
The C-Terminal Acidic Tail Modulates the Anticancer Properties of HMGB1

 , , , ,
, , , ,  ,
,  ,
,
Abstract
:1. Introduction
2. Results
2.1. Production and Characterization of HMGB1-ΔC
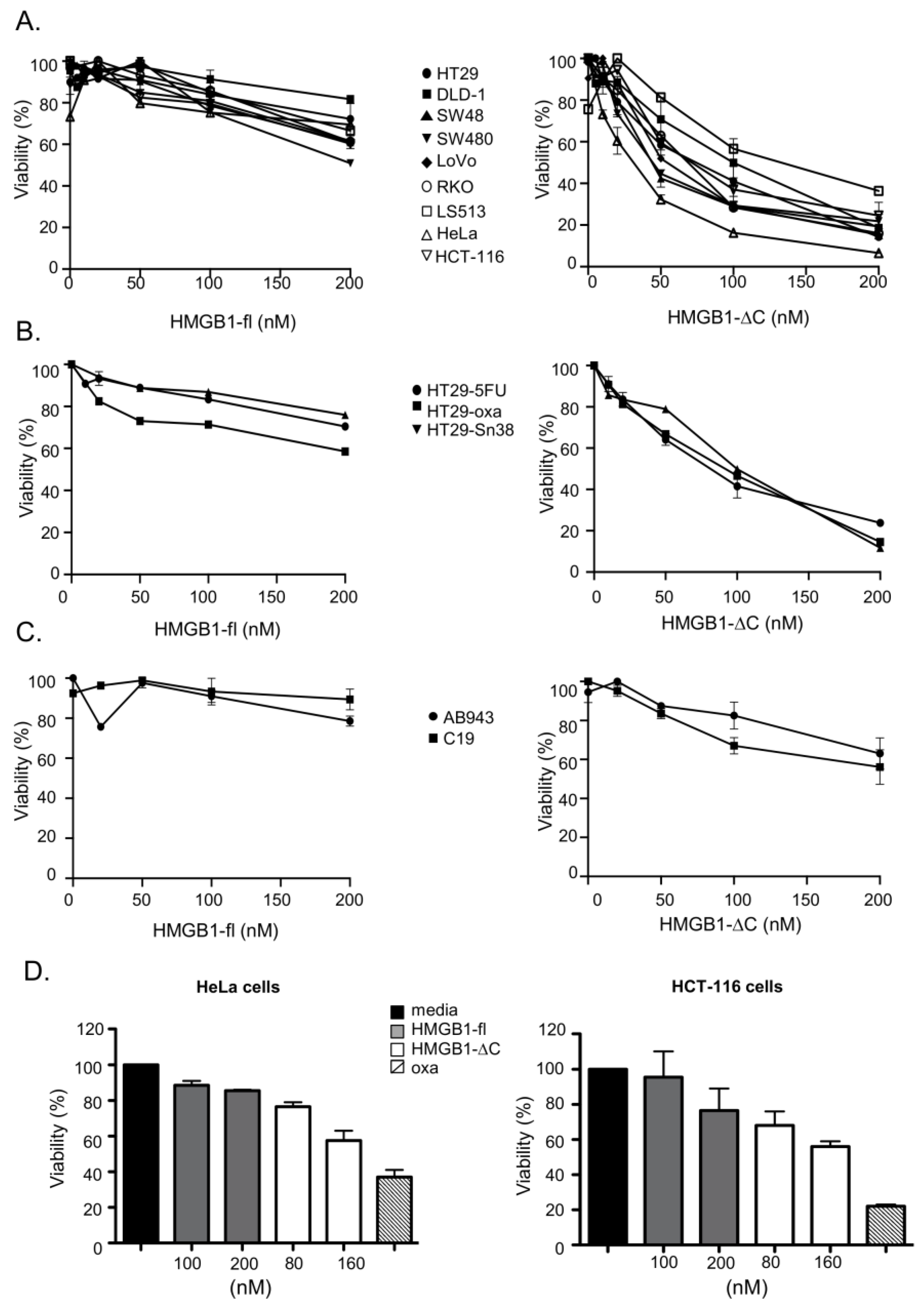
2.2. Comparison of the In Vitro Anticancer Activities of HMGB1-ΔC and HMGB1-fl on a Panel of Cancer Cell Lines in 2D/3D Cell Cultures
2.3. Cytotoxic Versus Cytostatic Activities of HMGB1-fl and HMGB1-ΔC
2.4. In Silico HMGB1-ΔC–PKM2 Docking
2.5. Expression Levels of PKM1/PKM2 mRNA in C19, HeLa, HT-29 and HCT-116 Cells
2.6. Metabolic Effects of HMGB1-fl and HMGB1-ΔC on C19, HeLa, HT-29 and HCT-116 Cells
3. Discussion
4. Materials and Methods
4.1. Cells
4.2. Plasmid Construction
4.3. Protein Expression and Purification
4.4. In-Gel Digestion
4.5. MALDI-TOF Mass Spectrometry Analysis of HMGB1-ΔC
4.6. 2D Cell Viability Assay
4.7. 3D Cell Viability Assay
4.8. Flow Cytometry
4.9. In Silico Protein Docking Studies
4.10. RNA Extraction and RT-qPCR Analysis
4.11. Measurement of Oxygen Consumption Rate (OCR) and Extracellular Acidification Rate (ECAR)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nowell, P.C. The Clonal Evolution of Tumor Cell Populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.; Maley, C.C. Clonal Evolution in Cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Escargueil, A.E.; Prado, S.; Dezaire, A.; Clairambault, J.; Larsen, A.K.; Soares, D.G. Genotype- or Phenotype-Targeting Anticancer Therapies? Lessons from Tumor Evolutionary Biology. Curr. Pharm. Des. 2016, 22, 6625–6644. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Wiese, E.K.; Hitosugi, T. Tyrosine Kinase Signaling in Cancer Metabolism: PKM2 Paradox in the Warburg Effect. Front. Cell Dev. Biol. 2018, 6, 79. [Google Scholar] [CrossRef]
- Eigenbrodt, E.; Basenau, D.; Holthusen, S.; Mazurek, S.; Fischer, G. Quantification of Tumor Type M2 Pyruvate Kinase (Tu M2-PK) in Human Carcinomas. Anticancer Res. 1997, 17, 3153–3156. [Google Scholar]
