
Cell Autophagy in NASH and NASH-Related Hepatocellular Carcinoma

 ,
, {kind=link}
Abstract
:1. Introduction
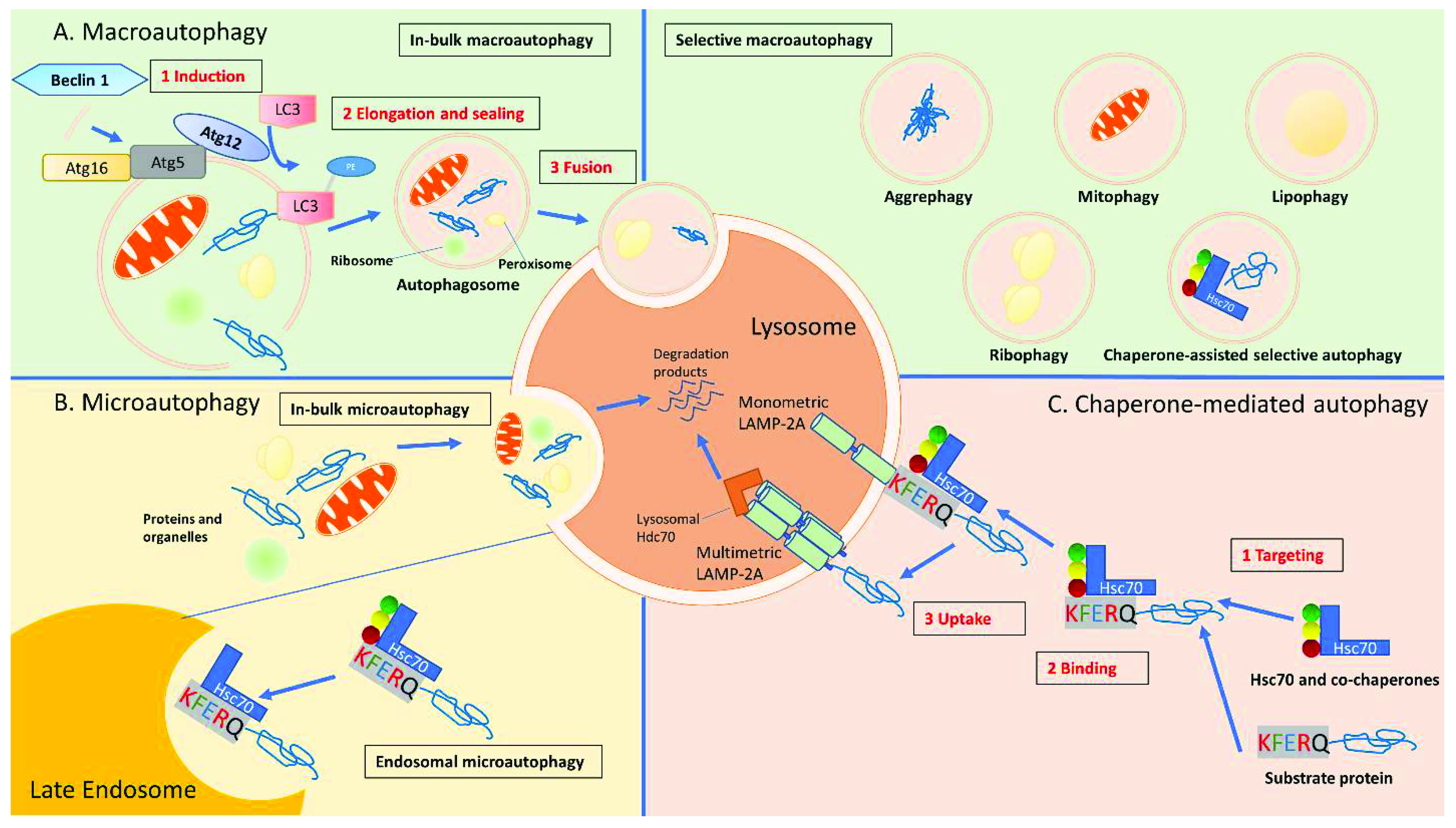
2. Macro-Autophagy (MA)
2.1. Macro-Autophagy (MA) Process
2.2. Regulation of Autophagosome Formation
2.2.1. Initiation
2.2.2. Nucleation
2.2.3. Elongation and Maturation
3. Micro-Autophagy (mA)
3.1. mA Process
3.2. Regulation of mA Activity
3.3. Types of mA
4. Chaperone-Mediated Autophagy (CMA)
4.1. CMA Process
4.2. CMA Activity
4.3. CMA Activity in Health and Disease
5. Autophagy in Liver Physiology
6. Autophagy in NASH and NASH-Related HCC
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, D.E.; Brunt, E.M.; Wilson, L.A.; Behling, C.; Guy, C.; Contos, M.; Cummings, O.; Yeh, M.; Gill, R.; Chalasani, N.; et al. Association of Histologic Disease Activity with Progression of Nonalcoholic Fatty Liver Disease. JAMA Netw. Open 2019, 2, e1912565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 2018, 67, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Bellentani, S.; Scaglioni, F.; Marino, M.; Bedogni, G. Epidemiology of non-alcoholic fatty liver disease. Dig. Dis. 2010, 28, 155–161. [Google Scholar] [CrossRef]
- Ludwig, J.; Viggiano, T.R.; McGill, D.B.; Oh, B.J. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin. Proc. 1980, 55, 434–438. [Google Scholar]
- Ascha, M.S.; Hanouneh, I.A.; Lopez, R.; Tamimi, T.A.; Feldstein, A.F.; Zein, N.N. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology 2010, 51, 1972–1978. [Google Scholar] [CrossRef]
- Said, A.; Ghufran, A. Epidemic of non-alcoholic fatty liver disease and hepatocellular carcinoma. World J. Clin. Oncol. 2017, 8, 429–436. [Google Scholar] [CrossRef]
- Kutlu, O.; Kaleli, H.N.; Ozer, E. Molecular Pathogenesis of Nonalcoholic Steatohepatitis- (NASH-) Related Hepatocellular Carcinoma. Can. J. Gastroenterol. Hepatol. 2018, 2018, 8543763. [Google Scholar] [CrossRef] [Green Version]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Obesity and nonalcoholic fatty liver disease: From pathophysiology to therapeutics. Metabolism 2019, 92, 82–97. [Google Scholar] [CrossRef]
- Finck, B.N. Targeting Metabolism, Insulin Resistance, and Diabetes to Treat Nonalcoholic Steatohepatitis. Diabetes 2018, 67, 2485–2493. [Google Scholar] [CrossRef] [Green Version]
- Adams, L.A.; Lymp, J.F.; St Sauver, J.; Sanderson, S.O.; Lindor, K.D.; Feldstein, A.; Angulo, P. The natural history of nonalcoholic fatty liver disease: A population-based cohort study. Gastroenterology 2005, 129, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Soderberg, C.; Marmur, J.; Eckes, K.; Glaumann, H.; Sallberg, M.; Frelin, L.; Rosenberg, P.; Stal, P.; Hultcrantz, R. Microvesicular fat, inter cellular adhesion molecule-1 and regulatory T-lymphocytes are of importance for the inflammatory process in livers with non-alcoholic steatohepatitis. APMIS 2011, 119, 412–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reid, D.T.; Reyes, J.L.; McDonald, B.A.; Vo, T.; Reimer, R.A.; Eksteen, B. Kupffer Cells Undergo Fundamental Changes during the Development of Experimental NASH and Are Critical in Initiating Liver Damage and Inflammation. PLoS ONE 2016, 11, e0159524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browning, J.D.; Kumar, K.S.; Saboorian, M.H.; Thiele, D.L. Ethnic differences in the prevalence of cryptogenic cirrhosis. Am. J. Gastroenterol. 2004, 99, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, S.H.; Harris, D.M.; Patrie, J.T.; Hespenheide, E.E. Is NASH underdiagnosed among African Americans? Am. J. Gastroenterol. 2002, 97, 1496–1500. [Google Scholar] [CrossRef]
- Schade, M.; Sanabria, J.A.; Mallick, A.; Aguilar, R.; Andryka, M.; Schlatzer, D.; Li, X.; Hazlett, F.E.; Kachman, M.; Raskind, A.; et al. Normalization of the α1-Na/K-ATPase/Src Signalosome Restored Microbiota Communities and Rescinded NASH Progression in the Mouse. Available online: https://www.preprints.org/manuscript/202109.0023 (accessed on 15 June 2022).
