
Functions of Breast Cancer Predisposition Genes: Implications for Clinical Management


Abstract
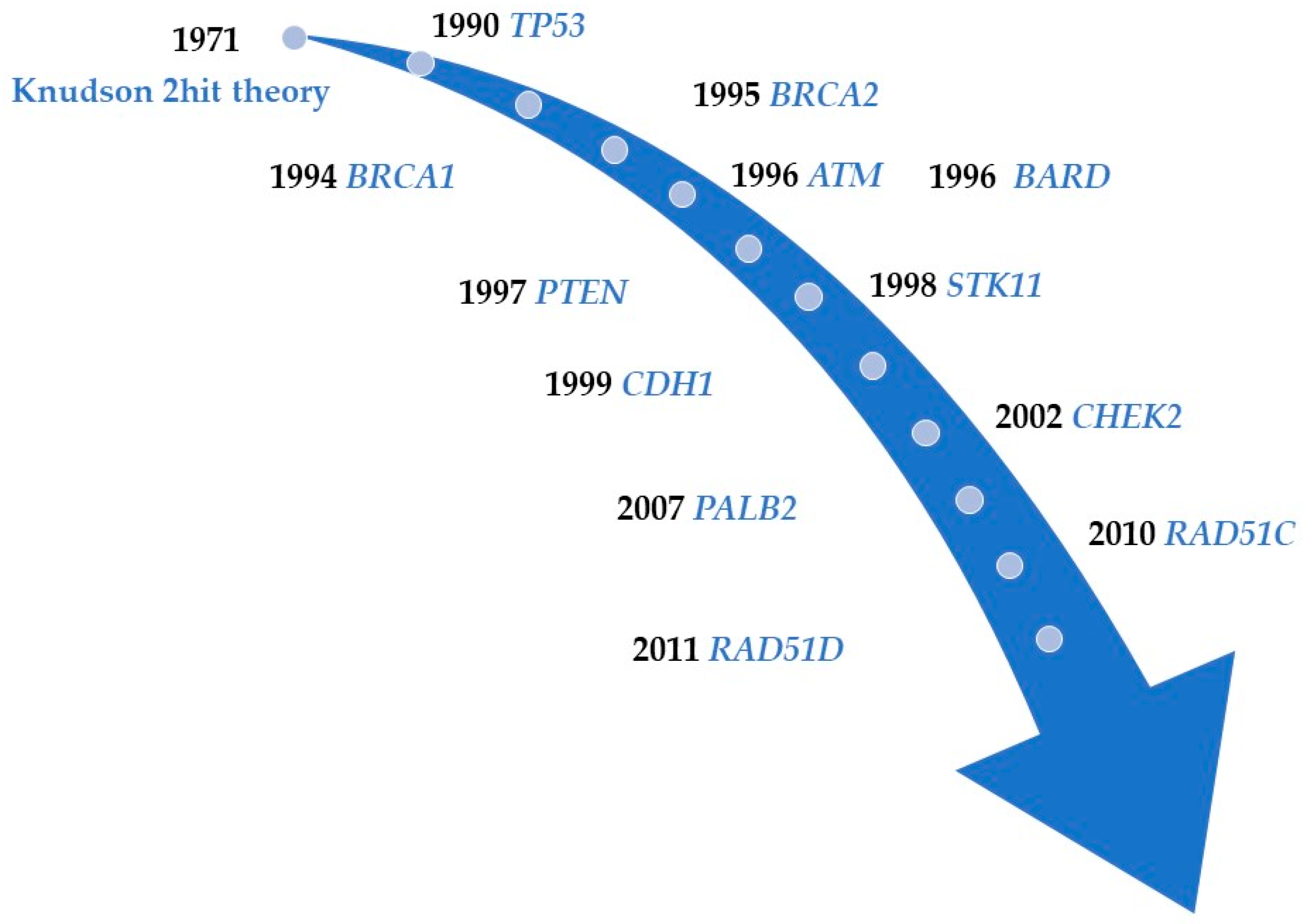
:1. Introduction
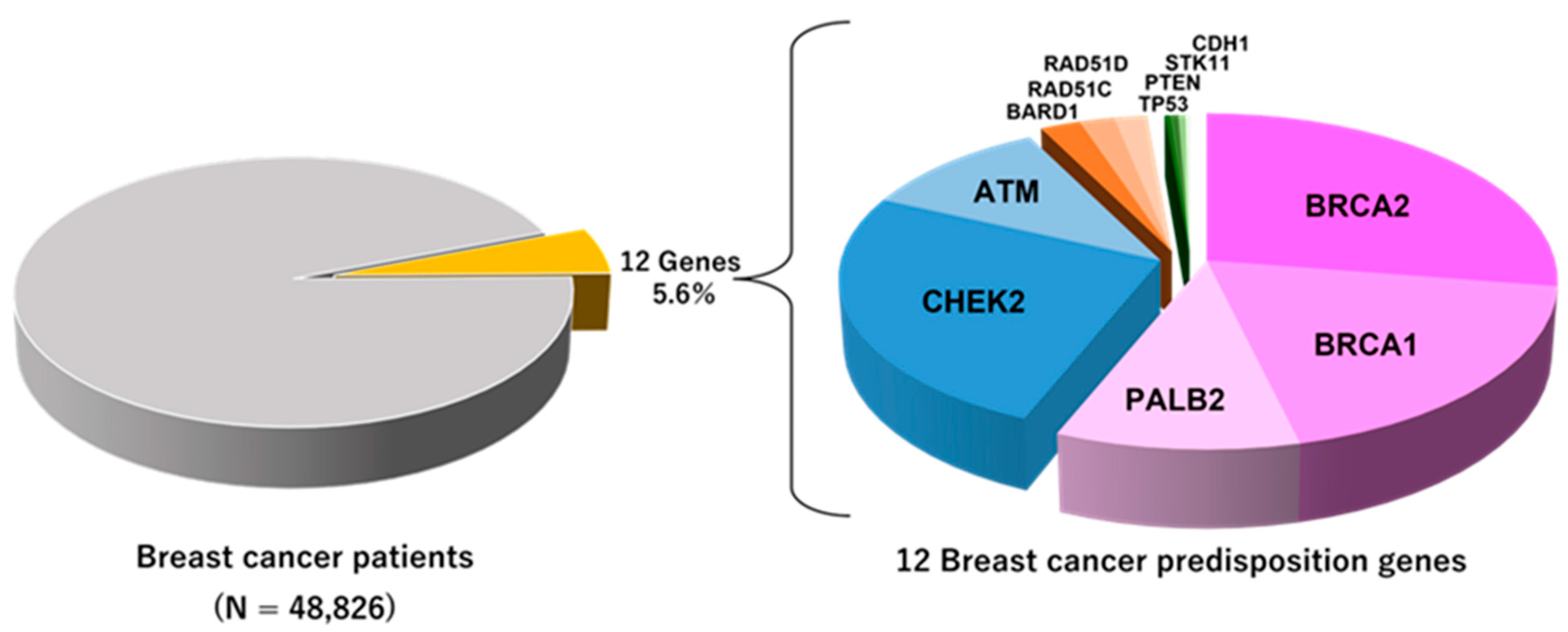
2. High-Penetrance Genes in Breast Cancer
2.1. BRCA1 and BRCA2
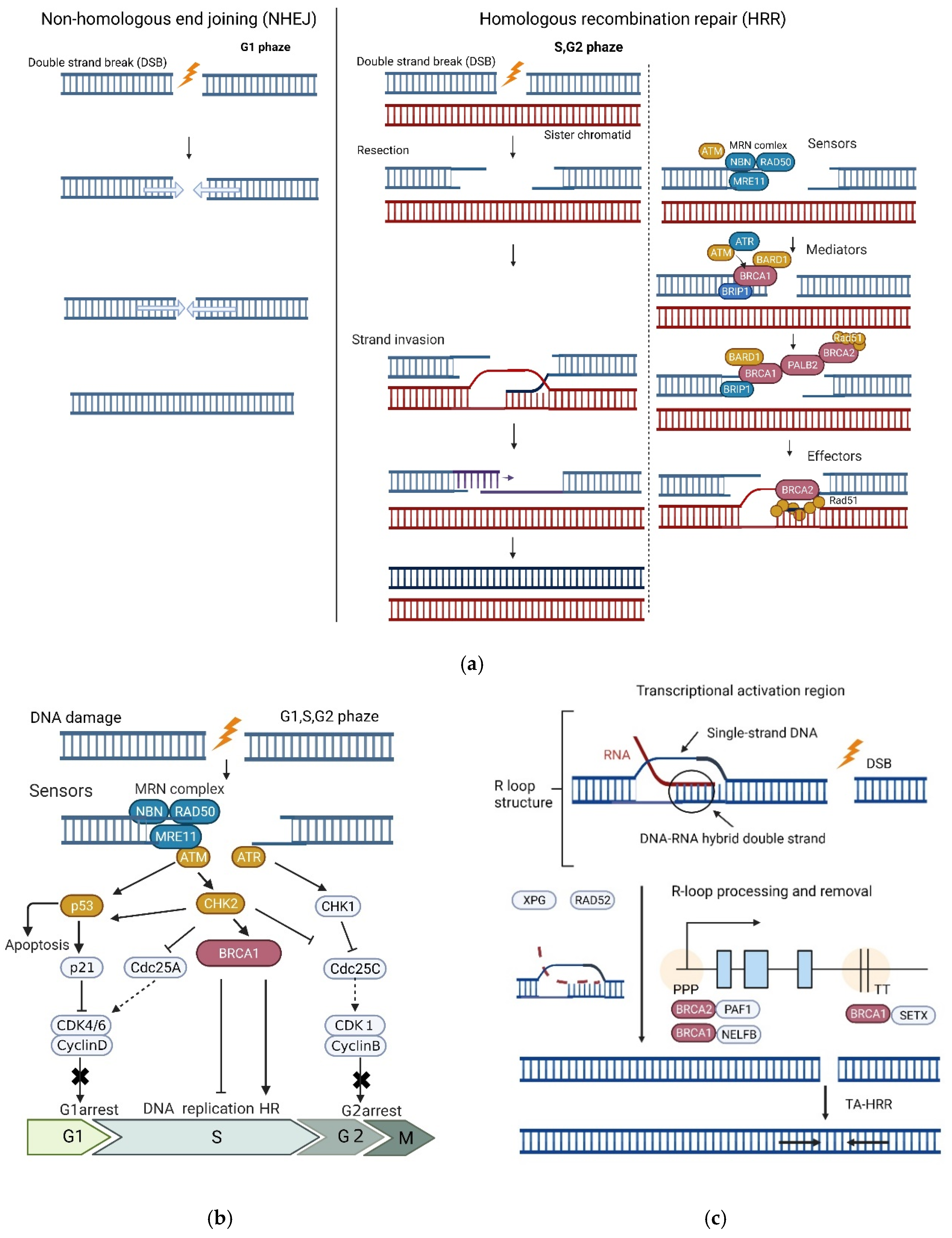
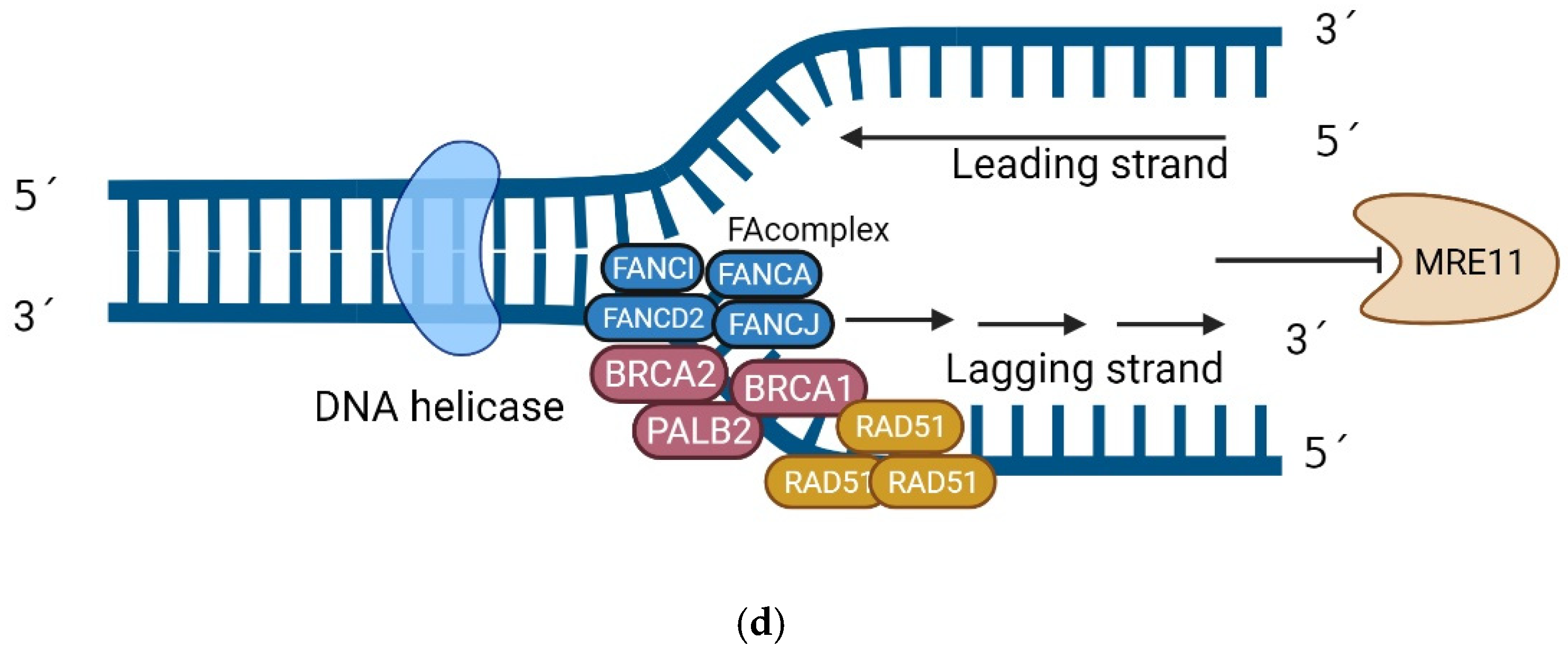
2.1.1. Functions
- HR Repair
- DNA Damage-induced Cell Cycle Checkpoints
- R-loop Processing and Transcription
- DNA Replication Fork Protection
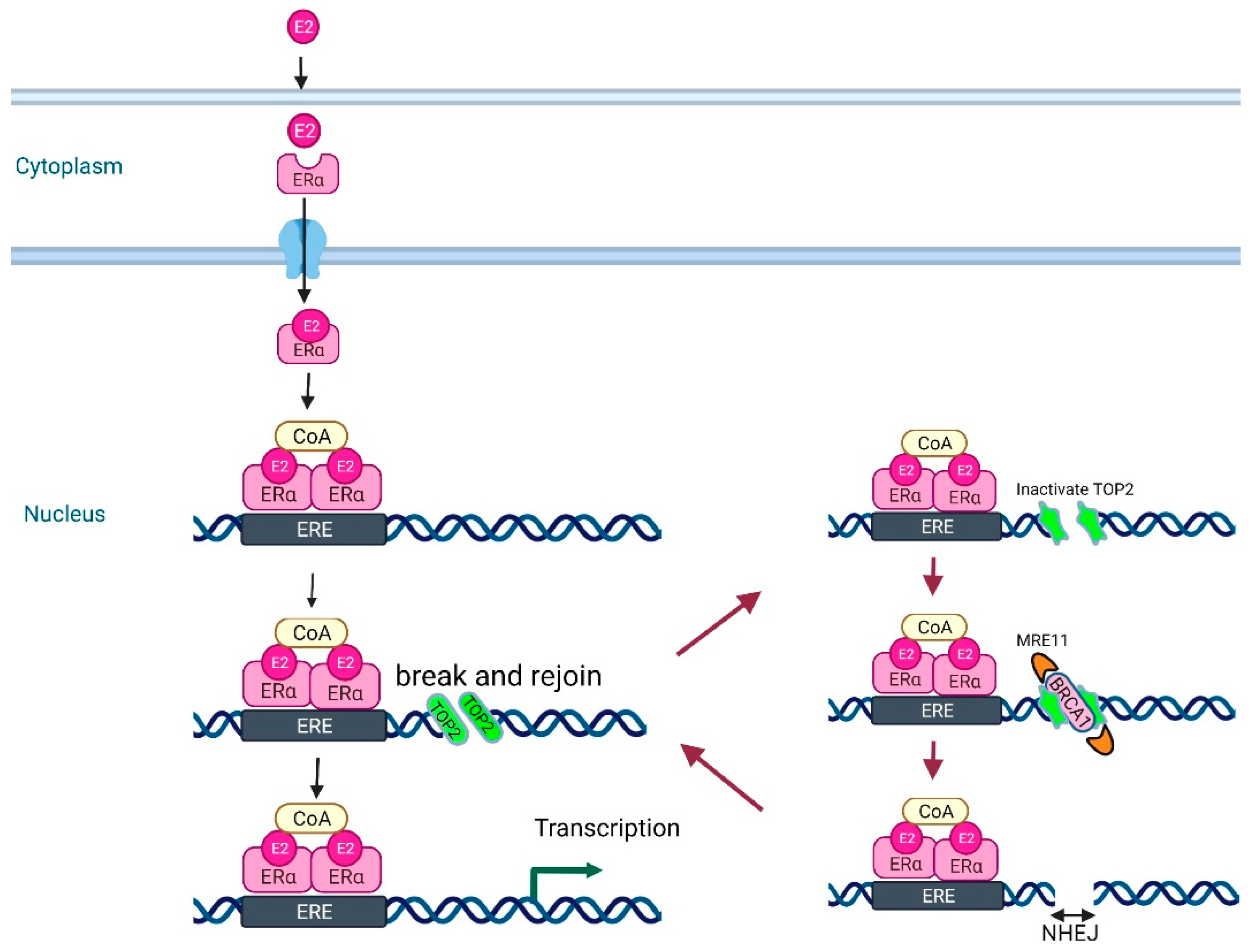
- Removal of Estrogen-induced Pathological Topoisomerase II–DNA Complexes to Ensure Genome Integrity
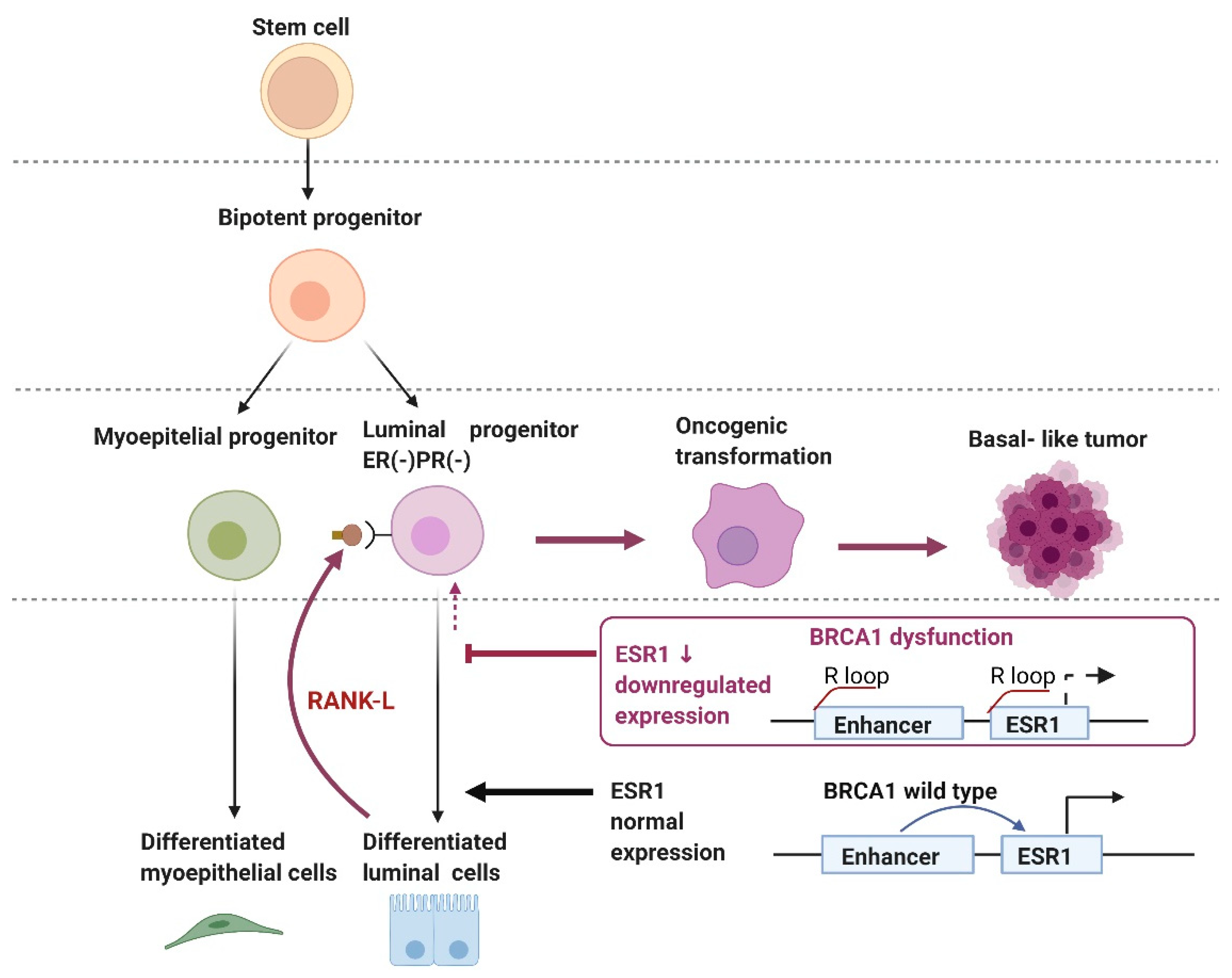
- R-loop Regulation of the Enhancer Region of ESR1 by BRCA1
2.1.2. Prevalence and Risk of Developing Cancers
2.1.3. Medical Management for Cancer Prevention
- Risk-Reducing Mastectomy (RRM)
- Risk-Reducing Salpingo-Oophorectomy (RRSO)
- Tamoxifen
- Denosumab: Anti-RANK-L Monoclonal Antibody
2.1.4. Treatment
- Anthracycline-based Chemotherapy/Taxanes
- Platinum-based Anticancer Agents
- CDK4/6 Inhibitors
- PARP Inhibitors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trial | Type of Study | Patients | Arms | Results |
|---|---|---|---|---|
| Olaparib | ||||
| Neoadjuvant setting | ||||
| GeparOLA [88] | Ph. II | 102 HER2 negative-BC pts with HRD tumors | Paclitaxel and olaparib followed by EC (n = 65) Paclitaxel and carboplatin followed by EC (n = 37) | Olaparib arm pCR = 55.1% Carboplatin arm pCR = 48.6% |
| Adjuvant setting | ||||
| OlympiA [85] | Ph. III RCT | 1836 pts with BRCA1/2 GPV post (neo)adjuvant chemotherapy | Olaparib 300 mg Placebo for 12 mo | Olaparib IDFS = 85.9% Placebo IDFS = 77.1% |
| Advanced or Metastatic Setting | ||||
| Tutt A, et al. [89] | Ph. II non-randomised | 54 MBC pts with BRCA1/2 GPV | Olaparib 400 mg Olaparib 100 mg | ORR = 41% ORR = 22% |
| Kaufman B, et al. [90] | Ph. II Single arm | 298 solid tumor pts (62 BC) with BRCA1/2 GPV >3 lines of chemotherapy for MBC | Olaparib 400 mg | RR = 12.9% (8/62 pts) |
| OlympiAD [84] | Ph. III RCT | 302 MBC pts with BRCA1/2 GPV <2 lines of chemotherapy for MBC | Olaparib 300 mg TPC | Olaparib ORR = 59.9% mPFS = 7.0 mo TPC ORR = 28.8% mPFS = 4.2 mo |
| Niraparib | ||||
| Advanced or Metastatic Setting | ||||
| Sandhu SK, et al. [91] | Ph. I dose-escalation | 100 solid tumors including 22 MBC | Niraparib 30–400 mg daily | Maximum-tolerated dose is 300 mg daily |
| Rucaparib | ||||
| Advanced or Metastatic Setting | ||||
| Drew Y, et al. [92] | Ph. II dose escalation IV → oral study | 78 solid tumors pts including 23 pts with BRCA1/2 GPV | Rucaparib IV 4–18 mg →oral 92–600 mg twice daily | Well-tolerated doses as oral 480 mg daily. |
| Wilson RH, et al. [93] | Ph. I dose-escalation in combination with chemotherapy | 85 pts with advanced solid tumors including 7 pts with BRCA1/2 GPV | Rucaparib IV12–24 mg →oral 80–360 mg + chemotherapy | Maximum-tolerated dose for the combination was oral 240 mg daily rucaparib and carboplatin |
| Miller K, et al. [94] | Ph. II RCT | 128 pts with TNBC or BRCA-associated BC (n = 22) with residual tumor post neoadjuvant chemotherapy | Cisplatin 75 mg/m2 ± Rucaparib 25–30 mg IV days 1 to 3 (4 cycles) → oral rucaparib 100 mg weekly | Cisplatin alone 2-yr DFS = 54.2% Cisplatin + rucaparib 2-yr DFS = 64.1% |
| Talazoparib i | ||||
| Neoadjuvant setting | ||||
| Litton JK, et al. [95] | Ph. II | 20 HER2 negative BC pts with BRCA1/2 GPV | Talazoparib 1 mg for 6 mo | RCB 0 (pCR) = 53% RCB 0/1 = 63% |
| Advanced or Metastatic Setting | ||||
| EMBRACA [86] | Ph. III RCT | 431 advanced/metastatic BC pts with BRCA1/2 GPV <3 lines of chemotherapy for MBC | Talazoparib 1 mg TPC | Talazoparib ORR = 62.6% mPFS = 8.6 mo TPC ORR = 27.2% mPFS = 5.6 mo |
| Veriparib | ||||
| Neoadjuvant setting | ||||
| I SPY2 [96] | Ph. II adaptive randomized trial | Stage II or III TNBC (n = 116) veriparib group (n = 72) including 12 pts with BRCA1/2 GPV control group (n = 44) including 3 pts with BRCA1/2 GPV | Carboplatin/paclitaxel + placebo (CP) Carboplatin/paclitaxel + veriparib 50 mg (VCP) All patients received followed by AC | CP pCR = 26% VCP pCR = 51% |
| BrighTNess [87] | Ph. III RCT | Stage II or III TNBC (n = 634) including 92 pts with BRCA1/2 GPV | Paclitaxel CP VCP All patients received followed by AC | Paclitaxel pCR = 31% CP pCR = 58% VCP pCR = 53% |
| Advanced or Metastatic Setting | ||||
| BROCADE [97] | Ph. II RCT | 290 advanced/metastatic BC with BRCA1/2 GPV | CP VCP Veliparib 120 mg + temozolomide (VT) | CP ORR = 61.3% mPFS = 12.3 mo VCP ORR = 78% mPFS = 14.1 mo VT ORR = 28.6% mPFS = 7.4 mo |
2.2. PALB2
2.2.1. Function
2.2.2. Prevalence and Risk of Developing Cancers
2.2.3. Medical Management for Cancer Prevention
2.2.4. Treatment
- Platinum-based Anticancer Agents
- PARP Inhibitors
3. Moderate-Risk Genes for Breast Cancer
3.1. ATM
3.1.1. Function
3.1.2. Prevalence and Risk of Developing Cancers
3.1.3. Medical Management for Cancer Prevention
3.1.4. Treatment
- Platinum-based Anticancer Agents
- CDK4/6 Inhibitors
- PARP Inhibitors
- Radiation Therapy
3.2. CHEK2
3.2.1. Function
3.2.2. Prevalence and Risk of Developing Cancers
3.2.3. Medical Management for Cancer Prevention
3.2.4. Treatment
- Anthracycline-based Chemotherapy/Tamoxifen
- Platinum-based Anticancer Agents
- PARP Inhibitors
3.3. BARD1
3.3.1. Function
3.3.2. Prevalence and Risk of Developing Cancers
3.3.3. Medical Management for Cancer Prevention
3.4. RAD51C/RAD51D
3.4.1. Function
3.4.2. Prevalence and Risk of Developing Cancer
3.4.3. Medical Management for Cancer Prevention
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Silvestri, V.; Leslie, G.; Barnes, D.R.; Agnarsson, B.A.; Aittomäki, K.; Alducci, E.; Andrulis, I.L.; Barkardottir, R.B.; Barroso, A.; Barrowdale, D.; et al. Characterization of the Cancer Spectrum in Men with Germline BRCA1 and BRCA2 Pathogenic Variants: Results from the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA). JAMA Oncol. 2020, 6, 1218–1230. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Hori, M.; Matsuda, T.; Shibata, A.; Katanoda, K.; Sobue, T.; Nishimoto, H. Cancer incidence and incidence rates in Japan in 2009: A study of 32 population-based cancer registries for the Monitoring of Cancer Incidence in Japan (MCIJ) project. Jpn. J. Clin. Oncol. 2015, 45, 884–891. [Google Scholar] [CrossRef]