- Lüftner, D.; Mesterharm, J.; Akrivakis, C.; Geppert, R.; Petrides, P.E.; Wernecke, K.D.; Possinger, K. Tumor Type M2 Pyruvate Kinase Expression in Advanced Breast Cancer. Anticancer Res. 2000, 20, 5077–5082. [Google Scholar] [PubMed]
- Anastasiou, D.; Yu, Y.; Israelsen, W.J.; Jiang, J.-K.; Boxer, M.B.; Hong, B.S.; Tempel, W.; Dimov, S.; Shen, M.; Jha, A.; et al. Pyruvate Kinase M2 Activators Promote Tetramer Formation and Suppress Tumorigenesis. Nat. Chem. Biol. 2012, 8, 839–847. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, J.; Phillips, J.J.; Zheng, S.; Wiencke, J.; Ronen, S.M.; Pieper, R.O. Pyruvate Kinase M2 Expression, but Not Pyruvate Kinase Activity, Is up-Regulated in a Grade-Specific Manner in Human Glioma. PLoS ONE 2013, 8, e57610. [Google Scholar] [CrossRef] [Green Version]
- Desai, S.; Ding, M.; Wang, B.; Lu, Z.; Zhao, Q.; Shaw, K.; Yung, W.K.A.; Weinstein, J.N.; Tan, M.; Yao, J. Tissue-Specific Isoform Switch and DNA Hypomethylation of the Pyruvate Kinase PKM Gene in Human Cancers. Oncotarget 2014, 5, 8202–8210. [Google Scholar] [CrossRef] [Green Version]
- Israelsen, W.J.; Vander Heiden, M.G. Pyruvate Kinase: Function, Regulation and Role in Cancer. Semin. Cell Dev. Biol. 2015, 43, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, G.; Mao, Q.; Xia, W.; Xu, Y.; Wang, J.; Xu, L.; Jiang, F. PKM2 and Cancer: The Function of PKM2 beyond Glycolysis. Oncol. Lett. 2016, 11, 1980–1986. [Google Scholar] [CrossRef] [Green Version]
- Elia, I.; Rossi, M.; Stegen, S.; Broekaert, D.; Doglioni, G.; van Gorsel, M.; Boon, R.; Escalona-Noguero, C.; Torrekens, S.; Verfaillie, C.; et al. Breast Cancer Cells Rely on Environmental Pyruvate to Shape the Metastatic Niche. Nature 2019, 568, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.Y.; Yoon, S.O.; Seol, S.Y.; Hong, S.W.; Kim, J.W.; Choi, S.H.; Cho, J.Y. Overexpression of the M2 Isoform of Pyruvate Kinase Is an Adverse Prognostic Factor for Signet Ring Cell Gastric Cancer. World J. Gastroenterol. 2012, 18, 4037–4043. [Google Scholar] [CrossRef] [PubMed]