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Wu, H.; Chen, S.; Ammar, A.B.; Xu, J.; Wu, Q.; Pan, K.; Zhang, J.; Hong, Y. Crosstalk between Macroautophagy and Chaperone-Mediated Autophagy: Implications for the Treatment of Neurological Diseases. Mol. Neurobiol. 2015, 52, 1284–1296. [Google Scholar] [CrossRef] [Green Version]
- Allaire, M.; Rautou, P.E.; Codogno, P.; Lotersztajn, S. Autophagy in liver diseases: Time for translation? J. Hepatol. 2019, 70, 985–998. [Google Scholar] [CrossRef] [Green Version]
- Scrivo, A.; Bourdenx, M.; Pampliega, O.; Cuervo, A.M. Selective autophagy as a potential therapeutic target for neurodegenerative disorders. Lancet Neurol. 2018, 17, 802–815. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [Green Version]
- Rautou, P.E.; Mansouri, A.; Lebrec, D.; Durand, F.; Valla, D.; Moreau, R. Autophagy in liver diseases. J. Hepatol. 2010, 53, 1123–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yorimitsu, T.; Klionsky, D.J. Autophagy: Molecular machinery for self-eating. Cell Death Differ. 2005, 12 (Suppl. 2), 1542–1552. [Google Scholar] [CrossRef] [Green Version]
- Madrigal-Matute, J.; Cuervo, A.M. Regulation of Liver Metabolism by Autophagy. Gastroenterology 2016, 150, 328–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Ocak, U.; Gao, L.; Tu, S.; Lenahan, C.J.; Zhang, J.; Shao, A. Selective autophagy as a therapeutic target for neurological diseases. Cell. Mol. Life Sci. 2021, 78, 1369–1392. [Google Scholar] [CrossRef] [PubMed]
- Khaminets, A.; Behl, C.; Dikic, I. Ubiquitin-Dependent and Independent Signals in Selective Autophagy. Trends Cell Biol. 2016, 26, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Khandia, R.; Dadar, M.; Munjal, A.; Dhama, K.; Karthik, K.; Tiwari, R.; Yatoo, M.I.; Iqbal, H.M.N.; Singh, K.P.; Joshi, S.K.; et al. A Comprehensive Review of Autophagy and Its Various Roles in Infectious, Non-Infectious, and Lifestyle Diseases: Current Knowledge and Prospects for Disease Prevention, Novel Drug Design, and Therapy. Cells 2019, 8, 674. [Google Scholar] [CrossRef] [Green Version]
- Reggiori, F.; Ungermann, C. Autophagosome Maturation and Fusion. J. Mol. Biol. 2017, 429, 486–496. [Google Scholar] [CrossRef]
- Hemelaar, J.; Lelyveld, V.S.; Kessler, B.M.; Ploegh, H.L. A single protease, Apg4B, is specific for the autophagy-related ubiquitin-like proteins GATE-16, MAP1-LC3, GABARAP, and Apg8L. J. Biol. Chem. 2003, 278, 51841–51850. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Klionsky, D.J. The regulation of autophagy—Unanswered questions. J. Cell Sci. 2011, 124, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Itakura, E.; Mizushima, N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010, 6, 764–776. [Google Scholar] [CrossRef] [Green Version]
- Yla-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E.L. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy 2009, 5, 1180–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, H.M.; Mizushima, N. At the end of the autophagic road: An emerging understanding of lysosomal functions in autophagy. Trends Biochem. Sci. 2014, 39, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Tooze, J.; Hollinshead, M.; Ludwig, T.; Howell, K.; Hoflack, B.; Kern, H. In exocrine pancreas, the basolateral endocytic pathway converges with the autophagic pathway immediately after the early endosome. J. Cell Biol. 1990, 111, 329–345. [Google Scholar] [CrossRef] [PubMed]
- Berg, T.O.; Fengsrud, M.; Stromhaug, P.E.; Berg, T.; Seglen, P.O. Isolation and characterization of rat liver amphisomes. Evidence for fusion of autophagosomes with both early and late endosomes. J. Biol. Chem. 1998, 273, 21883–21892. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [Green Version]
- Mehrpour, M.; Esclatine, A.; Beau, I.; Codogno, P. Overview of macroautophagy regulation in mammalian cells. Cell Res. 2010, 20, 748–762. [Google Scholar] [CrossRef]
- Hosokawa, N.; Sasaki, T.; Iemura, S.; Natsume, T.; Hara, T.; Mizushima, N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 2009, 5, 973–979. [Google Scholar] [CrossRef] [Green Version]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [Green Version]
