
Dispirooxindole-β-Lactams: Synthesis via Staudinger Ketene-Imine Cycloaddition and Biological Evaluation


 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis of Dispirooxindole-β-Lactams
2.2. Structural Determination of Compounds 5 and Other Reaction Products
2.3. Biological Investigations of Compounds 5
3. Materials and Methods
3.1. General Information
3.2. General Procedure for thePreparation of 6,6-Dimethyl-5,7-Dioxaspiro [2.5]Octane-4,8-Dione (2)
3.3. General Procedure for Preparation of Isatinimines 4a,b
3.4. General Procedure for Preparation of N-Aryl-2-Oxopyrrolidine-3-Carboxylic Acids 3a–e
3.5. General Procedure for Preparation of Dispirooxindole-β-Lactams 5a–f
3.6. Cell Lines
3.7. In Vitro Survival Assay (MTT Assay)
3.8. Plate Test and Determination of Mechanism of Action
3.9. Determination of Minimal Inhibitory Concentration
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Padma, V.V. An Overview of Targeted Cancer Therapy. Biomedicine 2015, 5, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Sheng, C.; Georg, G.I. Targeting Protein-Protein Interactions by Small Molecules; Springer: Singapore, 2018; ISBN 9789811307737. [Google Scholar]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small Molecules in Targeted Cancer Therapy: Advances, Challenges, and Future Perspectives. Signal Transduct. Target. Ther. 2021, 6, 1–48. [Google Scholar] [CrossRef] [PubMed]
- Mora-Ochomogo, M.; Lohans, C.T. β-Lactam antibiotic targets and resistance mechanisms: From covalent inhibitors to substrates. RSC Med. Chem. 2021, 12, 1623–1639. [Google Scholar] [CrossRef] [PubMed]
- Xing, B.; Rao, J.; Liu, R. Novel Beta-Lactam Antibiotics Derivatives: Their New Applications as Gene Reporters, Antitumor Prodrugs and Enzyme Inhibitors. Mini-Rev. Med. Chem. 2008, 8, 455–471. [Google Scholar] [CrossRef]
- Bedard, P.L.; Hyman, D.M.; Davids, M.S.; Siu, L.L. Small Molecules, Big Impact: 20 Years of Targeted Therapy in Oncology. Lancet 2020, 395, 1078–1088. [Google Scholar] [CrossRef]
- Carneiro, B.A.; El-Deiry, W.S. Targeting Apoptosis in Cancer Therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef]
- Liu, W.; Jin, W.; Zhu, S.; Chen, Y.; Liu, B. Targeting Regulated Cell Death (RCD) with Small-Molecule Compounds in Cancer Therapy: A Revisited Review of Apoptosis, Autophagy-Dependent Cell Death and Necroptosis. Drug Discov. Today 2022, 27, 612–625. [Google Scholar] [CrossRef]
- Pathak, A.; Tanwar, S.; Kumar, V.; Banarjee, B.D. Present and Future Prospect of Small Molecule & Related Targeted Therapy Against Human Cancer. Vivechan Int. J. Res. 2018, 9, 36–49. [Google Scholar]