- Macacu, A.; Autier, P.; Boniol, M.; Boyle, P. Active and passive smoking and risk of breast cancer: A meta-analysis. Breast Cancer Res. Treat. 2015, 154, 213–224. [Google Scholar] [CrossRef] [Green Version]
- Oze, I.; Ito, H.; Kasugai, Y.; Yamaji, T.; Kijima, Y.; Ugai, T.; Kasuga, Y.; Ouellette, T.K.; Taniyama, Y.; Koyanagi, Y.N.; et al. A Personal Breast Cancer Risk Stratification Model Using Common Variants and Environmental Risk Factors in Japanese Females. Cancers 2021, 13, 3796. [Google Scholar] [CrossRef]
- Gail, M.H. Twenty-five years of breast cancer risk models and their applications. J. Natl. Cancer Inst. 2015, 107, djv042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cintolo-Gonzalez, J.A.; Braun, D.; Blackford, A.L.; Mazzola, E.; Acar, A.; Plichta, J.K.; Griffin, M.; Hughes, K.S. Breast cancer risk models: A comprehensive overview of existing models, validation, and clinical applications. Breast Cancer Res. Treat. 2017, 164, 263–284. [Google Scholar] [CrossRef] [PubMed]
- Melchor, L.; Benítez, J. The complex genetic landscape of familial breast cancer. Hum. Genet. 2013, 132, 845–863. [Google Scholar] [CrossRef]
- Nielsen, F.C.; van Overeem Hansen, T.; Sørensen, C.S. Hereditary breast and ovarian cancer: New genes in confined pathways. Nat. Rev. Cancer 2016, 16, 599–612. [Google Scholar] [CrossRef]
- Loveday, C.; Turnbull, C.; Ramsay, E.; Hughes, D.; Ruark, E.; Frankum, J.R.; Bowden, G.; Kalmyrzaev, B.; Warren-Perry, M.; Snape, K.; et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat. Genet. 2011, 43, 879–882. [Google Scholar] [CrossRef]
- Ruscito, I.; Gasparri, M.L.; De Marco, M.P.; Costanzi, F.; Besharat, A.R.; Papadia, A.; Kuehn, T.; Gentilini, O.D.; Bellati, F.; Caserta, D. The Clinical and Pathological Profile of BRCA1 Gene Methylated Breast Cancer Women: A Meta-Analysis. Cancers 2021, 13, 1391. [Google Scholar] [CrossRef]
- Dorling, L.; Carvalho, S.; Allen, J.; González-Neira, A.; Luccarini, C.; Wahlström, C.; Pooley, K.A.; Parsons, M.T.; Fortuno, C.; Wang, Q.; et al. Breast Cancer Risk Genes—Association Analysis in More than 113,000 Women. N. Engl. J. Med. 2021, 384, 428–439. [Google Scholar] [CrossRef]
- Hu, C.; Hart, S.N.; Gnanaolivu, R.; Huang, H.; Lee, K.Y.; Na, J.; Gao, C.; Lilyquist, J.; Yadav, S.; Boddicker, N.J.; et al. A Population-Based Study of Genes Previously Implicated in Breast Cancer. N. Engl. J. Med. 2021, 384, 440–451. [Google Scholar] [CrossRef]
- Daly, M.B.; Pal, T.; Berry, M.P.; Buys, S.S.; Dickson, P.; Domchek, S.M.; Elkhanany, A.; Friedman, S.; Goggins, M.; Hutton, M.L.; et al. Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 77–102. [Google Scholar] [CrossRef] [PubMed]
- Piombino, C.; Cortesi, L.; Lambertini, M.; Punie, K.; Grandi, G.; Toss, A. Secondary Prevention in Hereditary Breast and/or Ovarian Cancer Syndromes Other Than BRCA. J. Oncol. 2020, 2020, 6384190. [Google Scholar] [CrossRef] [PubMed]
- Neiger, H.E.; Siegler, E.L.; Shi, Y. Breast Cancer Predisposition Genes and Synthetic Lethality. Int. J. Mol. Sci. 2021, 22, 5614. [Google Scholar] [CrossRef] [PubMed]
- Guilford, P.; Hopkins, J.; Harraway, J.; McLeod, M.; McLeod, N.; Harawira, P.; Taite, H.; Scoular, R.; Miller, A.; Reeve, A.E. E-cadherin germline mutations in familial gastric cancer. Nature 1998, 392, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Liaw, D.; Marsh, D.J.; Li, J.; Dahia, P.L.; Wang, S.I.; Zheng, Z.; Bose, S.; Call, K.M.; Tsou, H.C.; Peacocke, M.; et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat. Genet. 1997, 16, 64–67. [Google Scholar] [CrossRef]
- Hemminki, A.; Markie, D.; Tomlinson, I.; Avizienyte, E.; Roth, S.; Loukola, A.; Bignell, G.; Warren, W.; Aminoff, M.; Höglund, P.; et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 1998, 391, 184–187. [Google Scholar] [CrossRef]
- Malkin, D.; Li, F.P.; Strong, L.C.; Fraumeni, J.F., Jr.; Nelson, C.E.; Kim, D.H.; Kassel, J.; Gryka, M.A.; Bischoff, F.Z.; Tainsky, M.A.; et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990, 250, 1233–1238. [Google Scholar] [CrossRef]
- Hall, J.M.; Lee, M.K.; Newman, B.; Morrow, J.E.; Anderson, L.A.; Huey, B.; King, M.C. Linkage of early-onset familial breast cancer to chromosome 17q21. Science 1990, 250, 1684–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W.; et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wooster, R.; Bignell, G.; Lancaster, J.; Swift, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory, S.; Gumbs, C.; Micklem, G. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995, 378, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Boque-Sastre, R.; Soler, M.; Guil, S. Detection and Characterization of R Loop Structures. Methods Mol. Biol. 2017, 1543, 231–242. [Google Scholar] [CrossRef]
- Stok, C.; Kok, Y.P.; van den Tempel, N.; van Vugt, M. Shaping the BRCAness mutational landscape by alternative double-strand break repair, replication stress and mitotic aberrancies. Nucleic Acids Res. 2021, 49, 4239–4257. [Google Scholar] [CrossRef]
- Takaoka, M.; Miki, Y. BRCA1 gene: Function and deficiency. Int. J. Clin. Oncol. 2018, 23, 36–44. [Google Scholar] [CrossRef]
- Creeden, J.F.; Nanavaty, N.S.; Einloth, K.R.; Gillman, C.E.; Stanbery, L.; Hamouda, D.M.; Dworkin, L.; Nemunaitis, J. Homologous recombination proficiency in ovarian and breast cancer patients. BMC Cancer 2021, 21, 1154. [Google Scholar] [CrossRef]
- Wu, S.; Zhou, J.; Zhang, K.; Chen, H.; Luo, M.; Lu, Y.; Sun, Y.; Chen, Y. Molecular Mechanisms of PALB2 Function and Its Role in Breast Cancer Management. Front. Oncol. 2020, 10, 301. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Wei, C.; Li, J.; Xing, P.; Li, J.; Zheng, S.; Chen, X. Cell cycle-dependent control of homologous recombination. Acta Biochim. Biophys. Sin. 2017, 49, 655–668. [Google Scholar] [CrossRef] [Green Version]