- Furuya, N.; Yu, J.; Byfield, M.; Pattingre, S.; Levine, B. The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, autophagy and tumor suppressor function. Autophagy 2005, 1, 46–52. [Google Scholar] [CrossRef] [Green Version]
- Itakura, E.; Kishi, C.; Inoue, K.; Mizushima, N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol. Biol. Cell 2008, 19, 5360–5372. [Google Scholar] [CrossRef] [Green Version]
- Matsunaga, K.; Morita, E.; Saitoh, T.; Akira, S.; Ktistakis, N.T.; Izumi, T.; Noda, T.; Yoshimori, T. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J. Cell Biol. 2010, 190, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Burman, C.; Ktistakis, N.T. Regulation of autophagy by phosphatidylinositol 3-phosphate. FEBS Lett. 2010, 584, 1302–1312. [Google Scholar] [CrossRef] [Green Version]
- Romanov, J.; Walczak, M.; Ibiricu, I.; Schuchner, S.; Ogris, E.; Kraft, C.; Martens, S. Mechanism and functions of membrane binding by the Atg5-Atg12/Atg16 complex during autophagosome formation. EMBO J. 2012, 31, 4304–4317. [Google Scholar] [CrossRef]
- Matsushita, M.; Suzuki, N.N.; Obara, K.; Fujioka, Y.; Ohsumi, Y.; Inagaki, F. Structure of Atg5·Atg16, a complex essential for autophagy. J. Biol. Chem. 2007, 282, 6763–6772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weidberg, H.; Shvets, E.; Elazar, Z. Biogenesis and cargo selectivity of autophagosomes. Annu. Rev. Biochem. 2011, 80, 125–156. [Google Scholar] [CrossRef] [PubMed]
- Ohsumi, Y. Molecular dissection of autophagy: Two ubiquitin-like systems. Nat. Rev. Mol. Cell Biol. 2001, 2, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Shintani, T.; Mizushima, N.; Ogawa, Y.; Matsuura, A.; Noda, T.; Ohsumi, Y. Apg10p, a novel protein-conjugating enzyme essential for autophagy in yeast. EMBO J. 1999, 18, 5234–5241. [Google Scholar] [CrossRef] [Green Version]
- Geng, J.; Klionsky, D.J. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ‘Protein modifications: Beyond the usual suspects’ review series. EMBO Rep. 2008, 9, 859–864. [Google Scholar] [CrossRef] [Green Version]
- Hanada, T.; Noda, N.N.; Satomi, Y.; Ichimura, Y.; Fujioka, Y.; Takao, T.; Inagaki, F.; Ohsumi, Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J. Biol. Chem. 2007, 282, 37298–37302. [Google Scholar] [CrossRef] [Green Version]
- Kuma, A.; Mizushima, N.; Ishihara, N.; Ohsumi, Y. Formation of the approximately 350-kDa Apg12-Apg5·Apg16 multimeric complex, mediated by Apg16 oligomerization, is essential for autophagy in yeast. J. Biol. Chem. 2002, 277, 18619–18625. [Google Scholar] [CrossRef] [Green Version]
- Fujita, N.; Itoh, T.; Omori, H.; Fukuda, M.; Noda, T.; Yoshimori, T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 2008, 19, 2092–2100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, H.; Rubinsztein, D.C. Control of autophagy as a therapy for neurodegenerative disease. Nat. Rev. Neurol. 2011, 8, 108–117. [Google Scholar] [CrossRef]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.M.; Jung, C.H.; Seo, M.; Otto, N.M.; Grunwald, D.; Kim, K.H.; Moriarity, B.; Kim, Y.M.; Starker, C.; Nho, R.S.; et al. The ULK1 complex mediates MTORC1 signaling to the autophagy initiation machinery via binding and phosphorylating ATG14. Autophagy 2016, 12, 547–564. [Google Scholar] [CrossRef] [PubMed]
- de Duve, C.W.; Wattiaux, R. Functions of Lysosomes. Annu. Rev. Physiol. 1966, 28, 435–492. [Google Scholar] [CrossRef] [PubMed]
- Moeller, C.H.; Thompson, W.W. An ultrastructural study of the yeast tonoplast during the shift from exponential to stationary phase. J. Ultrastruct. Res. 1979, 68, 28–37. [Google Scholar] [CrossRef]
- Campbell, C.L.; Thorsness, P.E. Escape of mitochondrial DNA to the nucleus in yme1 yeast is mediated by vacuolar-dependent turnover of abnormal mitochondrial compartments. J. Cell Sci. 1998, 111 Pt 16, 2455–2464. [Google Scholar] [CrossRef]
- Tuttle, D.L.; Lewin, A.S.; Dunn, W.A., Jr. Selective autophagy of peroxisomes in methylotrophic yeasts. Eur. J. Cell Biol. 1993, 60, 283–290. [Google Scholar]