- Munisamy, M.; Mukherjee, N.; Thomas, L.; Pham, A.T.; Shakeri, A. Therapeutic Opportunities in Cancer Therapy: Targeting the P53-MDM2/MDMX Interactions-PubMed. Am. J. Cancer Res. 2021, 11, 5762–5781. [Google Scholar]
- Lees, A.; Sessler, T.; McDade, S. Dying to Survive—The P53 Paradox. Cancers 2021, 13, 3257. [Google Scholar] [CrossRef]
- Kwok, M.; Agathanggelou, A.; Davies, N.; Stankovic, T. Targeting the P53 Pathway in CLL: State of the Art and Future Perspectives. Cancers 2021, 13, 4681. [Google Scholar] [CrossRef]
- Chi, S.W.; Lee, S.H.; Kim, D.H.; Ahn, M.J.; Kim, J.S.; Woo, J.Y.; Torizawa, T.; Kainosho, M.; Han, K.H. Structural Details on Mdm2-P53 Interaction. J. Biol. Chem. 2005, 280, 38795–38802. [Google Scholar] [CrossRef] [Green Version]
- Uhrinova, S.; Uhrin, D.; Powers, H.; Watt, K.; Zheleva, D.; Fischer, P.; McInnes, C.; Barlow, P.N. Structure of Free MDM2 N-Terminal Domain Reveals Conformational Adjustments That Accompany P53-Binding. J. Mol. Biol. 2005, 350, 587–598. [Google Scholar] [CrossRef]
- Millard, M.; Pathania, D.; Grande, F.; Xu, S.; Neamati, N. Small-Molecule Inhibitors of P53-MDM2 Interaction: The 2006–2010 Update. Curr. Pharm. Des. 2011, 17, 536–559. [Google Scholar] [CrossRef]
- Konopleva, M.; Martinelli, G.; Daver, N.; Papayannidis, C.; Wei, A.; Higgins, B.; Ott, M.; Mascarenhas, J.; Andreeff, M. MDM2 Inhibition: An Important Step Forward in Cancer Therapy. Leukemia 2020, 34, 2858–2874. [Google Scholar] [CrossRef]
- Beloglazkina, A.; Zyk, N.; Majouga, A.; Beloglazkina, E. Recent Small-Molecule Inhibitors of the P53–MDM2 Protein–Protein Interaction. Molecules 2020, 25, 1211. [Google Scholar] [CrossRef] [Green Version]
- Pawge, G.; Khatik, G.L. P53 Regulated Senescence Mechanism and Role of Its Modulators in Age-Related Disorders. Biochem. Pharmacol. 2021, 190, 114651. [Google Scholar] [CrossRef]
- Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Qiu, S.; Ding, Y.; Gao, W.; Stuckey, J.; Krajewski, K.; Roller, P.P.; Tomita, Y.; et al. Structure-Based Design of Potent Non-Peptide MDM2 Inhibitors. J. Am. Chem. Soc. 2005, 127, 10130–10131. [Google Scholar] [CrossRef]
- Galliford, C.V.; Scheidt, K.A. Pyrrolidinyl-Spirooxindole Natural Products as Inspirations for the Development of Potential Therapeutic Agents. Angew. Chem. Int. Ed. 2007, 46, 8748–8758. [Google Scholar] [CrossRef]