- Sadeghi, F.; Asgari, M.; Matloubi, M.; Ranjbar, M.; Karkhaneh Yousefi, N.; Azari, T.; Zaki-Dizaji, M. Molecular contribution of BRCA1 and BRCA2 to genome instability in breast cancer patients: Review of radiosensitivity assays. Biol. Proced. Online 2020, 22, 23. [Google Scholar] [CrossRef]
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.K.; Choi, Y.L.; Kwon, M.; Park, P.J. Mechanisms and Consequences of Cancer Genome Instability: Lessons from Genome Sequencing Studies. Annu. Rev. Pathol. 2016, 11, 283–312. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Pao, G.M.; Huynh, A.M.; Suh, H.; Tonnu, N.; Nederlof, P.M.; Gage, F.H.; Verma, I.M. BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature 2011, 477, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Nussenzweig, A. Thwarting endogenous stress: BRCA protects against aldehyde toxicity. EMBO Mol. Med. 2017, 9, 1331–1333. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.L.W.; Chadha, S.; Liu, Y.; Gabasova, E.; Perera, D.; Ahmed, K.; Constantinou, S.; Renaudin, X.; Lee, M.; Aebersold, R.; et al. A Class of Environmental and Endogenous Toxins Induces BRCA2 Haploinsufficiency and Genome Instability. Cell 2017, 169, 1105–1118.e1115. [Google Scholar] [CrossRef] [Green Version]
- Tacconi, E.M.; Lai, X.; Folio, C.; Porru, M.; Zonderland, G.; Badie, S.; Michl, J.; Sechi, I.; Rogier, M.; Matía García, V.; et al. BRCA1 and BRCA2 tumor suppressors protect against endogenous acetaldehyde toxicity. EMBO Mol. Med. 2017, 9, 1398–1414. [Google Scholar] [CrossRef]
- Simhadri, S.; Vincelli, G.; Huo, Y.; Misenko, S.; Foo, T.K.; Ahlskog, J.; Sørensen, C.S.; Oakley, G.G.; Ganesan, S.; Bunting, S.F.; et al. PALB2 connects BRCA1 and BRCA2 in the G2/M checkpoint response. Oncogene 2019, 38, 1585–1596. [Google Scholar] [CrossRef]
- Yasuhara, T.; Kato, R.; Hagiwara, Y.; Shiotani, B.; Yamauchi, M.; Nakada, S.; Shibata, A.; Miyagawa, K. Human Rad52 Promotes XPG-Mediated R-loop Processing to Initiate Transcription-Associated Homologous Recombination Repair. Cell 2018, 175, 558–570.e511. [Google Scholar] [CrossRef] [Green Version]
- Venkitaraman, A.R. How do mutations affecting the breast cancer genes BRCA1 and BRCA2 cause cancer susceptibility? DNA Repair 2019, 81, 102668. [Google Scholar] [CrossRef]
- Zhang, X.; Chiang, H.C.; Wang, Y.; Zhang, C.; Smith, S.; Zhao, X.; Nair, S.J.; Michalek, J.; Jatoi, I.; Lautner, M.; et al. Attenuation of RNA polymerase II pausing mitigates BRCA1-associated R-loop accumulation and tumorigenesis. Nat. Commun. 2017, 8, 15908. [Google Scholar] [CrossRef]
- Allred, D.C.; Brown, P.; Medina, D. The origins of estrogen receptor alpha-positive and estrogen receptor alpha-negative human breast cancer. Breast Cancer Res. 2004, 6, 240–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.H.; Ding, D.C.; Chu, T.Y. Estradiol and Progesterone Induced Differentiation and Increased Stemness Gene Expression of Human Fallopian Tube Epithelial Cells. J. Cancer 2019, 10, 3028–3036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, S.; Jiang, G.; Cao, L.; Huang, J. Replication Fork Reversal and Protection. Front. Cell Dev. Biol. 2021, 9, 670392. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, K.; Miyabe, I.; Schalbetter, S.A.; Carr, A.M.; Murray, J.M. Recombination-restarted replication makes inverted chromosome fusions at inverted repeats. Nature 2013, 493, 246–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daza-Martin, M.; Starowicz, K.; Jamshad, M.; Tye, S.; Ronson, G.E.; MacKay, H.L.; Chauhan, A.S.; Walker, A.K.; Stone, H.R.; Beesley, J.F.J.; et al. Isomerization of BRCA1-BARD1 promotes replication fork protection. Nature 2019, 571, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Sasanuma, H.; Tsuda, M.; Morimoto, S.; Saha, L.K.; Rahman, M.M.; Kiyooka, Y.; Fujiike, H.; Cherniack, A.D.; Itou, J.; Callen Moreu, E.; et al. BRCA1 ensures genome integrity by eliminating estrogen-induced pathological topoisomerase II-DNA complexes. Proc. Natl. Acad. Sci. USA 2018, 115, E10642–E10651. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Yang, L.; Tanasa, B.; Hutt, K.; Ju, B.G.; Ohgi, K.; Zhang, J.; Rose, D.W.; Fu, X.D.; Glass, C.K.; et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell 2009, 139, 1069–1083. [Google Scholar] [CrossRef] [Green Version]
- Chiang, H.C.; Zhang, X.; Li, J.; Zhao, X.; Chen, J.; Wang, H.T.; Jatoi, I.; Brenner, A.; Hu, Y.; Li, R. BRCA1-associated R-loop affects transcription and differentiation in breast luminal epithelial cells. Nucleic Acids Res. 2019, 47, 5086–5099. [Google Scholar] [CrossRef] [Green Version]
- Prat, A.; Perou, C.M. Mammary development meets cancer genomics. Nat. Med. 2009, 15, 842–844. [Google Scholar] [CrossRef]
- Chiarelli, A.M.; Blackmore, K.M.; Muradali, D.; Done, S.J.; Majpruz, V.; Weerasinghe, A.; Mirea, L.; Eisen, A.; Rabeneck, L.; Warner, E. Performance Measures of Magnetic Resonance Imaging Plus Mammography in the High Risk Ontario Breast Screening Program. J. Natl. Cancer Inst. 2020, 112, 136–144. [Google Scholar] [CrossRef]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konstantinopoulos, P.A.; Ceccaldi, R.; Shapiro, G.I.; D’Andrea, A.D. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov. 2015, 5, 1137–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirasawa, A.; Imoto, I.; Naruto, T.; Akahane, T.; Yamagami, W.; Nomura, H.; Masuda, K.; Susumu, N.; Tsuda, H.; Aoki, D. Prevalence of pathogenic germline variants detected by multigene sequencing in unselected Japanese patients with ovarian cancer. Oncotarget 2017, 8, 112258–112267. [Google Scholar] [CrossRef] [Green Version]
- Nyberg, T.; Tischkowitz, M.; Antoniou, A.C. BRCA1 and BRCA2 pathogenic variants and prostate cancer risk: Systematic review and meta-analysis. Br. J. Cancer 2021, 126, 1067–1081. [Google Scholar] [CrossRef] [PubMed]
- Nyberg, T.; Frost, D.; Barrowdale, D.; Evans, D.G.; Bancroft, E.; Adlard, J.; Ahmed, M.; Barwell, J.; Brady, A.F.; Brewer, C.; et al. Prostate Cancer Risks for Male BRCA1 and BRCA2 Mutation Carriers: A Prospective Cohort Study. Eur. Urol. 2020, 77, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, J.; Ragone, A.; Lubinski, J.; Lynch, H.T.; Moller, P.; Ghadirian, P.; Foulkes, W.D.; Armel, S.; Eisen, A.; Neuhausen, S.L.; et al. The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. Br. J. Cancer 2012, 107, 2005–2009. [Google Scholar] [CrossRef]