- Muller, O.; Sattler, T.; Flotenmeyer, M.; Schwarz, H.; Plattner, H.; Mayer, A. Autophagic tubes: Vacuolar invaginations involved in lateral membrane sorting and inverse vesicle budding. J. Cell Biol. 2000, 151, 519–528. [Google Scholar] [CrossRef]
- Uttenweiler, A.; Schwarz, H.; Neumann, H.; Mayer, A. The vacuolar transporter chaperone (VTC) complex is required for microautophagy. Mol. Biol. Cell 2007, 18, 166–175. [Google Scholar] [CrossRef] [Green Version]
- Sattler, T.; Mayer, A. Cell-free reconstitution of microautophagic vacuole invagination and vesicle formation. J. Cell Biol. 2000, 151, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Sou, Y.S.; Waguri, S.; Iwata, J.; Ueno, T.; Fujimura, T.; Hara, T.; Sawada, N.; Yamada, A.; Mizushima, N.; Uchiyama, Y.; et al. The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol. Biol. Cell 2008, 19, 4762–4775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef]
- Uttenweiler, A.; Schwarz, H.; Mayer, A. Microautophagic vacuole invagination requires calmodulin in a Ca2+-independent function. J. Biol. Chem. 2005, 280, 33289–33297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Li, Z.; Hu, W.; Ren, H.; Tian, E.; Zhao, Y.; Lu, Q.; Huang, X.; Yang, P.; Li, X.; et al. C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell 2010, 141, 1042–1055. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Klionsky, D.J. Permeases recycle amino acids resulting from autophagy. Autophagy 2007, 3, 149–150. [Google Scholar] [CrossRef] [Green Version]
- Roberts, P.; Moshitch-Moshkovitz, S.; Kvam, E.; O’Toole, E.; Winey, M.; Goldfarb, D.S. Piecemeal microautophagy of nucleus in Saccharomyces cerevisiae. Mol. Biol. Cell 2003, 14, 129–141. [Google Scholar] [CrossRef] [Green Version]
- Todde, V.; Veenhuis, M.; van der Klei, I.J. Autophagy: Principles and significance in health and disease. Biochim. Biophys. Acta 2009, 1792, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Dalby, K.N.; Tekedereli, I.; Lopez-Berestein, G.; Ozpolat, B. Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer. Autophagy 2010, 6, 322–329. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M. Autophagy: Many paths to the same end. Mol. Cell Biochem. 2004, 263, 55–72. [Google Scholar] [CrossRef]
- Dubouloz, F.; Deloche, O.; Wanke, V.; Cameroni, E.; De Virgilio, C. The TOR and EGO protein complexes orchestrate microautophagy in yeast. Mol. Cell 2005, 19, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Iwata, A.; Christianson, J.C.; Bucci, M.; Ellerby, L.M.; Nukina, N.; Forno, L.S.; Kopito, R.R. Increased susceptibility of cytoplasmic over nuclear polyglutamine aggregates to autophagic degradation. Proc. Natl. Acad. Sci. USA 2005, 102, 13135–13140. [Google Scholar] [CrossRef] [Green Version]
- Schuck, S. Microautophagy—Distinct molecular mechanisms handle cargoes of many sizes. J. Cell Sci. 2020, 133, jcs246322. [Google Scholar] [CrossRef] [PubMed]
- Oku, M.; Maeda, Y.; Kagohashi, Y.; Kondo, T.; Yamada, M.; Fujimoto, T.; Sakai, Y. Evidence for ESCRT- and clathrin-dependent microautophagy. J. Cell Biol. 2017, 216, 3263–3274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahu, R.; Kaushik, S.; Clement, C.C.; Cannizzo, E.S.; Scharf, B.; Follenzi, A.; Potolicchio, I.; Nieves, E.; Cuervo, A.M.; Santambrogio, L. Microautophagy of cytosolic proteins by late endosomes. Dev. Cell 2011, 20, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Oku, M.; Sakai, Y. Three Distinct Types of Microautophagy Based on Membrane Dynamics and Molecular Machineries. Bioessays 2018, 40, e1800008. [Google Scholar] [CrossRef]
- Schafer, J.A.; Schessner, J.P.; Bircham, P.W.; Tsuji, T.; Funaya, C.; Pajonk, O.; Schaeff, K.; Ruffini, G.; Papagiannidis, D.; Knop, M.; et al. ESCRT machinery mediates selective microautophagy of endoplasmic reticulum in yeast. EMBO J. 2020, 39, e102586. [Google Scholar] [CrossRef]
- Nixon, R.A. Autophagy, amyloidogenesis and Alzheimer disease. J. Cell Sci. 2007, 120, 4081–4091. [Google Scholar] [CrossRef] [Green Version]
