.; Haertlé, T.; Siotto, M.C.; Saboury, A.A. Role of Copper in the Onset of Alzheimer’s Disease Compared to Other Metals. Front. Aging Neurosci. 2018, 9, 446. [Google Scholar] [CrossRef]
- Chen, C.; Jiang, X.; Li, Y.; Yu, H.; Li, S.; Zhang, Z.; Xu, H.; Yang, Y.; Liu, G.; Zhu, F.; et al. Low-dose oral copper treatment changes the hippocampal phosphoproteomic profile and perturbs mitochondrial function in a mouse model of Alzheimer’s disease. Free Radic. Biol. Med. 2019, 135, 144–156. [Google Scholar] [CrossRef]
- Zhao, J.; Shi, Q.; Tian, H.; Li, Y.; Liu, Y.; Xu, Z.; Robert, A.; Liu, Q.; Meunier, B. TDMQ20, a Specific Copper Chelator, Reduces Memory Impairments in Alzheimer’s Disease Mouse Models. ACS Chem. Neurosci. 2021, 12, 140–149. [Google Scholar] [CrossRef]
- Varadarajan, S.; Yatin, S.; Kanski, J.; Jahanshahi, F.; Butterfield, D.A. Methionine residue 35 is important in amyloid b peptide-associated free radical oxidative stress. Brain Res. Bull. 1999, 50, 133–141. [Google Scholar] [CrossRef]
- Weng, W.; Huang, W.; Tang, H.; Cheng, M.; Chen, K. The Differences of Serum Metabolites between Patients with Early-Stage Alzheimer’s Disease and Mild Cognitive Impairment. Front. Neurol. 2019, 10, 1223. [Google Scholar] [CrossRef]
- Kruman, I.I.; Kumaravel, T.S.; Lohani, A.; Pedersen, W.A.; Cutler, R.G.; Kruman, Y.; Haughey, N.; Lee, J.; Evans, M.; Mattson, M.P. Folic Acid Deficiency and Homocysteine Impair DNA Repair in Hippocampal Neurons and Sensitize Them to Amyloid Toxicity in Experimental Models of Alzheimer’s Disease. J. Neurosci. 2002, 22, 1752–1762. [Google Scholar] [CrossRef] [Green Version]
- Leermakers, E.T.M.; Moreira, E.M.; Kiefte-de Jong, J.C.; Darweesh, S.K.L.; Visser, T.; Voortman, T.; Bautista, P.K.; Chowdhury, R.; Gorman, D.; Bramer, W.M.; et al. Effects of choline on health across the life course: Asystematic review. Nutr. Rev. 2015, 73, 500–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moss, M.; Waring, R.H. The Plasma Cysteine/Sulphate Ratio: A Possible Clinical Biomarker. J. Nutr. Environ. Med. 2003, 13, 215–229. [Google Scholar] [CrossRef]
- Luo, Y.; Zhang, L.; Liu, W.; Yu, Y.; Tian, Y. A Single Biosensor for Evaluating the Levels of Copper Ion and L-Cysteine in a Live Rat Brain with Alzheimer’s Disease. Angew. Chem. Int. Ed. 2015, 54, 14053–14056. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Saharan, S.; Tripathi, M.; Murari, G. Brain Glutathione Levels—A Novel Biomarker for Mild Cognitive Impairment and Alzheimer’s Disease. Biol. Psychiatry 2015, 79, 71. [Google Scholar] [CrossRef]
- Quadri, P.; Fragiacomo, C.; Pezzati, R.; Zanda, E.; Forloni, G.; Tettamanti, M.; Lucca, U. Homocysteine, folate, and vitamin B-12 in mild cognitive impairment, Alzheimer disease, and vascular dementia. Am. J. Clin. Nutr. 2004, 80, 114–122. [Google Scholar] [CrossRef]
- Babiloni, C.; Bosco, P.; Ghidoni, R.; Del Percio, C.; Squitti, R.; Binetti, G.; Benussi, L.; Ferri, R.; Frisoni, G.; Lanuzza, B.; et al. Homocysteine and electroencephalographic rhythms in Alzheimer disease: A multicentric study. Neuroscience 2007, 145, 942–954. [Google Scholar] [CrossRef]
- Gorgone, G.; Ursini, F.; Altamura, C.; Bressi, F.; Tombini, M.; Curcio, G.; Chiovenda, P.; Squitti, R.; Silvestrini, M.; Ientile, R.; et al. Hyperhomocysteinemia, intima-media thickness and C677T MTHFR gene polymorphism: A correlation study in patients with cognitive impairment. Atherosclerosis 2009, 206, 309–313. [Google Scholar] [CrossRef]