- Yu, B.; Zheng, Y.-C.; Shi, X.-J.; Qi, P.-P.; Liu, H.-M. Natural Product-Derived Spirooxindole Fragments Serve as Privileged Substructures for Discovery of New Anticancer Agents. Anticancer. Agents Med. Chem. 2016, 16, 1315–1324. [Google Scholar] [CrossRef]
- Burgess, A.; Chia, K.M.; Haupt, S.; Thomas, D.; Haupt, Y.; Lim, E. Clinical Overview of MDM2/X-Targeted Therapies. Front. Oncol. 2016, 6, 1–7. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Fang, D.D.; Li, Y.; Tang, Y.; Ji, J.; Wang, H.; Karim, R.; Rosas, C.; Huang, Y.; Zhai, Y. A Phase Ib/II Study of APG-115 in Combination with Pembrolizumab in Patients with Unresectable or Metastatic Melanomas or Advanced Solid Tumors. Ann. Oncol. 2019, 30, i2. [Google Scholar] [CrossRef]
- de Weger, V.A.; de Jonge, M.; Langenberg, M.H.G.; Schellens, J.H.M.; Lolkema, M.; Varga, A.; Demers, B.; Thomas, K.; Hsu, K.; Tuffal, G.; et al. A Phase I Study of the HDM2 Antagonist SAR405838 Combined with the MEK Inhibitor Pimasertib in Patients with Advanced Solid Tumours. Br. J. Cancer 2019, 120, 286–293. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.L.; Zhang, Z.M.; Li, X.L.; Tao, Y.F.; Wu, S.Y.; Fang, F.; Xie, Y.; Liao, X.M.; Li, G.; Wu, D.; et al. MI-773, a Breaker of the MDM2/P53 Axis, Exhibits Anticancer Effects in Neuroblastoma via Downregulation of INSM1. Oncol. Lett. 2021, 22, 1–13. [Google Scholar] [CrossRef]
- Ribeiro, C.J.A.; Nunes, R.C.; Amaral, J.D.; Gonçalves, L.M.; Rodrigues, C.M.P.; Moreira, R.; Santos, M.M.M. Spirotriazoline Oxindoles: A Novel Chemical Scaffold with in Vitro Anticancer Properties. Eur. J. Med. Chem. 2017, 140, 494–509. [Google Scholar] [CrossRef]
- Filatov, V.; Kukushkin, M.; Kuznetsova, J.; Skvortsov, D.; Tafeenko, V.; Zyk, N.; Majouga, A.; Beloglazkina, E. Synthesis of 1,3-Diaryl-Spiro[Azetidine-2,3′-Indoline]-2′,4-Diones: Via the Staudinger Reaction: Cis-Or Trans -Diastereoselectivity with Different Addition Modes. RSC Adv. 2020, 10, 14122–14133. [Google Scholar] [CrossRef] [Green Version]
- O’Boyle, N.M.; Carr, M.; Greene, L.M.; Bergin, O.; Nathwani, S.M.; McCabe, T.; Lloyd, D.G.; Zisterer, D.M.; Meegan, M.J. Synthesis and Evaluation of Azetidinone Analogues of Combretastatin A-4 as Tubulin Targeting Agents. J. Med. Chem. 2010, 53, 8569–8584. [Google Scholar] [CrossRef]
- Malebari, A.M.; Greene, L.M.; Nathwani, S.M.; Fayne, D.; O’Boyle, N.M.; Wang, S.; Twamley, B.; Zisterer, D.M.; Meegan, M.J. β-Lactam Analogues of Combretastatin A-4 Prevent Metabolic Inactivation by Glucuronidation in Chemoresistant HT-29 Colon Cancer Cells. Eur. J. Med. Chem. 2017, 130, 261–285. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 32, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Haldar, P.; Barman, G.; Ray, J.K. Sodium Borohydride-Iodine Mediated Reduction of γ-Lactam Carboxylic Acids Followed by DDQ Mediated Oxidative Aromatisation: A Simple Approach towards N-Aryl-Formylpyrroles and 1,3-Diaryl-Formylpyrroles. Tetrahedron 2007, 63, 3049–3056. [Google Scholar] [CrossRef]