- Zhan, C.; Shi, Y.; Lu, C.; Wang, Q. Pyruvate Kinase M2 Is Highly Correlated with the Differentiation and the Prognosis of Esophageal Squamous Cell Cancer. Dis. Esophagus 2013, 26, 746–753. [Google Scholar] [CrossRef]
- Gdynia, G.; Keith, M.; Kopitz, J.; Bergmann, M.; Fassl, A.; Weber, A.N.R.; George, J.; Kees, T.; Zentgraf, H.-W.; Wiestler, O.D.; et al. Danger Signaling Protein HMGB1 Induces a Distinct Form of Cell Death Accompanied by Formation of Giant Mitochondria. Cancer Res. 2010, 70, 8558–8568. [Google Scholar] [CrossRef] [Green Version]
- Gdynia, G.; Sauer, S.W.; Kopitz, J.; Fuchs, D.; Duglova, K.; Ruppert, T.; Miller, M.; Pahl, J.; Cerwenka, A.; Enders, M.; et al. The HMGB1 Protein Induces a Metabolic Type of Tumour Cell Death by Blocking Aerobic Respiration. Nat. Commun. 2016, 7, 10764. [Google Scholar] [CrossRef]
- Kang, R.; Zhang, Q.; Zeh, H.J.; Lotze, M.T.; Tang, D. HMGB1 in Cancer: Good, Bad, or Both? Clin. Cancer Res. 2013, 19, 4046–4057. [Google Scholar] [CrossRef] [Green Version]
- Janko, C.; Filipović, M.; Munoz, L.E.; Schorn, C.; Schett, G.; Ivanović-Burmazović, I.; Herrmann, M. Redox Modulation of HMGB1-Related Signaling. Antioxid. Redox Signal. 2014, 20, 1075–1085. [Google Scholar] [CrossRef] [Green Version]
- Sims, G.P.; Rowe, D.C.; Rietdijk, S.T.; Herbst, R.; Coyle, A.J. HMGB1 and RAGE in Inflammation and Cancer. Annu. Rev. Immunol. 2010, 28, 367–388. [Google Scholar] [CrossRef] [PubMed]
- Diener, K.R.; Al-Dasooqi, N.; Lousberg, E.L.; Hayball, J.D. The Multifunctional Alarmin HMGB1 with Roles in the Pathophysiology of Sepsis and Cancer. Immunol. Cell Biol. 2013, 91, 443–450. [Google Scholar] [CrossRef] [PubMed]
- He, S.-J.; Cheng, J.; Feng, X.; Yu, Y.; Tian, L.; Huang, Q. The Dual Role and Therapeutic Potential of High-Mobility Group Box 1 in Cancer. Available online: https://pubmed.ncbi.nlm.nih.gov/28969092/ (accessed on 2 December 2020).