- Ma, F.; Wu, T.; Zhao, J.; Ji, L.; Song, A.; Zhang, M.; Huang, G. Plasma Homocysteine and Serum Folate and Vitamin B12 Levels in Mild Cognitive Impairment and Alzheimer’s Disease: A Case-Control Study. Nutrients 2017, 9, 725. [Google Scholar] [CrossRef] [Green Version]
- Pietrzik, K.; Brönstrup, A. Vitamins B12, B6 and folate as determinants of homocysteine concentration in the healthy population. Eur. J. Pediatr. 1998, 157 (Suppl. 2), S135–S138. [Google Scholar] [CrossRef]
- Irizarry, M.C.; Gurol, M.E.; Raju, S.; Diaz-Arrastia, R.; Locascio, J.J.; Tennis, M.; Hyman, B.T.; Growdon, J.H.; Greenberg, S.M.; Bottiglieri, T. Association of homocysteine with plasma amyloid protein in aging and neurodegenerative disease. Neurology 2005, 65, 1402–1408. [Google Scholar] [CrossRef]
- Tinelli, C.; Di Pino, A.; Ficulle, E.; Marcelli, S.; Feligioni, M. Hyperhomocysteinemia as a Risk Factor and Potential Nutraceutical Target for Certain Pathologies. Front. Nutr. 2019, 6, 49. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.; Grabowski, P.; Rehman, I. Alzheimer’s disease pathogenesis: Is there a role for folate? Mech. Ageing Dev. 2018, 174, 86–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellott, T.J.; Huleatt, O.M.; Shade, B.N.; Pender, S.M.; Liu, Y.B.; Slack, B.E.; Blusztajn, J.K. Perinatal Choline Supplementation Reduces Amyloidosis and Increases Choline Acetyltransferase Expression in the Hippocampus of the APPswePS1dE9 Alzheimer’s Disease Model Mice. PLoS ONE 2017, 12, e0170450. [Google Scholar] [CrossRef] [Green Version]
- Wilde, M.C.; Vellas, B.; Giraulta, E.; Yavuza, A.C.; Sijbena, J.W. Lower brain and blood nutrient status in Alzheimer’s disease: Results from meta-analyses. Alzheimers Dement. 2017, 3, 416–431. [Google Scholar] [CrossRef]
- Caballero, F.; Fernandez, A.; Matias, N.; Martínez, L.; Fucho, R.; Elena, M.; Caballeria, J.; Morales, A.; Fernández-Checa, J.C.; García-Ruiz, C. Specific contribution of methionine and choline in nutritional nonalcoholic steatohepatitis: Impact on mitochondrial S-adenosyl-L-methionine and glutathione. J. Biol. Chem. 2010, 285, 18528–18536. [Google Scholar] [CrossRef] [Green Version]
- Novotny, J.A. Molybdenum Nutriture in Humans. J. Evid.-Based Complement. Altern. Med. 2011, 16, 164–168. [Google Scholar] [CrossRef]
- Richards, J.M.; Swislocki, N.I. Activation of Adenylate Cyclase by Molybdate. J. Biol. Chem. 1979, 254, 6857–6860. Available online: https://www.jbc.org/article/S0021-925850251-9/pdf (accessed on 20 November 2020). [CrossRef]
- Richards, J.M.; Swislocki, N.I. Mechanism of molybdate activation of adenylate cyclase. Biochim. Biophys. Acta 1981, 678, 180–186. [Google Scholar] [CrossRef]
- Sim, A.T.R.; White, M.D.; Denborough, M.A. Effects of adenylate cyclase activators on porcine skeletal muscle in malignant hyperpyrexia. Br. J. Anaesth. 1987, 59, 1557–1562. [Google Scholar] [CrossRef]
- Rajagopalan, K.V. Molybdenum: An essential trace element in human nutrition. Ann. Rev. Nutr. 1988, 8, 401–427. [Google Scholar] [CrossRef]
- Coelho, F.C.; Vieira, C.; Mosquim, P.R.; Cassini, S.T.A. Nitrogênio e molibdênionasculturas do milho e do feijãoemmonocultivos e emconsórcios: Efeitossobre o feijão. Rev. Ceres 1998, 45, 393–407. [Google Scholar]