- Basu, S.; Barawkar, D.A.; Ramdas, V.; Waman, Y.; Patel, M.; Panmand, A.; Kumar, S.; Thorat, S.; Bonagiri, R.; Jadhav, D.; et al. A2Badenosine Receptor Antagonists: Design, Synthesis and Biological Evaluation of Novel Xanthine Derivatives. Eur. J. Med. Chem. 2017, 127, 986–996. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, T.; Seenisamy, J.; Becker, F.; Blume, B.; Bomke, J.; Dietz, M.; Eckert, U.; Friese-Hamim, M.; Gunera, J.; Hansen, K.; et al. Identification of Methionine Aminopeptidase-2 (MetAP-2) Inhibitor M8891: A Clinical Compound for the Treatment of Cancer. J. Med. Chem. 2019, 62, 11119–11134. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Chen, F.; Dong, Y.; Liu, Y.; Gong, P. Discovery of Novel Dual C-Met/HDAC Inhibitors as a Promising Strategy for Cancer Therapy. Bioorg. Chem. 2020, 101, 103970. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Danishefsky, S. Homoconjugate Addition of Nucleophiles To Cyclopropane-1,1-Dicarboxylate Derivatives: 2-Oxo-1-Phenyl-3-Pyrrolidinecarboxylic Acid. Org. Synth. 1981, 60, 66. [Google Scholar] [CrossRef]
- Zhu, X.; Gan, P. A Novel Synthesis of 1-Aminocyclopropane-1-Carboxylic Acid (ACC). Synth. Commun. 1998, 28, 3159–3162. [Google Scholar] [CrossRef]
- Tóth, G.; Tamás, T.; Borbély, I. Synthesis of 5-(2-Chloroalkyl)-2,2-Dimethyl-1,3-Dioxane-4,6-Diones. Synth. Commun. 2002, 32, 3659–3665. [Google Scholar] [CrossRef]
- Filatov, V.E.; Kuznetsova, J.; Petrovskaya, L.; Yuzabchuk, D.; Tafeenko, V.A.; Zyk, N.V.; Beloglazkina, E.K. Cis-Diastereoselective Synthesis of Spirooxindolo-β-Lactams by Staudinger Cycloaddition with TsCl as Activating Co-Reagent. ACS Omega 2021, 6, 22740–22751. [Google Scholar] [CrossRef]
- Laikov, D.N.; Ustynyuk, Y.A. PRIRODA-04: A Quantum-Chemical Program Suite. New Possibilities in the Study of Molecular Systems with the Application of Parallel Computing. Russ. Chem. Bull. 2005, 54, 820–826. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Baba, T.; Ara, T.; Hasegawa, M.; Takai, Y.; Okumura, Y.; Baba, M.; Datsenko, K.A.; Tomita, M.; Wanner, B.L.; Mori, H. Construction of Escherichia coli K-12 in-Frame, Single-Gene Knockout Mutants: The Keio Collection. Mol. Syst. Biol. 2006, 2. [Google Scholar] [CrossRef] [Green Version]