- Li, J.; Kokkola, R.; Tabibzadeh, S.; Yang, R.; Ochani, M.; Qiang, X.; Harris, H.E.; Czura, C.J.; Wang, H.; Ulloa, L.; et al. Structural Basis for the Proinflammatory Cytokine Activity of High Mobility Group Box 1. Mol. Med. 2003, 9, 37–45. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Billiar, T.R.; Lotze, M.T. A Janus Tale of Two Active High Mobility Group Box 1 (HMGB1) Redox States. Mol. Med. 2012, 18, 1360–1362. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Bianchi, M.E.; Knapp, S. Thermodynamics of HMGB1 Interaction with Duplex DNA. Biochemistry 2001, 40, 10254–10261. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zeng, M.; Wang, W.; Tang, J. The HMGB1 Acidic Tail Regulates HMGB1 DNA Binding Specificity by a Unique Mechanism. Biochem. Biophys. Res. Commun. 2007, 360, 14–19. [Google Scholar] [CrossRef]
- Musumeci, D.; Roviello, G.N.; Montesarchio, D. An Overview on HMGB1 Inhibitors as Potential Therapeutic Agents in HMGB1-Related Pathologies. Pharmacol. Ther. 2014, 141, 347–357. [Google Scholar] [CrossRef]
- Ramstein, J.; Locker, D.; Bianchi, M.E.; Leng, M. Domain-Domain Interactions in High Mobility Group 1 Protein (HMG1). Eur. J. Biochem. 1999, 260, 692–700. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.; Lippard, S.J. Nature of Full-Length HMGB1 Binding to Cisplatin-Modified DNA. Biochemistry 2003, 42, 2664–2671. [Google Scholar] [CrossRef]
- Stott, K.; Watson, M.; Howe, F.S.; Grossmann, J.G.; Thomas, J.O. Tail-Mediated Collapse of HMGB1 Is Dynamic and Occurs via Differential Binding of the Acidic Tail to the A and B Domains. J. Mol. Biol. 2010, 403, 706–722. [Google Scholar] [CrossRef]
- Knapp, S.; Müller, S.; Digilio, G.; Bonaldi, T.; Bianchi, M.E.; Musco, G. The Long Acidic Tail of High Mobility Group Box 1 (HMGB1) Protein Forms an Extended and Flexible Structure That Interacts with Specific Residues within and between the HMG Boxes. Biochemistry 2004, 43, 11992–11997. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, S.; Nishino, H.; Saito, K.; Kimura, K.; Shirakawa, H.; Yoshida, M. Stimulation of Transcription in Cultured Cells by High Mobility Group Protein 1: Essential Role of the Acidic Carboxyl-Terminal Region. Biochemistry 1994, 33, 14690–14695. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Chou, H.; Kawase, T.; Shirakawa, H.; Yoshida, M. Acidic C-Tail of HMGB1 Is Required for Its Target Binding to Nucleosome Linker DNA and Transcription Stimulation. Biochemistry 2004, 43, 9901–9908. [Google Scholar] [CrossRef] [PubMed]
- Bonaldi, T.; Längst, G.; Strohner, R.; Becker, P.B.; Bianchi, M.E. The DNA Chaperone HMGB1 Facilitates ACF/CHRAC-Dependent Nucleosome Sliding. EMBO J. 2002, 21, 6865–6873. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Friggeri, A.; Liu, G.; Abraham, E. The C-Terminal Acidic Tail Is Responsible for the Inhibitory Effects of HMGB1 on Efferocytosis. J. Leukoc. Biol. 2010, 88, 973–979. [Google Scholar] [CrossRef] [Green Version]
- Gong, W.; Li, Y.; Chao, F.; Huang, G.; He, F. Amino Acid Residues 201-205 in C-Terminal Acidic Tail Region Plays a Crucial Role in Antibacterial Activity of HMGB1. J. Biomed. Sci. 2009, 16, 83. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, H.; Mason, J.M.; Levine, J.; Yu, M.; Ulloa, L.; Czura, C.J.; Tracey, K.J.; Yang, H. Recombinant HMGB1 with Cytokine-Stimulating Activity. J. Immunol. Methods 2004, 289, 211–223. [Google Scholar] [CrossRef]
- Thierry, S.; Gozlan, J.; Jaulmes, A.; Boniface, R.; Nasreddine, N.; Strauss, F.; Maréchal, V. High-Mobility Group Box 1 Protein Induces HIV-1 Expression from Persistently Infected Cells. AIDS 2007, 21, 283–292. [Google Scholar] [CrossRef]
- Ayadi, M.; Bouygues, A.; Ouaret, D.; Ferrand, N.; Chouaib, S.; Thiery, J.-P.; Muchardt, C.; Sabbah, M.; Larsen, A.K. Chronic Chemotherapeutic Stress Promotes Evolution of Stemness and WNT/Beta-Catenin Signaling in Colorectal Cancer Cells: Implications for Clinical Use of WNT-Signaling Inhibitors. Oncotarget 2015, 6, 18518–18533. [Google Scholar] [CrossRef] [Green Version]
- Perry, S.W.; Norman, J.P.; Barbieri, J.; Brown, E.B.; Gelbard, H.A. Mitochondrial Membrane Potential Probes and the Proton Gradient: A Practical Usage Guide. BioTechniques 2011, 50, 98–115. [Google Scholar] [CrossRef]
- Feoktistova, M.; Geserich, P.; Leverkus, M. Crystal Violet Assay for Determining Viability of Cultured Cells. Available online: https://pubmed.ncbi.nlm.nih.gov/27037069/ (accessed on 2 December 2020).