- Khadanand, K.; Kirk, M.L. Mo and W Cofactors and the Reactions they Catalyze. Met. Ions Life Sci. 2020, 23, 20. [Google Scholar] [CrossRef]
- Schwarz, G.; Mendel, R.R. Molybdenum co-factor biosynthesis and molybdenum enzymes. Annu. Rev. Plant Biol. 2006, 57, 623–647. [Google Scholar] [CrossRef]
- Scientific Committee on Food, European Commission. Opinion of the Scientific Committee on Food on the Tolerable Upper Intake Level of Molybdenum; SCF/SC/NUT/UPPERLEV/22; Scientific Committee on Food, European Commission: Brussels, Belgium, 2000; pp. 1–15. [Google Scholar]
- Baj, J.; Forma, A.; Sitarz, E.; Karakuła, K.; Flieger, W.; Sitarz, M.; Grochowski, C.; Maciejewski, R.; Karakula-Juchnowicz, H. Beyond the Mind-Serum Trace Element Levels in Schizophrenic Patients: A Systematic Review. Int. J. Mol. Sci. 2020, 21, 9566. [Google Scholar] [CrossRef]
- Giussani, A.; Arogunjo, A.M.; Claire Cantone, M.; Tavola, F.; Veronese, I. Rates of intestinal absorption of molybdenum in humans. Appl. Radiat. Isot. 2006, 64, 639–644. [Google Scholar] [CrossRef]
- Johnson, L.E. Molybdenum Deficiency. Merck Manual Professional Version. Available online: https://www.merckmanuals.com/professional/nutritional-disorders/mineral-deficiency-and-toxicity/molybdenum-deficiency# (accessed on 20 November 2021).
- Ardan, T.; Kovaceva, J.; Cejková, J. Comparative histochemical and immunohistochemical study on xanthine oxidoreductase/xanthine oxidase in mammalian corneal epithelium. Acta Histochem. 2004, 106, 69–75. [Google Scholar] [CrossRef]
- Hille, R. Molybdenum-containing hydroxylases. Arch. Biochem. Biophys. 2005, 433, 107–116. [Google Scholar] [CrossRef]
- Harrison, R. Structure and function of xanthine oxidoreductase: Where are we now? Free Radic. Biol. Med. 2002, 33, 774–797. [Google Scholar] [CrossRef]
- Sebesta, I.; Stiburkova, B.; Krijt, J. Hereditary xanthinuria is not so rare disorder of purine metabolism. Nucleosides Nucleotides Nucleic Acids 2018, 37, 324–328. [Google Scholar] [CrossRef]
- Alonso-Andrés, P.; Albasanz, J.L.; Ferrer, I.; Martín, M. Purine-related metabolites and their converting enzymes are altered in frontal, parietal and temporal cortex at early stages of Alzheimer’s disease pathology. Brain Pathol. 2018, 28, 933–946. [Google Scholar] [CrossRef] [Green Version]
- Du, N.; Xu, D.; Hou, X.; Song, X.; Liu, C.; Chen, Y.; Wang, Y.; Li, X. Inverse Association between Serum Uric Acid Levels and Alzheimer’s Disease Risk. Mol. Neurobiol. 2016, 53, 2594–2599. [Google Scholar] [CrossRef]
- Tana, C.; Ticinesi, A.; Prati, B.; Nouvenne, A.; Meschi, T. Uric Acid and Cognitive Function in Older Individuals. Nutrients 2018, 10, 975. [Google Scholar] [CrossRef] [Green Version]
- Latourte, A.; Bardin, T.; Richette, P. Uric acid and cognitive decline: A double-edge sword? Curr. Opin. Rheumatol. 2018, 30, 183–187. [Google Scholar] [CrossRef]
- Griffith, O.W. Mammalian sulfur amino acid metabolism: An overview. Methods Enzymol. 1987, 143, 366–376. [Google Scholar] [CrossRef]
- Mellis, A.; Misko, A.L.; Arjune, S.; Liang, Y.; Erdélyi, K.; Ditrói, T.; Kaczmarek, A.T.; Nagy, P.; Schwarz, G. The role of glutamate oxaloacetate transaminases in sulfite biosynthesis and H2S metabolism. Redox Biol. 2021, 38, 101800. [Google Scholar] [CrossRef]
- Abe, K.; Kimura, H. The Possible Role of Hydrogen Sulfide as an Endogenous Neuromodulator. J. Neurosci. 1996, 16, 1066–1071. [Google Scholar] [CrossRef] [Green Version]