- Risch, H.A.; McLaughlin, J.R.; Cole, D.E.; Rosen, B.; Bradley, L.; Fan, I.; Tang, J.; Li, S.; Zhang, S.; Shaw, P.A.; et al. Population BRCA1 and BRCA2 mutation frequencies and cancer penetrances: A kin-cohort study in Ontario, Canada. J. Natl. Cancer Inst. 2006, 98, 1694–1706. [Google Scholar] [CrossRef]
- Canto, M.I.; Almario, J.A.; Schulick, R.D.; Yeo, C.J.; Klein, A.; Blackford, A.; Shin, E.J.; Sanyal, A.; Yenokyan, G.; Lennon, A.M.; et al. Risk of Neoplastic Progression in Individuals at High Risk for Pancreatic Cancer Undergoing Long-term Surveillance. Gastroenterology 2018, 155, 740–751.e742. [Google Scholar] [CrossRef] [Green Version]
- Fiesco-Roa, M.O.; Giri, N.; McReynolds, L.J.; Best, A.F.; Alter, B.P. Genotype-phenotype associations in Fanconi anemia: A literature review. Blood Rev. 2019, 37, 100589. [Google Scholar] [CrossRef]
- Li, X.; You, R.; Wang, X.; Liu, C.; Xu, Z.; Zhou, J.; Yu, B.; Xu, T.; Cai, H.; Zou, Q. Effectiveness of Prophylactic Surgeries in BRCA1 or BRCA2 Mutation Carriers: A Meta-analysis and Systematic Review. Clin. Cancer Res. 2016, 22, 3971–3981. [Google Scholar] [CrossRef] [Green Version]
- Evans, D.G.; Ingham, S.L.; Baildam, A.; Ross, G.L.; Lalloo, F.; Buchan, I.; Howell, A. Contralateral mastectomy improves survival in women with BRCA1/2-associated breast cancer. Breast Cancer Res. Treat. 2013, 140, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Heemskerk-Gerritsen, B.A.; Rookus, M.A.; Aalfs, C.M.; Ausems, M.G.; Collée, J.M.; Jansen, L.; Kets, C.M.; Keymeulen, K.B.; Koppert, L.B.; Meijers-Heijboer, H.E.; et al. Improved overall survival after contralateral risk-reducing mastectomy in BRCA1/2 mutation carriers with a history of unilateral breast cancer: A prospective analysis. Int. J. Cancer 2015, 136, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Heemskerk-Gerritsen, B.A.; Menke-Pluijmers, M.B.; Jager, A.; Tilanus-Linthorst, M.M.; Koppert, L.B.; Obdeijn, I.M.; van Deurzen, C.H.; Collée, J.M.; Seynaeve, C.; Hooning, M.J. Substantial breast cancer risk reduction and potential survival benefit after bilateral mastectomy when compared with surveillance in healthy BRCA1 and BRCA2 mutation carriers: A prospective analysis. Ann. Oncol. 2013, 24, 2029–2035. [Google Scholar] [CrossRef] [PubMed]
- Ingham, S.L.; Sperrin, M.; Baildam, A.; Ross, G.L.; Clayton, R.; Lalloo, F.; Buchan, I.; Howell, A.; Evans, D.G. Risk-reducing surgery increases survival in BRCA1/2 mutation carriers unaffected at time of family referral. Breast Cancer Res. Treat. 2013, 142, 611–618. [Google Scholar] [CrossRef]
- Eleje, G.U.; Eke, A.C.; Ezebialu, I.U.; Ikechebelu, J.I.; Ugwu, E.O.; Okonkwo, O.O. Risk-reducing bilateral salpingo-oophorectomy in women with BRCA1 or BRCA2 mutations. Cochrane Database Syst. Rev. 2018, 8, Cd012464. [Google Scholar] [CrossRef]
- Xu, L.; Zhao, Y.; Chen, Z.; Wang, Y.; Chen, L.; Wang, S. Tamoxifen and risk of contralateral breast cancer among women with inherited mutations in BRCA1 and BRCA2: A meta-analysis. Breast Cancer 2015, 22, 327–334. [Google Scholar] [CrossRef]
- Nolan, E.; Vaillant, F.; Branstetter, D.; Pal, B.; Giner, G.; Whitehead, L.; Lok, S.W.; Mann, G.B.; Rohrbach, K.; Huang, L.Y.; et al. RANK ligand as a potential target for breast cancer prevention in BRCA1-mutation carriers. Nat. Med. 2016, 22, 933–939. [Google Scholar] [CrossRef]
- Kotsopoulos, J.; Singer, C.; Narod, S.A. Can we prevent BRCA1-associated breast cancer by RANKL inhibition? Breast Cancer Res. Treat. 2017, 161, 11–16. [Google Scholar] [CrossRef]
- Fedier, A.; Steiner, R.A.; Schwarz, V.A.; Lenherr, L.; Haller, U.; Fink, D. The effect of loss of Brca1 on the sensitivity to anticancer agents in p53-deficient cells. Int. J. Oncol. 2003, 22, 1169–1173. [Google Scholar] [CrossRef]
- Sylvain, V.; Lafarge, S.; Bignon, Y.J. Dominant-negative activity of a Brca1 truncation mutant: Effects on proliferation, tumorigenicity in vivo, and chemosensitivity in a mouse ovarian cancer cell line. Int. J. Oncol. 2002, 20, 845–853. [Google Scholar] [CrossRef]
- Nakamura, S.; Aoki, D.; Miki, Y. Hereditary Breast and Ovarian Cancer; Springer: Singapore, 2021; ISBN 978-981-16-4521-1. [Google Scholar]
- Lafarge, S.; Sylvain, V.; Ferrara, M.; Bignon, Y.J. Inhibition of BRCA1 leads to increased chemoresistance to microtubule-interfering agents, an effect that involves the JNK pathway. Oncogene 2001, 20, 6597–6606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tassone, P.; Tagliaferri, P.; Perricelli, A.; Blotta, S.; Quaresima, B.; Martelli, M.L.; Goel, A.; Barbieri, V.; Costanzo, F.; Boland, C.R.; et al. BRCA1 expression modulates chemosensitivity of BRCA1-defective HCC1937 human breast cancer cells. Br. J. Cancer 2003, 88, 1285–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arun, B.; Bayraktar, S.; Liu, D.D.; Gutierrez Barrera, A.M.; Atchley, D.; Pusztai, L.; Litton, J.K.; Valero, V.; Meric-Bernstam, F.; Hortobagyi, G.N.; et al. Response to neoadjuvant systemic therapy for breast cancer in BRCA mutation carriers and noncarriers: A single-institution experience. J. Clin. Oncol. 2011, 29, 3739–3746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Abo, M.; Dejsuphong, D.; Hirota, K.; Yonetani, Y.; Yamazoe, M.; Kurumizaka, H.; Takeda, S. Compensatory functions and interdependency of the DNA-binding domain of BRCA2 with the BRCA1-PALB2-BRCA2 complex. Cancer Res. 2014, 74, 797–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lord, C.J.; Garrett, M.D.; Ashworth, A. Targeting the double-strand DNA break repair pathway as a therapeutic strategy. Clin. Cancer Res. 2006, 12, 4463–4468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrski, T.; Gronwald, J.; Huzarski, T.; Grzybowska, E.; Budryk, M.; Stawicka, M.; Mierzwa, T.; Szwiec, M.; Wisniowski, R.; Siolek, M.; et al. Pathologic complete response rates in young women with BRCA1-positive breast cancers after neoadjuvant chemotherapy. J. Clin. Oncol. 2010, 28, 375–379. [Google Scholar] [CrossRef]