- Eastman, A. Improving Anticancer Drug Development Begins with Cell Culture: Misinformation Perpetrated by the Misuse of Cytotoxicity Assays. Oncotarget 2017, 8, 8854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Zheng, Y.; Xia, Y.; Ji, H.; Chen, X.; Guo, F.; Lyssiotis, C.A.; Aldape, K.; Cantley, L.C.; Lu, Z. ERK1/2-Dependent Phosphorylation and Nuclear Translocation of PKM2 Promotes the Warburg Effect. Nat. Cell Biol. 2012, 14, 1295–1304. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Zhao, X.; Huang, L.; Zhang, T.; Yang, F.; Xie, L.; Song, S.; Miao, P.; Zhao, L.; Sun, X.; et al. Proviral Insertion in Murine Lymphomas 2 (PIM2) Oncogene Phosphorylates Pyruvate Kinase M2 (PKM2) and Promotes Glycolysis in Cancer Cells. J. Biol. Chem. 2013, 288, 35406–35416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, L.; Xu, Y.-P.; Zhao, D.; Li, F.-L.; Wang, W.; Sasaki, N.; Jiang, Y.; Zhou, X.; Li, T.-T.; Guan, K.-L.; et al. Mitogenic and Oncogenic Stimulation of K433 Acetylation Promotes PKM2 Protein Kinase Activity and Nuclear Localization. Mol. Cell 2013, 52, 340–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.S.; Kim, D.J.; Koo, H.; Jang, S.H.; You, Y.-M.; Cho, J.H.; Yang, S.-J.; Yu, E.S.; Jung, Y.; Lee, D.C.; et al. AKT-Induced PKM2 Phosphorylation Signals for IGF-1-Stimulated Cancer Cell Growth. Oncotarget 2016, 7, 48155–48167. [Google Scholar] [CrossRef] [Green Version]
- Hitosugi, T.; Kang, S.; Vander Heiden, M.G.; Chung, T.-W.; Elf, S.; Lythgoe, K.; Dong, S.; Lonial, S.; Wang, X.; Chen, G.Z.; et al. Tyrosine Phosphorylation Inhibits PKM2 to Promote the Warburg Effect and Tumor Growth. Sci. Signal 2009, 2, ra73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spadiut, O.; Capone, S.; Krainer, F.; Glieder, A.; Herwig, C. Microbials for the Production of Monoclonal Antibodies and Antibody Fragments. Trends Biotechnol. 2014, 32, 54. [Google Scholar] [CrossRef] [Green Version]
- Venereau, E.; Casalgrandi, M.; Schiraldi, M.; Antoine, D.J.; Cattaneo, A.; De Marchis, F.; Liu, J.; Antonelli, A.; Preti, A.; Raeli, L.; et al. Mutually Exclusive Redox Forms of HMGB1 Promote Cell Recruitment or Proinflammatory Cytokine Release. Available online: https://pubmed.ncbi.nlm.nih.gov/22869893/ (accessed on 2 December 2020).