- Eto, K.; Asada, T.; Arima, K.; Makifuchi, T.; Kimura, H. Brain hydrogen sulfide is severely decreased in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2002, 293, 1485–1488. [Google Scholar] [CrossRef]
- Velayutham, M.; Hemann, C.F.; Cardounel, A.J.; Zweiera, J.L. Sulfite oxidase activity of cytochrome c: Role of hydrogen peroxide. Biochem. Biophys. Rep. 2016, 5, 96–104. [Google Scholar] [CrossRef] [Green Version]
- Bosetti, F.; Brizzi, F.; Barogi, S.; Mancuso, M.; Siciliano, G.; Tendi, E.A.; Murri, L.; Rapoport, S.L.; Solaini, G. Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer’s disease. Neurobiol. Aging 2002, 23, 371–376. [Google Scholar] [CrossRef]
- Maurer, I.; Zierz, S.; Möller, H. A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol. Aging 2000, 21, 455–462. [Google Scholar] [CrossRef]
- Kuper, J.; Llamas, A.; Hecht, H.; Mendel, R.R.; Schwarz, G. Structure of the molybdopterin-bound Cnx1G domain links molybdenum and copper metabolism. Nature 2004, 430, 803–806. [Google Scholar] [CrossRef] [PubMed]
- Alonzo-Martínez, M.C.; Cazorla, E.; Cánovas, E.; Anniuk, K.; Cores, A.E.; Serrano, A.M. Molybdenum Cofactor Deficiency: Mega Cisterna Magna in Two Consecutive Pregnancies and Review of the Literature. Appl. Clin. Genet. 2020, 13, 49–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misko, A.L.; Liang, Y.; Kohl, J.B.; Eichler, F. Delineating the phenotypic spectrum of sulfite oxidase and molybdenum co-factor deficiency. Neurol. Genet. 2020, 6, e486. [Google Scholar] [CrossRef] [PubMed]
- Berry, T.; Abohamza, E.; Moustafa, A.A. A disease-modifying treatment for Alzheimer’s disease: Focus on the trans-sulfuration pathway. Rev. Neurosci. 2020, 31, 319–334. [Google Scholar] [CrossRef]
- Xia, Z.; Storm, D. Role of Circadian Rhythm and REM sleep for memory consolidation. Neurosci. Res. 2017, 118, 13–20. [Google Scholar] [CrossRef]
- Yamamoto, M.; Gotz, M.E.; Ozawa, H.; Luckhaus, C.; Saito, T.; Rosler, M.; Riederer, P. Hippocampal level of neural speci¢c adenylyl cyclase type I is decreased in Alzheimer’s disease. Biochim. Biophys. Acta 2000, 1535, 60–68. [Google Scholar] [CrossRef] [Green Version]
- Kelly, M.P. Cyclic nucleotide signaling changes associated with normal aging and age-related diseases of the brain. Cell. Signal. 2018, 42, 281–291. [Google Scholar] [CrossRef]
- Nonaka, N.; Banks, A.W.; Shioda, S. Pituitary adenylate cyclase-activating polypeptide: Protective effects in stroke and dementia. Peptides 2020, 130, 17033. [Google Scholar] [CrossRef]
- Abumrad, N.N.; Schneider, A.J.; Steel, D.; Rogers, L.S. Amino acid intolerance during prolonged total parenteral nutrition reversed by molybdate therapy. Am. J. Clin. Nutr. 1981, 34, 2551–2559. [Google Scholar] [CrossRef]
- Hajjar, I.; Liu, C.; Jones, D.P.; Uppal, K. Untargeted metabolomics reveal dysregulations in sugar, methionine, and tyrosine pathways in the prodromal state of AD. Alzheimers Dement. 2020, 12, e12064. [Google Scholar] [CrossRef]
- Atwal, P.S.; Scaglia, F. Molybdenum co-factor deficiency. Mol. Genet. Metab. 2016, 117, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhou, X.; Shi, G.; Yu, Y. Molybdenum disulfide nanosheets based fluorescent “off-to-on” probe for targeted monitoring and inhibition of Î2-amyloid oligomers. Analyst 2020, 145, 6369–6377. [Google Scholar] [CrossRef] [PubMed]