- Martin-Aparicio, E.; Yamamoto, A.; Hernandez, F.; Hen, R.; Avila, J.; Lucas, J.J. Proteasomal-dependent aggregate reversal and absence of cell death in a conditional mouse model of Huntington’s disease. J. Neurosci. 2001, 21, 8772–8781. [Google Scholar] [CrossRef] [Green Version]
- Takikita, S.; Myerowitz, R.; Zaal, K.; Raben, N.; Plotz, P.H. Murine muscle cell models for Pompe disease and their use in studying therapeutic approaches. Mol. Genet. Metab. 2009, 96, 208–217. [Google Scholar] [CrossRef] [Green Version]
- Bejarano, E.; Cuervo, A.M. Chaperone-mediated autophagy. Proc. Am. Thorac. Soc. 2010, 7, 29–39. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M. Chaperone-mediated autophagy: Selectivity pays off. Trends Endocrinol. Metab. 2010, 21, 142–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, H.L.; Terlecky, S.R.; Plant, C.P.; Dice, J.F. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 1989, 246, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Salvador, N.; Aguado, C.; Horst, M.; Knecht, E. Import of a cytosolic protein into lysosomes by chaperone-mediated autophagy depends on its folding state. J. Biol. Chem. 2000, 275, 27447–27456. [Google Scholar] [CrossRef]
- Chiang, H.L.; Dice, J.F. Peptide sequences that target proteins for enhanced degradation during serum withdrawal. J. Biol. Chem. 1988, 263, 6797–6805. [Google Scholar] [CrossRef]
- Dice, J.F. Altered degradation of proteins microinjected into senescent human fibroblasts. J. Biol. Chem. 1982, 257, 14624–14627. [Google Scholar] [CrossRef]
- Dice, J.F. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem. Sci. 1990, 15, 305–309. [Google Scholar] [CrossRef]
- Park, C.; Suh, Y.; Cuervo, A.M. Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat. Commun. 2015, 6, 6823. [Google Scholar] [CrossRef] [Green Version]
- Agarraberes, F.A.; Dice, J.F. A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J. Cell Sci. 2001, 114, 2491–2499. [Google Scholar] [CrossRef]
- Bandyopadhyay, U.; Kaushik, S.; Varticovski, L.; Cuervo, A.M. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol. Cell Biol. 2008, 28, 5747–5763. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M.; Terlecky, S.R.; Dice, J.F.; Knecht, E. Selective binding and uptake of ribonuclease A and glyceraldehyde-3-phosphate dehydrogenase by isolated rat liver lysosomes. J. Biol. Chem. 1994, 269, 26374–26380. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Dice, J.F. A receptor for the selective uptake and degradation of proteins by lysosomes. Science 1996, 273, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Dice, J.F. Unique properties of lamp2a compared to other lamp2 isoforms. J. Cell Sci. 2000, 113 Pt 24, 4441–4450. [Google Scholar] [CrossRef] [PubMed]
- Valdor, R.; Mocholi, E.; Botbol, Y.; Guerrero-Ros, I.; Chandra, D.; Koga, H.; Gravekamp, C.; Cuervo, A.M.; Macian, F. Chaperone-mediated autophagy regulates T cell responses through targeted degradation of negative regulators of T cell activation. Nat. Immunol. 2014, 15, 1046–1054. [Google Scholar] [CrossRef] [Green Version]
- Kiffin, R.; Christian, C.; Knecht, E.; Cuervo, A.M. Activation of chaperone-mediated autophagy during oxidative stress. Mol. Biol. Cell 2004, 15, 4829–4840. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, J.V.; Fofo, H.; Bejarano, E.; Bento, C.F.; Ramalho, J.S.; Girao, H.; Pereira, P. STUB1/CHIP is required for HIF1A degradation by chaperone-mediated autophagy. Autophagy 2013, 9, 1349–1366. [Google Scholar] [CrossRef] [Green Version]
- Hubbi, M.E.; Gilkes, D.M.; Hu, H.; Kshitiz, A.I.; Semenza, G.L. Cyclin-dependent kinases regulate lysosomal degradation of hypoxia-inducible factor 1alpha to promote cell-cycle progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3325–E3334. [Google Scholar] [CrossRef] [Green Version]