- Tung, N.; Arun, B.; Hacker, M.R.; Hofstatter, E.; Toppmeyer, D.L.; Isakoff, S.J.; Borges, V.; Legare, R.D.; Isaacs, C.; Wolff, A.C.; et al. TBCRC 031: Randomized Phase II Study of Neoadjuvant Cisplatin Versus Doxorubicin-Cyclophosphamide in Germline BRCA Carriers with HER2-Negative Breast Cancer (the INFORM trial). J. Clin. Oncol. 2020, 38, 1539–1548. [Google Scholar] [CrossRef]
- Caramelo, O.; Silva, C.; Caramelo, F.; Frutuoso, C.; Almeida-Santos, T. The effect of neoadjuvant platinum-based chemotherapy in BRCA mutated triple negative breast cancers-systematic review and meta-analysis. Hered. Cancer Clin. Pract. 2019, 17, 11. [Google Scholar] [CrossRef] [Green Version]
- Tutt, A.; Tovey, H.; Cheang, M.C.U.; Kernaghan, S.; Kilburn, L.; Gazinska, P.; Owen, J.; Abraham, J.; Barrett, S.; Barrett-Lee, P.; et al. Carboplatin in BRCA1/2-mutated and triple-negative breast cancer BRCAness subgroups: The TNT Trial. Nat. Med. 2018, 24, 628–637. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Lin, Y.; Sun, X.J.; Wang, B.Y.; Wang, Z.H.; Luo, J.F.; Wang, L.P.; Zhang, S.; Cao, J.; Tao, Z.H.; et al. Biomarker assessment of the CBCSG006 trial: A randomized phase III trial of cisplatin plus gemcitabine compared with paclitaxel plus gemcitabine as first-line therapy for patients with metastatic triple-negative breast cancer. Ann. Oncol. 2018, 29, 1741–1747. [Google Scholar] [CrossRef]
- Safonov, A.; Bandlamudi, C.; de Lara, P.; Ferraro, E.; Derakhshan, F.; Will, M.; Donoghue, M.; Selenica, P.; Drago, J.; Rosen, E.; et al. BRCA2 Linked to Inferior Outcomes of Treatment with CDK4/6 Inhibitors Plus Endocrine Therapy. In Proceedings of the SABCS 2021, San Antonio, TX, USA, 13 December 2021; Volume Abstract GS4-08. [Google Scholar]
- Bruno, L.; Ostinelli, A.; Waisberg, F.; Enrico, D.; Ponce, C.; Rivero, S.; Blanco, A.; Zarba, M.; Loza, M.; Fabiano, V.; et al. Cyclin-Dependent Kinase 4/6 Inhibitor Outcomes in Patients With Advanced Breast Cancer Carrying Germline Pathogenic Variants in DNA Repair-Related Genes. JCO Precis. Oncol. 2022, 6, e2100140. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Tutt, A.N.J.; Garber, J.E.; Kaufman, B.; Viale, G.; Fumagalli, D.; Rastogi, P.; Gelber, R.D.; de Azambuja, E.; Fielding, A.; Balmaña, J.; et al. Adjuvant Olaparib for Patients with BRCA1- or BRCA2-Mutated Breast Cancer. N. Engl. J. Med. 2021, 384, 2394–2405. [Google Scholar] [CrossRef] [PubMed]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef]
- Loibl, S.; O’Shaughnessy, J.; Untch, M.; Sikov, W.M.; Rugo, H.S.; McKee, M.D.; Huober, J.; Golshan, M.; von Minckwitz, G.; Maag, D.; et al. Addition of the PARP inhibitor veliparib plus carboplatin or carboplatin alone to standard neoadjuvant chemotherapy in triple-negative breast cancer (BrighTNess): A randomised, phase 3 trial. Lancet Oncol. 2018, 19, 497–509. [Google Scholar] [CrossRef]
- Fasching, P.A.; Link, T.; Hauke, J.; Seither, F.; Jackisch, C.; Klare, P.; Schmatloch, S.; Hanusch, C.; Huober, J.; Stefek, A.; et al. Neoadjuvant paclitaxel/olaparib in comparison to paclitaxel/carboplatinum in patients with HER2-negative breast cancer and homologous recombination deficiency (GeparOLA study). Ann. Oncol. 2021, 32, 49–57. [Google Scholar] [CrossRef]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet 2010, 376, 235–244. [Google Scholar] [CrossRef]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Sandhu, S.K.; Schelman, W.R.; Wilding, G.; Moreno, V.; Baird, R.D.; Miranda, S.; Hylands, L.; Riisnaes, R.; Forster, M.; Omlin, A.; et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: A phase 1 dose-escalation trial. Lancet Oncol. 2013, 14, 882–892. [Google Scholar] [CrossRef]
- Drew, Y.; Ledermann, J.; Hall, G.; Rea, D.; Glasspool, R.; Highley, M.; Jayson, G.; Sludden, J.; Murray, J.; Jamieson, D.; et al. Phase 2 multicentre trial investigating intermittent and continuous dosing schedules of the poly(ADP-ribose) polymerase inhibitor rucaparib in germline BRCA mutation carriers with advanced ovarian and breast cancer. Br. J. Cancer 2016, 114, 723–730. [Google Scholar] [CrossRef] [Green Version]
- Wilson, R.H.; Evans, T.J.; Middleton, M.R.; Molife, L.R.; Spicer, J.; Dieras, V.; Roxburgh, P.; Giordano, H.; Jaw-Tsai, S.; Goble, S.; et al. A phase I study of intravenous and oral rucaparib in combination with chemotherapy in patients with advanced solid tumours. Br. J. Cancer 2017, 116, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Kalra, M.; Tong, Y.; Jones, D.R.; Walsh, T.; Danso, M.A.; Ma, C.X.; Silverman, P.; King, M.C.; Badve, S.S.; Perkins, S.M.; et al. Cisplatin +/− rucaparib after preoperative chemotherapy in patients with triple-negative or BRCA mutated breast cancer. NPJ Breast Cancer 2021, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Litton, J.K.; Scoggins, M.E.; Hess, K.R.; Adrada, B.E.; Murthy, R.K.; Damodaran, S.; DeSnyder, S.M.; Brewster, A.M.; Barcenas, C.H.; Valero, V.; et al. Neoadjuvant Talazoparib for Patients with Operable Breast Cancer With a Germline BRCA Pathogenic Variant. J. Clin. Oncol. 2020, 38, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Rugo, H.S.; Olopade, O.I.; DeMichele, A.; Yau, C.; van ’t Veer, L.J.; Buxton, M.B.; Hogarth, M.; Hylton, N.M.; Paoloni, M.; Perlmutter, J.; et al. Adaptive Randomization of Veliparib-Carboplatin Treatment in Breast Cancer. N. Engl. J. Med. 2016, 375, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Han, H.S.; Diéras, V.; Robson, M.; Palácová, M.; Marcom, P.K.; Jager, A.; Bondarenko, I.; Citrin, D.; Campone, M.; Telli, M.L.; et al. Veliparib with temozolomide or carboplatin/paclitaxel versus placebo with carboplatin/paclitaxel in patients with BRCA1/2 locally recurrent/metastatic breast cancer: Randomized phase II study. Ann. Oncol. 2018, 29, 154–161. [Google Scholar] [CrossRef]
- Xia, B.; Sheng, Q.; Nakanishi, K.; Ohashi, A.; Wu, J.; Christ, N.; Liu, X.; Jasin, M.; Couch, F.J.; Livingston, D.M. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol. Cell 2006, 22, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Erkko, H.; Xia, B.; Nikkilä, J.; Schleutker, J.; Syrjäkoski, K.; Mannermaa, A.; Kallioniemi, A.; Pylkäs, K.; Karppinen, S.M.; Rapakko, K.; et al. A recurrent mutation in PALB2 in Finnish cancer families. Nature 2007, 446, 316–319. [Google Scholar] [CrossRef]