- Manna, S.; Karmakar, P.; Kisan, B.; Mishra, M.; Barooah, N.; Bhasikuttan, A.C.; Mohant, J. Fibril-induced neurodegenerative disorders in an Ab-mutant Drosophila model: Therapeutic targeting using ammonium molybdate. Chem. Commun. 2021, 57, 8488. [Google Scholar] [CrossRef]
- Kulwich, R.; Hansard, S.L.; Comar, C.L.; Davis, G.K. Copper, Molybendurn and Zinc Interrelationships in Rats and Swine. Exp. Biol. Med. 1953, 84, 2. [Google Scholar] [CrossRef]
- Halverson, A.W.; Phifer, J.H.; Monty, K.J. A mechanism for the copper-molybdenum interrelationship. J. Nutr. 1960, 71, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Gray, L.F.; Daniel, L.J. Effect of the Copper Status of the Rat on the Copper-Molybdenum-Sulfate Interaction. J. Nutr. 1964, 84, 31–37. [Google Scholar] [CrossRef]
- Suttle, N.F. Copper Imbalances in Ruminants and Humans: Unexpected Common Ground. Adv. Nutr. 2012, 3, 666–674. [Google Scholar] [CrossRef] [Green Version]
- Dowdy, R.P.; Matrone, G. A Copper-Molybdenum Complex: Its effects and movement in the piglet and sheep. J. Nutr. 1968, 95, 197–201. [Google Scholar] [CrossRef]
- Dowdy, R.P.; Kunz, G.A.; Sauberlich, H.E. Effect of a Copper-Molybdenum Compound upon Copper Metabolism in the Rat. J. Nutr. 1969, 99, 491–496. [Google Scholar] [CrossRef] [Green Version]
- Arthur, D. Interrelationships of Molybdenum and Copper in the Diet of the Guinea Pig. J. Nutr. 1965, 87, 69–76. [Google Scholar] [CrossRef]
- Hadizadeh, M.; Keyhani, E.; Keyhani, J.; Khodadadi, C. Functional and structural alterations induced by copper in xanthine oxidase. Acta Biochim. Biophys. Sin. 2009, 41, 603–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendel, R.R.; Kruse, T. Cell biology of molybdenum in plants and humans. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2012, 1823, 1568–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.T.; Karasawa, A.; Yamanaka, K. Binding of copper to albumin and participation of cysteine in vivo and in vitro. Arch. Biochem. Biophys. 1989, 273, 572–577. [Google Scholar] [CrossRef]
- Mahoney, J.P.; Sandberg, A.A.; Gubler, C.J.; Cartwright, G.E.; Wintrobe, M.M. Uric Acid Metabolism in Hepatolenticular Degeneration. Exp. Biol. Med. 1955, 88, 427–430. [Google Scholar] [CrossRef]
- Seelig, M.S. Proposed role of copper—Molybdenum interaction in iron-deficiency and iron-storage diseases. Am. J. Clin. Nutr. 1973, 26, 657–672. [Google Scholar] [CrossRef] [PubMed]
- Bar-Or, D.; Rael, L.; Thomas, G.; Kraus, J. Inhibitory Effect of Copper on Cystathionine β-Synthase Activity: Protective Effect of an Analog of the Human Albumin N-Terminus. Protein Pept. Lett. 2005, 12, 271–273. [Google Scholar] [CrossRef]
- Vasquez, E.F.A.; Herrera, A.P.N.; Santiago, G.S. Interaçãocobre, molibdênio e enxofreemruminantes. Cienc. Rural 2001, 31, 1101–1106. [Google Scholar] [CrossRef]
- Paglia, G.; Miedico, O.; Cristofano, A.; Vitale, M.; Angiolillo, A.; Chiaravalle, A.E.; Corso, G.; Costanzo, A.D. Distinctive Pattern of Serum Elements during the Progression of Alzheimer’s. Dis. Sci. Rep. 2016, 9, 22769. [Google Scholar] [CrossRef]
- Squitti, R.; Pasqualetti, P.; Polimanti, R.; Salustri, C.; Moffa, F.; Cassetta, E.; Lupoi, D.; Ventriglia, M.; Cortesi, M.; Siotto, M.; et al. Metal-score as a potential non-invasive diagnostic test for Alzheimer’s disease. Curr. Alzheimer Res. 2013, 10, 191–198. [Google Scholar] [CrossRef]