- Bandyopadhyay, U.; Sridhar, S.; Kaushik, S.; Kiffin, R.; Cuervo, A.M. Identification of regulators of chaperone-mediated autophagy. Mol. Cell 2010, 39, 535–547. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Murashige, D.S.; Humphrey, S.J.; James, D.E. A Positive Feedback Loop between Akt and mTORC2 via SIN1 Phosphorylation. Cell Rep. 2015, 12, 937–943. [Google Scholar] [CrossRef] [Green Version]
- Arias, E.; Koga, H.; Diaz, A.; Mocholi, E.; Patel, B.; Cuervo, A.M. Lysosomal mTORC2/PHLPP1/Akt Regulate Chaperone-Mediated Autophagy. Mol. Cell 2015, 59, 270–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endicott, S.J.; Ziemba, Z.J.; Beckmann, L.J.; Boynton, D.N.; Miller, R.A. Inhibition of class I PI3K enhances chaperone-mediated autophagy. J. Cell Biol. 2020, 219, e202001031. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.L.; Suh, Y.; Cuervo, A.M. Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab. 2014, 20, 417–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuervo, A.M.; Knecht, E.; Terlecky, S.R.; Dice, J.F. Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am. J. Physiol. 1995, 269 Pt 1, C1200–C1208. [Google Scholar] [CrossRef]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Kon, M.; Kiffin, R.; Koga, H.; Chapochnick, J.; Macian, F.; Varticovski, L.; Cuervo, A.M. Chaperone-mediated autophagy is required for tumor growth. Sci. Transl. Med. 2011, 3, 109ra117. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Cuervo, A.M. AMPK-dependent phosphorylation of lipid droplet protein PLIN2 triggers its degradation by CMA. Autophagy 2016, 12, 432–438. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M.; Hu, W.; Lim, B.; Dice, J.F. IkappaB is a substrate for a selective pathway of lysosomal proteolysis. Mol. Biol. Cell 1998, 9, 1995–2010. [Google Scholar] [CrossRef]
- Zhang, L.; Sun, Y.; Fei, M.; Tan, C.; Wu, J.; Zheng, J.; Tang, J.; Sun, W.; Lv, Z.; Bao, J.; et al. Disruption of chaperone-mediated autophagy-dependent degradation of MEF2A by oxidative stress-induced lysosome destabilization. Autophagy 2014, 10, 1015–1035. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.M.; Yang, Q.; Xie, X.Q.; Liao, C.Y.; Lin, H.; Liu, T.T.; Yin, L.; Shu, H.B. Sumoylation Promotes the Stability of the DNA Sensor cGAS and the Adaptor STING to Regulate the Kinetics of Response to DNA Virus. Immunity 2016, 45, 555–569. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M.; Dice, J.F. Age-related decline in chaperone-mediated autophagy. J. Biol. Chem. 2000, 275, 31505–31513. [Google Scholar] [CrossRef] [Green Version]
- Kiffin, R.; Kaushik, S.; Zeng, M.; Bandyopadhyay, U.; Zhang, C.; Massey, A.C.; Martinez-Vicente, M.; Cuervo, A.M. Altered dynamics of the lysosomal receptor for chaperone-mediated autophagy with age. J. Cell Sci. 2007, 120, 782–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Cuervo, A.M. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat. Med. 2008, 14, 959–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venugopal, B.; Mesires, N.T.; Kennedy, J.C.; Curcio-Morelli, C.; Laplante, J.M.; Dice, J.F.; Slaugenhaupt, S.A. Chaperone-mediated autophagy is defective in mucolipidosis type IV. J. Cell Physiol. 2009, 219, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Bose, J.K.; Majumder, P.; Lee, K.H.; Huang, J.T.; Huang, J.K.; Shen, C.K. Metabolism and mis-metabolism of the neuropathological signature protein TDP-43. J. Cell Sci. 2014, 127 Pt 14, 3024–3038. [Google Scholar] [CrossRef] [Green Version]
- Xilouri, M.; Brekk, O.R.; Polissidis, A.; Chrysanthou-Piterou, M.; Kloukina, I.; Stefanis, L. Impairment of chaperone-mediated autophagy induces dopaminergic neurodegeneration in rats. Autophagy 2016, 12, 2230–2247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef]
- Saha, T. LAMP2A overexpression in breast tumors promotes cancer cell survival via chaperone-mediated autophagy. Autophagy 2012, 8, 1643–1656. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.B.; Nin, D.S.; Tam, J.; Khan, M. Role of chaperone mediated autophagy (CMA) in the degradation of misfolded N-CoR protein in non-small cell lung cancer (NSCLC) cells. PLoS ONE 2011, 6, e25268. [Google Scholar] [CrossRef] [Green Version]