- Nepomuceno, T.C.; De Gregoriis, G.; de Oliveira, F.M.B.; Suarez-Kurtz, G.; Monteiro, A.N.; Carvalho, M.A. The Role of PALB2 in the DNA Damage Response and Cancer Predisposition. Int. J. Mol. Sci. 2017, 18, 1886. [Google Scholar] [CrossRef] [Green Version]
- Ducy, M.; Sesma-Sanz, L.; Guitton-Sert, L.; Lashgari, A.; Gao, Y.; Brahiti, N.; Rodrigue, A.; Margaillan, G.; Caron, M.C.; Côté, J.; et al. The Tumor Suppressor PALB2: Inside Out. Trends Biochem. Sci. 2019, 44, 226–240. [Google Scholar] [CrossRef]
- Toh, M.R.; Low, C.E.; Chong, S.T.; Chan, S.H.; Ishak, N.D.B.; Courtney, E.; Kolinjivadi, A.M.; Rodrigue, A.; Masson, J.Y.; Ngeow, J. Missense PALB2 germline variant disrupts nuclear localization of PALB2 in a patient with breast cancer. Fam. Cancer 2020, 19, 123–131. [Google Scholar] [CrossRef]
- Antoniou, A.C.; Casadei, S.; Heikkinen, T.; Barrowdale, D.; Pylkäs, K.; Roberts, J.; Lee, A.; Subramanian, D.; De Leeneer, K.; Fostira, F.; et al. Breast-cancer risk in families with mutations in PALB2. N. Engl. J. Med. 2014, 371, 497–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lilyquist, J.; LaDuca, H.; Polley, E.; Davis, B.T.; Shimelis, H.; Hu, C.; Hart, S.N.; Dolinsky, J.S.; Couch, F.J.; Goldgar, D.E. Frequency of mutations in a large series of clinically ascertained ovarian cancer cases tested on multi-gene panels compared to reference controls. Gynecol. Oncol. 2017, 147, 375–380. [Google Scholar] [CrossRef]
- Yang, X.; Leslie, G.; Doroszuk, A.; Schneider, S.; Allen, J.; Decker, B.; Dunning, A.M.; Redman, J.; Scarth, J.; Plaskocinska, I.; et al. Cancer Risks Associated with Germline PALB2 Pathogenic Variants: An International Study of 524 Families. J. Clin. Oncol. 2020, 38, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Villarroel, M.C.; Rajeshkumar, N.V.; Garrido-Laguna, I.; De Jesus-Acosta, A.; Jones, S.; Maitra, A.; Hruban, R.H.; Eshleman, J.R.; Klein, A.; Laheru, D.; et al. Personalizing cancer treatment in the age of global genomic analyses: PALB2 gene mutations and the response to DNA damaging agents in pancreatic cancer. Mol. Cancer Ther. 2011, 10, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pennington, K.P.; Walsh, T.; Harrell, M.I.; Lee, M.K.; Pennil, C.C.; Rendi, M.H.; Thornton, A.; Norquist, B.M.; Casadei, S.; Nord, A.S.; et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin. Cancer Res. 2014, 20, 764–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaac, D.; Karapetyan, L.; Tamkus, D. Association of Germline PALB2 Mutation and Response to Platinum-Based Chemotherapy in Metastatic Breast Cancer: A Case Series. JCO Precis. Oncol. 2018, 2, 1–5. [Google Scholar] [CrossRef]
- Tung, N.M.; Robson, M.E.; Ventz, S.; Santa-Maria, C.A.; Nanda, R.; Marcom, P.K.; Shah, P.D.; Ballinger, T.J.; Yang, E.S.; Vinayak, S.; et al. TBCRC 048: Phase II Study of Olaparib for Metastatic Breast Cancer and Mutations in Homologous Recombination-Related Genes. J. Clin. Oncol. 2020, 38, 4274–4282. [Google Scholar] [CrossRef]
- Gruber, J.J.; Afghahi, A.; Hatton, A.; Scott, D.; McMillan, A.; Ford, J.M.; Telli, M.L. Talazoparib beyond BRCA: A phase II trial of talazoparib monotherapy in BRCA1 and BRCA2 wild-type patients with advanced HER2-negative breast cancer or other solid tumors with a mutation in homologous recombination (HR) pathway genes. J. Clin. Oncol. 2019, 37, 3006. [Google Scholar] [CrossRef]
- Sun, Y.; McCorvie, T.J.; Yates, L.A.; Zhang, X. Structural basis of homologous recombination. Cell. Mol. Life Sci. 2020, 77, 3–18. [Google Scholar] [CrossRef] [Green Version]
- Savitsky, K.; Bar-Shira, A.; Gilad, S.; Rotman, G.; Ziv, Y.; Vanagaite, L.; Tagle, D.A.; Smith, S.; Uziel, T.; Sfez, S.; et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 1995, 268, 1749–1753. [Google Scholar] [CrossRef]
- Athma, P.; Rappaport, R.; Swift, M. Molecular genotyping shows that ataxia-telangiectasia heterozygotes are predisposed to breast cancer. Cancer Genet. Cytogenet. 1996, 92, 130–134. [Google Scholar] [CrossRef]
- Pandita, T.K.; Lieberman, H.B.; Lim, D.S.; Dhar, S.; Zheng, W.; Taya, Y.; Kastan, M.B. Ionizing radiation activates the ATM kinase throughout the cell cycle. Oncogene 2000, 19, 1386–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marabelli, M.; Cheng, S.C.; Parmigiani, G. Penetrance of ATM Gene Mutations in Breast Cancer: A Meta-Analysis of Different Measures of Risk. Genet. Epidemiol. 2016, 40, 425–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Os, N.J.; Roeleveld, N.; Weemaes, C.M.; Jongmans, M.C.; Janssens, G.O.; Taylor, A.M.; Hoogerbrugge, N.; Willemsen, M.A. Health risks for ataxia-telangiectasia mutated heterozygotes: A systematic review, meta-analysis and evidence-based guideline. Clin. Genet. 2016, 90, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.J.; Bernhisel, R.; Hughes, E.; Larson, K.; Rosenthal, E.T.; Singh, N.A.; Lancaster, J.M.; Kurian, A.W. Germline Pathogenic Variants in the Ataxia Telangiectasia Mutated (ATM) Gene are Associated with High and Moderate Risks for Multiple Cancers. Cancer Prev. Res. 2021, 14, 433–440. [Google Scholar] [CrossRef]
- Putti, S.; Giovinazzo, A.; Merolle, M.; Falchetti, M.L.; Pellegrini, M. ATM Kinase Dead: From Ataxia Telangiectasia Syndrome to Cancer. Cancers 2021, 13, 5498. [Google Scholar] [CrossRef]
- Aguirre, A.J.; Nowak, J.A.; Camarda, N.D.; Moffitt, R.A.; Ghazani, A.A.; Hazar-Rethinam, M.; Raghavan, S.; Kim, J.; Brais, L.K.; Ragon, D.; et al. Real-time Genomic Characterization of Advanced Pancreatic Cancer to Enable Precision Medicine. Cancer Discov. 2018, 8, 1096–1111. [Google Scholar] [CrossRef] [Green Version]
- Haricharan, S.; Punturi, N.; Singh, P.; Holloway, K.R.; Anurag, M.; Schmelz, J.; Schmidt, C.; Lei, J.T.; Suman, V.; Hunt, K.; et al. Loss of MutL Disrupts CHK2-Dependent Cell-Cycle Control through CDK4/6 to Promote Intrinsic Endocrine Therapy Resistance in Primary Breast Cancer. Cancer Discov. 2017, 7, 1168–1183. [Google Scholar] [CrossRef] [Green Version]