- Squitti, R.; Ghidoni, R.; Siotto, M.; Ventriglia, M.; Benussi, L.; Paterlini, A.; Magri, M.; Binetti, G.; Cassetta, E.; Caprara, D.; et al. Value of serum nonceruloplasmin copper for prediction of mild cognitive impairment conversion to Alzheimer disease. Ann. Neurol. 2014, 75, 574–580. [Google Scholar] [CrossRef]
- Smith, B.S.W.; Wright, H. Copper: Molybdenum interaction: Effect of dietary molybdenum on the binding of copper to plasma proteins in sheep. J. Comp. Pathol. 1975, 85, 299–305. [Google Scholar] [CrossRef]
- Post, L.O.; Bailey, E.M., Jr.; Williams, S.; Applegate, C.; Jones, B.; Herrman, T.J. Risk Assessment of Copper and Molybdenum and Other Minerals in Feed Ingredients and Finished Feeds. J. Regul. Sci. 2019, 7, 1–9. [Google Scholar] [CrossRef]
- Badgley, M.A.; Kremer, D.M.; Maurer, H.C.; DelGiorno, K.E.; Lee, H.; Purohit, V.; Sagalovskiy, I.R.; Ma, A.; Kapilian, J.; Firl, C.E.M.; et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 2020, 368, 85–89. [Google Scholar] [CrossRef]
- Paul, B.D. Neuroprotective Roles of the Reverse Transsulfuration Pathway in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 659402. [Google Scholar] [CrossRef]
- Grings, M.; Moura, A.P.; Amaral, A.U.; Parmeggiani, B.; Gasparotto, J.; Moreira, J.C.F.; Gelain, D.P.; Wyse, A.T.S.; Wajner, M.; Leipnitz, G. Sulfite disrupts brain mitochondrial energy homeostasis and induces mitochondrial permeability transition pore opening via thiol group modification. Biochim. Biophys. Acta 2014, 1842, 1413–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, W.; Eichler, F.S.; Hoda, S.; Lee, M.S.; Baris, H.; Hanley, C.A.P.; Grant, E.; Krishnamoorthy, K.S.; Shih, V.E. Isolated Sulfite Oxidase Deficiency: A Case Report with a Novel Mutation and Review of the Literature. Pediatrics 2005, 116, 757. [Google Scholar] [CrossRef]
- Cummings, J.; Ritter, A.; Zhong, K. Clinical Trials for Disease-Modifying Therapies in Alzheimer’s Disease: A Primer, Lessons Learned, and a Blueprint for the Future. J. Alzheimers Dis. 2018, 64, S3–S22. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Yang, Z.; Gu, Y.; Liu, H.; Wang, P. The effectiveness of eye tracking in the diagnosis of cognitive disorders: A systematic review and meta-analysis. PLoS ONE 2021, 16, e0254059. [Google Scholar] [CrossRef]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimers Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Sun, F. Aducanumab: The first targeted Alzheimer’s therapy. Drug Discov. Ther. 2021, 15, 166–168. [Google Scholar] [CrossRef]
- Cummings, J.; Aisen, P.; Apostolova, L.G.; Atri, A.; Salloway, S.; Weiner, M.J. Aducanumab: Appropriate Use Recommendations. Prev. Alzheimers Dis. 2021, 8, 398–410. [Google Scholar] [CrossRef]
- Fillit, H.; Green, A. Aducanumab and the FDA—Where are we now? Nat. Rev. Neurol. 2021, 17, 129–130. [Google Scholar] [CrossRef] [PubMed]
- Rabinovici, G.D. Controversy and Progress in Alzheimer’s Disease—FDA Approval of Aducanumab. N. Engl. J. Med. 2021, 385, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Baldari, S.; Di Rocco, G.; Toietta, G. Current Biomedical Use of Copper Chelation Therapy. Int. J. Mol. Sci. 2020, 21, 1069. [Google Scholar] [CrossRef] [Green Version]
- Montes, S.; Rivera-Mancia, S.; Diaz-Ruiz, A.; Tristan-Lopez, L.; Rios, C. Copper and Copper Proteins in Parkinson’s Disease. Oxidative Med. Cell. Longev. 2014, 2014, 147251. [Google Scholar] [CrossRef] [Green Version]