- Chava, S.; Lee, C.; Aydin, Y.; Chandra, P.K.; Dash, A.; Chedid, M.; Thung, S.N.; Moroz, K.; Wu, T.; Nayak, N.C.; et al. Chaperone-mediated autophagy compensates for impaired macroautophagy in the cirrhotic liver to promote hepatocellular carcinoma. Oncotarget 2017, 8, 40019–40036. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Yang, J.; Fan, X.; Hu, S.; Zhou, F.; Dong, J.; Zhang, S.; Shang, Y.; Jiang, X.; Guo, H.; et al. Chaperone-mediated autophagy regulates proliferation by targeting RND3 in gastric cancer. Autophagy 2016, 12, 515–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, W.; Zhang, L.; Jiao, H.; Guan, L.; Zha, J.; Li, X.; Wu, M.; Wang, Z.; Han, J.; You, H. Chaperone-mediated autophagy prevents apoptosis by degrading BBC3/PUMA. Autophagy 2015, 11, 1623–1635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, T.L.; Huang, G.J.; Wang, H.J.; Chen, J.L.; Hsu, H.P.; Lu, T.J. Hispolon promotes MDM2 downregulation through chaperone-mediated autophagy. Biochem. Biophys. Res. Commun. 2010, 398, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.R.; Menck, C.F.M.; Cuervo, A.M. Chaperone-mediated autophagy prevents cellular transformation by regulating MYC proteasomal degradation. Autophagy 2017, 13, 928–940. [Google Scholar] [CrossRef]
- Ke, P.Y. Diverse Functions of Autophagy in Liver Physiology and Liver Diseases. Int. J. Mol. Sci. 2019, 20, 300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Codogno, P.; Meijer, A.J. Autophagy in the liver. J. Hepatol. 2013, 59, 389–391. [Google Scholar] [CrossRef] [Green Version]
- Pfeifer, U. Cellular autophagy: Glycogen segregation in early stages of a partial liver atrophy. Virchows Arch. B Cell Pathol. 1970, 5, 242–253. [Google Scholar] [CrossRef]
- Schworer, C.M.; Mortimore, G.E. Glucagon-induced autophagy and proteolysis in rat liver: Mediation by selective deprivation of intracellular amino acids. Proc. Natl. Acad. Sci. USA 1979, 76, 3169–3173. [Google Scholar] [CrossRef] [Green Version]
- Schworer, C.M.; Shiffer, K.A.; Mortimore, G.E. Quantitative relationship between autophagy and proteolysis during graded amino acid deprivation in perfused rat liver. J. Biol. Chem. 1981, 256, 7652–7658. [Google Scholar] [CrossRef]
- Efeyan, A.; Zoncu, R.; Chang, S.; Gumper, I.; Snitkin, H.; Wolfson, R.L.; Kirak, O.; Sabatini, D.D.; Sabatini, D.M. Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival. Nature 2013, 493, 679–683. [Google Scholar] [CrossRef] [Green Version]
- Meijer, A.J.; Codogno, P. Autophagy: Regulation and role in disease. Crit. Rev. Clin. Lab. Sci. 2009, 46, 210–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czaja, M.J. Functions of autophagy in hepatic and pancreatic physiology and disease. Gastroenterology 2011, 140, 1895–1908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendy, R.; Grasso, P. Autophagy in acute liver damage produced in the rat by dimethylnitrosamine. Chem. Biol. Interact. 1972, 5, 401–413. [Google Scholar] [CrossRef]
- Hidvegi, T.; Ewing, M.; Hale, P.; Dippold, C.; Beckett, C.; Kemp, C.; Maurice, N.; Mukherjee, A.; Goldbach, C.; Watkins, S.; et al. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science 2010, 329, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Lavallard, V.J.; Gual, P. Autophagy and non-alcoholic fatty liver disease. Biomed. Res. Int. 2014, 2014, 120179. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.K.K.; Zhang, L.; Chan, M.T.V. Autophagy, NAFLD and NAFLD-Related HCC. Adv. Exp. Med. Biol. 2018, 1061, 127–138. [Google Scholar] [CrossRef]