- Anurag, M.; Punturi, N.; Hoog, J.; Bainbridge, M.N.; Ellis, M.J.; Haricharan, S. Comprehensive Profiling of DNA Repair Defects in Breast Cancer Identifies a Novel Class of Endocrine Therapy Resistance Drivers. Clin. Cancer Res. 2018, 24, 4887–4899. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zou, W.; Zhang, J.; Zhang, Y.; Xu, Q.; Li, S.; Chen, C. Mechanisms of CDK4/6 Inhibitor Resistance in Luminal Breast Cancer. Front. Pharmacol. 2020, 11, 580251. [Google Scholar] [CrossRef]
- Bang, Y.J.; Im, S.A.; Lee, K.W.; Cho, J.Y.; Song, E.K.; Lee, K.H.; Kim, Y.H.; Park, J.O.; Chun, H.G.; Zang, D.Y.; et al. Randomized, Double-Blind Phase II Trial with Prospective Classification by ATM Protein Level to Evaluate the Efficacy and Tolerability of Olaparib Plus Paclitaxel in Patients With Recurrent or Metastatic Gastric Cancer. J. Clin. Oncol. 2015, 33, 3858–3865. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.J.; Xu, R.H.; Chin, K.; Lee, K.W.; Park, S.H.; Rha, S.Y.; Shen, L.; Qin, S.; Xu, N.; Im, S.A.; et al. Olaparib in combination with paclitaxel in patients with advanced gastric cancer who have progressed following first-line therapy (GOLD): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1637–1651. [Google Scholar] [CrossRef]
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Survival with Olaparib in Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 383, 2345–2357. [Google Scholar] [CrossRef]
- Meyer, A.; John, E.; Dörk, T.; Sohn, C.; Karstens, J.H.; Bremer, M. Breast cancer in female carriers of ATM gene alterations: Outcome of adjuvant radiotherapy. Radiother. Oncol. 2004, 72, 319–323. [Google Scholar] [CrossRef]
- McDuff, S.G.R.; Bellon, J.R.; Shannon, K.M.; Gadd, M.A.; Dunn, S.; Rosenstein, B.S.; Ho, A.Y. ATM Variants in Breast Cancer: Implications for Breast Radiation Therapy Treatment Recommendations. Int. J. Radiat. Oncol. Biol. Phys. 2021, 110, 1373–1382. [Google Scholar] [CrossRef]
- Lowry, K.P.; Geuzinge, H.A.; Stout, N.K.; Alagoz, O.; Hampton, J.; Kerlikowske, K.; de Koning, H.J.; Miglioretti, D.L.; van Ravesteyn, N.T.; Schechter, C.; et al. Breast Cancer Screening Strategies for Women With ATM, CHEK2, and PALB2 Pathogenic Variants: A Comparative Modeling Analysis. JAMA Oncol. 2022, 8, 587–596. [Google Scholar] [CrossRef]
- Narayan, A.K.; Visvanathan, K.; Harvey, S.C. Comparative effectiveness of breast MRI and mammography in screening young women with elevated risk of developing breast cancer: A retrospective cohort study. Breast Cancer Res. Treat. 2016, 158, 583–589. [Google Scholar] [CrossRef]
- Bell, D.W.; Varley, J.M.; Szydlo, T.E.; Kang, D.H.; Wahrer, D.C.; Shannon, K.E.; Lubratovich, M.; Verselis, S.J.; Isselbacher, K.J.; Fraumeni, J.F.; et al. Heterozygous germ line hCHK2 mutations in Li-Fraumeni syndrome. Science 1999, 286, 2528–2531. [Google Scholar] [CrossRef]
- Apostolou, P.; Papasotiriou, I. Current perspectives on CHEK2 mutations in breast cancer. Breast Cancer Targets Ther. 2017, 9, 331–335. [Google Scholar] [CrossRef] [Green Version]
- Dufault, M.R.; Betz, B.; Wappenschmidt, B.; Hofmann, W.; Bandick, K.; Golla, A.; Pietschmann, A.; Nestle-Krämling, C.; Rhiem, K.; Hüttner, C.; et al. Limited relevance of the CHEK2 gene in hereditary breast cancer. Int. J. Cancer 2004, 110, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Meijers-Heijboer, H.; van den Ouweland, A.; Klijn, J.; Wasielewski, M.; de Snoo, A.; Oldenburg, R.; Hollestelle, A.; Houben, M.; Crepin, E.; van Veghel-Plandsoen, M.; et al. Low-penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat. Genet. 2002, 31, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Toss, A.; Tenedini, E.; Piombino, C.; Venturelli, M.; Marchi, I.; Gasparini, E.; Barbieri, E.; Razzaboni, E.; Domati, F.; Caggia, F.; et al. Clinicopathologic Profile of Breast Cancer in Germline ATM and CHEK2 Mutation Carriers. Genes 2021, 12, 616. [Google Scholar] [CrossRef] [PubMed]
- Peleg Hasson, S.; Menes, T.; Sonnenblick, A. Comparison of Patient Susceptibility Genes Across Breast Cancer: Implications for Prognosis and Therapeutic Outcomes. Pharmgenom. Pers. Med. 2020, 13, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Knappskog, S.; Chrisanthar, R.; Løkkevik, E.; Anker, G.; Østenstad, B.; Lundgren, S.; Risberg, T.; Mjaaland, I.; Leirvaag, B.; Miletic, H.; et al. Low expression levels of ATM may substitute for CHEK2/TP53 mutations predicting resistance towards anthracycline and mitomycin chemotherapy in breast cancer. Breast Cancer Res. 2012, 14, R47. [Google Scholar] [CrossRef] [Green Version]
- Kriege, M.; Jager, A.; Hollestelle, A.; Berns, E.M.; Blom, J.; Meijer-van Gelder, M.E.; Sieuwerts, A.M.; van den Ouweland, A.; Collée, J.M.; Kroep, J.R.; et al. Sensitivity to systemic therapy for metastatic breast cancer in CHEK2 1100delC mutation carriers. J. Cancer Res. Clin. Oncol. 2015, 141, 1879–1887. [Google Scholar] [CrossRef] [Green Version]
- Cimmino, F.; Formicola, D.; Capasso, M. Dualistic Role of BARD1 in Cancer. Genes 2017, 8, 375. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.C.; Wang, Z.W.; Tsan, J.T.; Spillman, M.A.; Phung, A.; Xu, X.L.; Yang, M.C.; Hwang, L.Y.; Bowcock, A.M.; Baer, R. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat. Genet. 1996, 14, 430–440. [Google Scholar] [CrossRef]
- Alenezi, W.M.; Fierheller, C.T.; Recio, N.; Tonin, P.N. Literature Review of BARD1 as a Cancer Predisposing Gene with a Focus on Breast and Ovarian Cancers. Genes 2020, 11, 856. [Google Scholar] [CrossRef]
- Baumann, P.; West, S.C. Role of the human RAD51 protein in homologous recombination and double-stranded-break repair. Trends Biochem. Sci. 1998, 23, 247–251. [Google Scholar] [CrossRef]
- Ochiai, K.; Ishiguro-Oonuma, T.; Yoshikawa, Y.; Udagawa, C.; Kato, Y.; Watanabe, M.; Bonkobara, M.; Morimatsu, M.; Omi, T. Polymorphisms of canine BRCA2 BRC repeats affecting interaction with RAD51. Biomed. Res. 2015, 36, 155–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meindl, A.; Hellebrand, H.; Wiek, C.; Erven, V.; Wappenschmidt, B.; Niederacher, D.; Freund, M.; Lichtner, P.; Hartmann, L.; Schaal, H.; et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat. Genet. 2010, 42, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Dicks, E.; Ramus, S.J.; Tyrer, J.P.; Intermaggio, M.P.; Hayward, J.; Edlund, C.K.; Conti, D.; Harrington, P.; Fraser, L.; et al. Contribution of Germline Mutations in the RAD51B, RAD51C, and RAD51D Genes to Ovarian Cancer in the Population. J. Clin. Oncol. 2015, 33, 2901–2907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurian, A.W.; Hughes, E.; Handorf, E.A.; Gutin, A.; Allen, B.; Hartman, A.-R.; Hall, M.J. Breast and Ovarian Cancer Penetrance Estimates Derived from Germline Multiple-Gene Sequencing Results in Women. JCO Precis. Oncol. 2017, 2017, 1–12. [Google Scholar] [CrossRef]