- Pal, A.; Kumar, A.; Prasad, R. Predictive association of copper metabolism proteins with Alzheimer’s disease and Parkinson’s disease: A preliminary perspective. BioMetals 2014, 27, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Mezzaroba, L.; Alfieri, D.F.; Simão, A.N.C.; Reiche, E.M.V. The role of zinc, copper, manganese and iron in neurodegenerative diseases. Neuro Toxicol. 2019, 74, 230–241. [Google Scholar] [CrossRef]
- Tosato, M.; Marco, V.D. Metal Chelation Therapy and Parkinson’s Disease: A Critical Review on the Thermodynamics of Complex Formation between Relevant Metal Ions and Promising or Established Drugs. Biomolecules 2019, 9, 269. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Zhao, J.; Chen, Y.; Ma, F.; Huang, G.; Li, W. Baseline folic acid status affects the effectiveness of folic acid supplements in cognitively relevant outcomes in older adults: A systematic review. Aging Ment. Health 2021, 26, 457–463. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Lower Activity or Synthesis of XO, AO, SUOX or Moco, or Mo Deficiency | Alzheimer’s Disease | |
|---|---|---|
| Similarities | ||
| Sulfate | Low urinary SO42− content [9,10]. | |
| Methionine | High plasma Met [105]. | |
| Cysteine | Sulfite loading stops the conversion of Met to Cys, and this causes Cys levels to fall [105]. | |
| Uric acid | Serum and urinary uric acid contents are reduced [107]. | |
| Event | Research Results |
|---|---|
| NCp–Cu accumulation in serum or plasma. |
|
| Cu and Mo are antagonists, so excessive Cu cause Mo deficiency and vice versa. |
|
| Products of enzymes’ activity that have Mo as a co-factor are in lower content in AD than in healthy people | |
| Altered S amino acids metabolism occurs in AD |
|
| Mo contents |
| Serum Levels | Brain Tissue Levels | |
|---|---|---|
| Similarities | ||
| Cu | High content of nCp–Cu in serum [36]. | In brain tissues, the Cu content is usually low [36,68]. |
| Evidence suggesting a low level of Mo in AD brain tissues: | ||
| Mo | Serum Mo content increased progressively, passing from healthy subjects (HS) through subjective memory complaint, mild cognitive impairment up to AD, and the difference between HS and AD was statistically significant [124]. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coelho, F.C.; Cerchiaro, G.; Araújo, S.E.S.; Daher, J.P.L.; Cardoso, S.A.; Coelho, G.F.; Guimarães, A.G. Is There a Connection between the Metabolism of Copper, Sulfur, and Molybdenum in Alzheimer’s Disease? New Insights on Disease Etiology. Int. J. Mol. Sci. 2022, 23, 7935. https://doi.org/10.3390/ijms23147935
Coelho FC, Cerchiaro G, Araújo SES, Daher JPL, Cardoso SA, Coelho GF, Guimarães AG. Is There a Connection between the Metabolism of Copper, Sulfur, and Molybdenum in Alzheimer’s Disease? New Insights on Disease Etiology. International Journal of Molecular Sciences. 2022; 23(14):7935. https://doi.org/10.3390/ijms23147935
Chicago/Turabian StyleCoelho, Fábio Cunha, Giselle Cerchiaro, Sheila Espírito Santo Araújo, João Paulo Lima Daher, Silvia Almeida Cardoso, Gustavo Fialho Coelho, and Arthur Giraldi Guimarães. 2022. "Is There a Connection between the Metabolism of Copper, Sulfur, and Molybdenum in Alzheimer’s Disease? New Insights on Disease Etiology" International Journal of Molecular Sciences 23, no. 14: 7935. https://doi.org/10.3390/ijms23147935