- Petitprez, A.; Poindessous, V.; Ouaret, D.; Regairaz, M.; Bastian, G.; Guérin, E.; Escargueil, A.E.; Larsen, A.K. Acquired Irinotecan Resistance Is Accompanied by Stable Modifications of Cell Cycle Dynamics Independent of MSI Status. Int. J. Oncol. 2013, 42, 1644–1653. [Google Scholar] [CrossRef] [Green Version]
- Jaulmes, A.; Thierry, S.; Janvier, B.; Raymondjean, M.; Maréchal, V. Activation of SPLA2-IIA and PGE2 Production by High Mobility Group Protein B1 in Vascular Smooth Muscle Cells Sensitized by IL-1beta. FASEB J. 2006, 20, 1727–1729. [Google Scholar] [CrossRef]
- Marchand, C.; Le Maréchal, P.; Meyer, Y.; Miginiac-Maslow, M.; Issakidis-Bourguet, E.; Decottignies, P. New Targets of Arabidopsis Thioredoxins Revealed by Proteomic Analysis. Proteomics 2004, 4, 2696–2706. [Google Scholar] [CrossRef]
- Marchand, C.H.; Fermani, S.; Rossi, J.; Gurrieri, L.; Tedesco, D.; Henri, J.; Sparla, F.; Trost, P.; Lemaire, S.D.; Zaffagnini, M. Structural and Biochemical Insights into the Reactivity of Thioredoxin H1 from Chlamydomonas Reinhardtii. Antioxidants 2019, 8, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, Z.; Borde, C.; Marchand, C.H.; Lemaire, S.D.; Busson, P.; Gozlan, J.-M.; Escargueil, A.; Maréchal, V. Detection of IgG Directed against a Recombinant Form of Epstein-Barr Virus BALF0/1 Protein in Patients with Nasopharyngeal Carcinoma. Protein Expr. Purif. 2019, 162, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Koeppel, F.; Poindessous, V.; Lazar, V.; Raymond, E.; Sarasin, A.; Larsen, A.K. Irofulven Cytotoxicity Depends on Transcription-Coupled Nucleotide Excision Repair and Is Correlated with XPG Expression in Solid Tumor Cells. Available online: https://pubmed.ncbi.nlm.nih.gov/15328203/ (accessed on 2 December 2020).
- Gabdoulline, R.R.; Wade, R.C. Brownian Dynamics Simulation of Protein-Protein Diffusional Encounter. Methods 1998, 14, 329–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 Web Portal for Protein Modeling, Prediction and Analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Sali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Shen, M.-Y.; Sali, A. Statistical Potential for Assessment and Prediction of Protein Structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A Program to Check the Stereochemical Quality of Protein Structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Czodrowski, P.; Li, H.; Nielsen, J.E.; Jensen, J.H.; Klebe, G.; Baker, N.A. PDB2PQR: Expanding and Upgrading Automated Preparation of Biomolecular Structures for Molecular Simulations. Nucleic Acids Res. 2007, 35, W522–W525. [Google Scholar] [CrossRef]
- Homeyer, N.; Horn, A.H.C.; Lanig, H.; Sticht, H. AMBER Force-Field Parameters for Phosphorylated Amino Acids in Different Protonation States: Phosphoserine, Phosphothreonine, Phosphotyrosine, and Phosphohistidine. J. Mol. Model. 2006, 12, 281–289. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of Nanosystems: Application to Microtubules and the Ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Forward (5′->3′) | Reverse (5′->3′) |
|---|---|---|
| Exon 8–9/9 | ATGCAGCACCTGATAGCTCGTGA | TGCCAGACTCCGTCAGAACTATCA |
| Exon 8–10/10 | ATGCAGCACCTGGTCTGCTCACC | CATTCATGGCAAAGTTCACCCGGA |
| Exon 2 | CCATGTCGAAGCCCCATAGT | GCTGGGCCAATGGTACAGAT |
| L19 | ATGTATCACAGCCTGTACCTG | CGTGCTTCCTTGGTCTTAGAC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borde, C.; Dillard, C.; L’Honoré, A.; Quignon, F.; Hamon, M.; Marchand, C.H.; Faccion, R.S.; Costa, M.G.S.; Pramil, E.; Larsen, A.K.; et al. The C-Terminal Acidic Tail Modulates the Anticancer Properties of HMGB1. Int. J. Mol. Sci. 2022, 23, 7865. https://doi.org/10.3390/ijms23147865
Borde C, Dillard C, L’Honoré A, Quignon F, Hamon M, Marchand CH, Faccion RS, Costa MGS, Pramil E, Larsen AK, et al. The C-Terminal Acidic Tail Modulates the Anticancer Properties of HMGB1. International Journal of Molecular Sciences. 2022; 23(14):7865. https://doi.org/10.3390/ijms23147865
Chicago/Turabian StyleBorde, Chloé, Clémentine Dillard, Aurore L’Honoré, Frédérique Quignon, Marion Hamon, Christophe H. Marchand, Roberta Soares Faccion, Maurício G. S. Costa, Elodie Pramil, Annette K. Larsen, and et al. 2022. "The C-Terminal Acidic Tail Modulates the Anticancer Properties of HMGB1" International Journal of Molecular Sciences 23, no. 14: 7865. https://doi.org/10.3390/ijms23147865