- Gonzalez-Rodriguez, A.; Mayoral, R.; Agra, N.; Valdecantos, M.P.; Pardo, V.; Miquilena-Colina, M.E.; Vargas-Castrillon, J.; Lo Iacono, O.; Corazzari, M.; Fimia, G.M.; et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014, 5, e1179. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Li, P.; Fu, S.; Calay, E.S.; Hotamisligil, G.S. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010, 11, 467–478. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.Y.; Han, J.; Cao, S.Y.; Hong, T.; Zhuo, D.; Shi, J.; Liu, Z.; Cao, W. Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: Inhibition of FoxO1-dependent expression of key autophagy genes by insulin. J. Biol. Chem. 2009, 284, 31484–31492. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; De Cegli, R.; Mansueto, G.; Saha, P.K.; Vetrini, F.; Visvikis, O.; Huynh, T.; Carissimo, A.; Palmer, D.; Klisch, T.J.; et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 2013, 15, 647–658. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.W.; Zhang, H.; Li, M.; Xiong, X.; Chen, X.; Chen, X.; Dong, X.C.; Yin, X.M. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J. Hepatol. 2013, 58, 993–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amir, M.; Czaja, M.J. Autophagy in nonalcoholic steatohepatitis. Expert Rev. Gastroenterol. Hepatol. 2011, 5, 159–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [Green Version]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Z.B.; Shi, Y.H.; Zhou, J.; Qiu, S.J.; Xu, Y.; Dai, Z.; Shi, G.M.; Wang, X.Y.; Ke, A.W.; Wu, B.; et al. Association of autophagy defect with a malignant phenotype and poor prognosis of hepatocellular carcinoma. Cancer Res. 2008, 68, 9167–9175. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, S.; Hikita, H.; Tatsumi, T.; Sakamori, R.; Nozaki, Y.; Sakane, S.; Shiode, Y.; Nakabori, T.; Saito, Y.; Hiramatsu, N.; et al. Rubicon inhibits autophagy and accelerates hepatocyte apoptosis and lipid accumulation in nonalcoholic fatty liver disease in mice. Hepatology 2016, 64, 1994–2014. [Google Scholar] [CrossRef]
- Takakura, K.; Oikawa, T.; Nakano, M.; Saeki, C.; Torisu, Y.; Kajihara, M.; Saruta, M. Recent Insights into the Multiple Pathways Driving Non-alcoholic Steatohepatitis-Derived Hepatocellular Carcinoma. Front. Oncol. 2019, 9, 762. [Google Scholar] [CrossRef] [Green Version]
- Inokuchi-Shimizu, S.; Park, E.J.; Roh, Y.S.; Yang, L.; Zhang, B.; Song, J.; Liang, S.; Pimienta, M.; Taniguchi, K.; Wu, X.; et al. TAK1-mediated autophagy and fatty acid oxidation prevent hepatosteatosis and tumorigenesis. J. Clin. Investig. 2014, 124, 3566–3578. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Hou, J.; Wang, X.; Jiang, R.; Yin, Y.; Ji, J.; Deng, L.; Huang, X.; Wang, K.; Sun, B. PTPRO-mediated autophagy prevents hepatosteatosis and tumorigenesis. Oncotarget 2015, 6, 9420–9433. [Google Scholar] [CrossRef] [Green Version]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012, 485, 251–255. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C.; et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, M. Liver autophagy: Physiology and pathology. J. Biochem. 2012, 152, 5–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Han, W.; Sui, X.; Fang, Y.; Pan, H. Autophagy: A novel therapeutic target for hepatocarcinoma (Review). Oncol. Lett. 2014, 7, 1345–1351. [Google Scholar] [CrossRef]
- Czaja, M.J. Function of Autophagy in Nonalcoholic Fatty Liver Disease. Dig. Dis. Sci. 2016, 61, 1304–1313. [Google Scholar] [CrossRef]
- Udoh, U.S.; Banerjee, M.; Rajan, P.K.; Sanabria, J.D.; Smith, G.; Schade, M.; Sanabria, J.A.; Nakafuku, Y.; Sodhi, K.; Pierre, S.V.; et al. Tumor-Suppressor Role of the alpha1-Na/K-ATPase Signalosome in NASH Related Hepatocellular Carcinoma. Int. J. Mol. Sci. 2022, 23, 7359. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Udoh, U.-A.S.; Rajan, P.K.; Nakafuku, Y.; Finley, R.; Sanabria, J.R. Cell Autophagy in NASH and NASH-Related Hepatocellular Carcinoma. Int. J. Mol. Sci. 2022, 23, 7734. https://doi.org/10.3390/ijms23147734
Udoh U-AS, Rajan PK, Nakafuku Y, Finley R, Sanabria JR. Cell Autophagy in NASH and NASH-Related Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2022; 23(14):7734. https://doi.org/10.3390/ijms23147734
Chicago/Turabian StyleUdoh, Utibe-Abasi S., Pradeep Kumar Rajan, Yuto Nakafuku, Robert Finley, and Juan Ramon Sanabria. 2022. "Cell Autophagy in NASH and NASH-Related Hepatocellular Carcinoma" International Journal of Molecular Sciences 23, no. 14: 7734. https://doi.org/10.3390/ijms23147734