- Gallagher, S.; Hughes, E.; Wagner, S.; Tshiaba, P.; Rosenthal, E.; Roa, B.B.; Kurian, A.W.; Domchek, S.M.; Garber, J.; Lancaster, J.; et al. Association of a Polygenic Risk Score with Breast Cancer Among Women Carriers of High- and Moderate-Risk Breast Cancer Genes. JAMA Netw. Open 2020, 3, e208501. [Google Scholar] [CrossRef]






| Susceptibility | Gene | BC Lifetime Risk 1 | Prevalence of GPVs | Odds Ratio (95% CI) | ||||
|---|---|---|---|---|---|---|---|---|
| Women with BC | Controls | All BC | ER-Positive BC | ER-Negative BC | Triple-Negative BC | |||
| High | BRCA1 | >60% | 1.05% 2 | 0.11% 2 | 10.57 (8.02–13.93) 2 | 3.92 (2.82–5.43) 2 | 35.32 (26.21–47.60) 2 | 56.80 (41.18–78.34) 2 |
| 0.85% 3 | 0.11% 3 | 7.62 (5.33–11.27) 3 | 3.39 (2.17–5.45) 3 | 26.33 (17.28–41.52) 3 | 42.88 (26.56–71.25) 3 | |||
| BRCA2 | >60% | 1.54% 2 | 0.26% 2 | 5.85 (4.85–7.06) 2 | 5.69 (4.65–6.96) 2 | 7.53 (5.89–9.62) 2 | 11.19 (8.27–15.16) 2 | |
| 1.29% 3 | 0.24% 3 | 5.23 (4.09–6.77) 3 | 4.66 (3.52–6.23) 3 | 8.89 (6.36–12.47) 3 | 9.70 (5.97–15.47) 3 | |||
| PALB2 | 41–60% | 0.56% 2 | 0.10% 2 | 5.02 (3.73–6.76) 2 | 4.45 (3.23–6.14) 2 | 6.72 (4.54–9.95) 2 | 10.36 (6.42–16.71) 2 | |
| 0.46% 3 | 0.12% 3 | 3.83 (2.68–5.63) 3 | 3.13 (2.02–4.96) 3 | 9.22 (5.63–15.25) 3 | 13.03 (7.08–23.75) 3 | |||
| Moderate | CHEK2 | 15–40% | 1.44% 1 | 0.62% 2 | 2.54 (2.21–2.91) 2 | 2.67 (2.30–3.11) 2 | 1.64 (1.25–2.16) 2 | 1.06 (0.63–1.76) 2 |
| 1.08% 3 | 0.42% 3 | 2.47 (2.02–3.05) 3 | 2.60 (2.05–3.31) 3 | 1.40 (0.83–2.25) 3 | 1.63 (0.72–3.20) 3 | |||
| ATM | 15–40% | 0.60% 2 | 0.29% 2 | 2.10 (1.71–2.57) 2 | 2.33 (1.87–2.91) 2 | 1.01 (0.64–1.59) 2 | 0.91 (0.42–1.95) 2 | |
| 0.78% 3 | 0.41% 3 | 1.82 (1.46–2.27) 3 | 1.96 (1.52–2.53) 3 | 1.04 (0.59–1.72) 3 | 0.50 (0.12–1.36) 3 | |||
| BARD1 | 15–40% | 0.12% 2 | 0.06% 2 | 2.09 (1.35–3.23) 2 | 1.40 (0.81–2.42) 2 | 5.99 (3.51–10.21) 2 | 9.29 (4.58–18.85) 2 | |
| 0.15% 3 | 0.11% 3 | 1.37 (0.87–2.16) 3 | 0.91 (0.49–1.64) 3 | 2.52 (1.18–5.00) 3 | 3.18 (1.16–7.42) 3 | |||
| RAD51C | 15–40% | 0.11% 2 | 0.05% 2 | 1.93 (1.20–3.11) 2 | 1.31 (0.74–2.30) 2 | 3.99 (2.20–7.26) 2 | 5.71 (2.69–12.13) 2 | |
| 0.13% 3 | 0.11% 3 | 1.20 (0.75–1.93) 3 | 0.83 (0.44–1.54) 3 | 2.19 (0.97–4.49) 3 | NA3 | |||
| RAD51D | 15–40% | 0.10% 2 | 0.04% 2 | 1.80 (1.11–2.93) 2 | 1.52 (0.87–2.65) 2 | 2.92 (1.47–5.78) 2 | 6.01 (2.73–13.24) 2 | |
| 0.08% 3 | 0.04% 3 | 1.72 (0.88–3.51) 3 | 1.61 (0.71–3.70) 3 | 3.93 (1.40–10.29) 3 | NA 3 | |||
| Syndrome | TP53 | >60% | 0.01% 2 | 0.003% 2 | 3.06 (0.63–14.91) 2 | 1.95 (0.32–11.82) 2 | 5.42 (0.75–39.24) 2 | NA 2 |
| 0.06% 3 | 0.01% 3 | NA 3 | NA 3 | NA 3 | NA 3 | |||
| PTEN | 40–60% | 0.02% 2 | 0.01% 2 | 2.25 (0.85–6.00) 2 | 2.42 (0.84–6.97) 2 | NA 2 | NA 2 | |
| 0.02% 3 | 0.01% 3 | NA3 | NA3 | NA 3 | NA 3 | |||
| STK11 | 40–60% | 0.01% 2 | 0.009% 2 | 1.60 (0.48–5.28) 2 | 1.56 (0.35–7.03) 2 | NA 2 | NA 2 | |
| CDH1 | 41–60% | 0.02% 2 | 0.02% 2 | 0.86 (0.37–1.98) 2 | 1.05 (0.42–2.63) 2 | 1.11 (0.24–5.10) 2 | 1.44 (0.18–11.28) 2 | |
| 0.05% 3 | 0.02% 3 | 2.50 (1.01–7.07) 3 | 3.37 (1.24–10.72) 3 | NA 3 | NA 3 | |||
| Susceptibility | Gene | Risk-Reducing Surgery | BC Screening | BC Treatment | Other Cancer Risks | |
|---|---|---|---|---|---|---|
| RRM | RRSO | |||||
| High | BRCA1 | Discuss option of RRM | Recommend RRSO | Age 25 y: annual breast MRI Age 30–75 y: additional mammogram | Platinum agents and PARP inhibitors | Ovary, pancreas, and prostate |
| BRCA2 | Ovary, pancreas, prostate, and melanoma | |||||
| PALB2 | Evidence insufficient, manage based on family history | Age 30 y: annual mammogram and breast MRI | Ovary and pancreas | |||
| Moderate | ATM | Evidence insufficient for RRM; manage based on family history | Age 40 y: annual mammogram and consider breast MRI | Heterozygous ATM GPV should not lead to a recommendation to avoid RT | Ovary and pancreas | |
| CHEK2 | (Insufficient evidence) | Colon | ||||
| BARD1 | (Insufficient evidence) | |||||
| RAD51C | Insufficient data; manage based on family history | Consider RRSO | Insufficient data; managed based on family history | Ovary | ||
| RAD51D | ||||||
| Syndrome | TP53 | Discuss option of RRM | Age 20 y: annual breast MRI Age 30–75 y: additional mammogram | Therapeutic RT for cancer should be avoided when possible; diagnostic radiation should be minimized to the extent feasible without sacrificing accuracy | Adrenocortical gland, central nervous system, bone, and soft tissue | |
| PTEN | Age 30–75 y: annual mammogram and breast MRI | (Insufficient evidence) | Thyroid, kidney, endometrium, and colon | |||
| CDH1 | Age 30 y: annual mammogram and consider breast MRI | Stomach | ||||
| STK11 | Evidence insufficient for RRM; manage based on family history | No established data | Age 30 y: annual mammogram and breast MRI | Colon, stomach, small bowel, pancreas, cervix, uterus, ovary, testis, and lung | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoshimura, A.; Imoto, I.; Iwata, H. Functions of Breast Cancer Predisposition Genes: Implications for Clinical Management. Int. J. Mol. Sci. 2022, 23, 7481. https://doi.org/10.3390/ijms23137481
Yoshimura A, Imoto I, Iwata H. Functions of Breast Cancer Predisposition Genes: Implications for Clinical Management. International Journal of Molecular Sciences. 2022; 23(13):7481. https://doi.org/10.3390/ijms23137481
Chicago/Turabian StyleYoshimura, Akiyo, Issei Imoto, and Hiroji Iwata. 2022. "Functions of Breast Cancer Predisposition Genes: Implications for Clinical Management" International Journal of Molecular Sciences 23, no. 13: 7481. https://doi.org/10.3390/ijms23137481