- Orelle, C.; Carlson, S.; Kaushal, B.; Almutairi, M.M.; Liu, H.; Ochabowicz, A.; Quan, S.; Pham, V.C.; Squires, C.L.; Murphy, B.T.; et al. Tools for Characterizing Bacterial Protein Synthesis Inhibitors. Antimicrob. Agents Chemother. 2013, 57, 5994–6004. [Google Scholar] [CrossRef] [Green Version]
- Sulavik, M.C.; Houseweart, C.; Cramer, C.; Jiwani, N.; Murgolo, N.; Greene, J.; Didomenico, B.; Shaw, K.J.; Miller, G.H.; Hare, R.; et al. Antibiotic Susceptibility Profiles of Escherichia coli Strains Lacking Multidrug Efflux Pump Genes. Antimicrob. Agents Chemother. 2001, 45, 1126–1136. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; McCandlish, A.C.; Gronenberg, L.S.; Chng, S.S.; Silhavy, T.J.; Kahne, D. Identification of a Protein Complex That Assembles Lipopolysaccharide in the Outer Membrane of Escherichia coli. Proc. Natl. Acad. Sci. USA 2006, 103, 11754–11759. [Google Scholar] [CrossRef] [Green Version]
- Osterman, I.A.; Komarova, E.S.; Shiryaev, D.I.; Korniltsev, I.A.; Khven, I.M.; Lukyanov, D.A.; Tashlitsky, V.N.; Serebryakova, M.V.; Efremenkova, O.V.; Ivanenkov, Y.A.; et al. Sorting out Antibiotics’ Mechanisms of Action: A Double Fluorescent Protein Reporter for High-Throughput Screening of Ribosome and DNA Biosynthesis Inhibitors. Antimicrob. Agents Chemother. 2016, 60, 7481–7489. [Google Scholar] [CrossRef] [Green Version]
- Tietze, L.F.; Eicher, T. Reaktionen und Synthesen Im Organisch-Chemischen Praktikum und Forschungslaboratorium; Georg Thieme Verlag: Stuttgart, Germany; New York, NY, USA, 1991; ISBN 3527308741. [Google Scholar]
- Griffioen, G.; Van Dooren, T.; De La Parra, V.R.; Marchand, A.; Allasia, S.; Kilonda, A.; Chaltin, P. Indole Amide Derivatives and Related Compounds for Use in the Treatment of Neurodegenerative Diseases. U.S. Patent No. WO2010142801A1, 11 June 2009. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
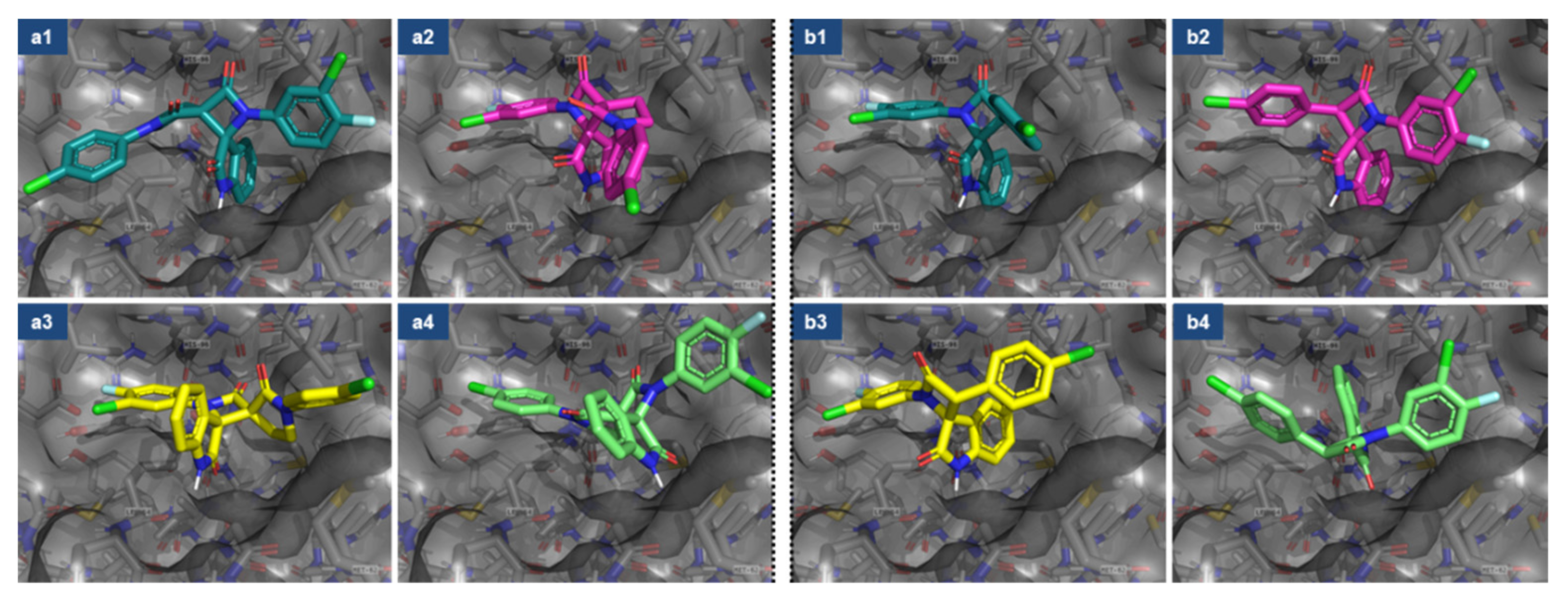
| Compound | Figure 2 Entry# | Stereoisomer | Affinity (kcal/mol) |
|---|---|---|---|
 | a1 | (3R,3′S) | −10.1 |
| a2 | (3S,3′R) | −9.1 | |
| a3 | (3R,3′R) | −8.8 | |
| a4 | (3S,3′S) | −8.1 | |
 | b1 | (2R,3S) | −9.2 |
| b2 | (2S,3R) | −9.4 | |
| b3 | (2R,3R) | −8.1 | |
| b4 | (2S,3S) | −7.3 |
| Bond | (Å) | Angle; Torsion | (°) |
|---|---|---|---|
| N(2)-C(23) | 1.410(3) | C(11)-N(2)-C(3) | 95.8(2) |
| N(2)-C(11) | 1.348(4) | N(2)-C(11)-C(12) | 93.2(2) |
| N(2)-C(3) | 1.467(3) | N(2)-C(3)-C(12) | 86.3(2) |
| C(3)-C(12) | 1.596(4) | C(11)-C(12)-C(3) | 83.8(2) |
| C(11)-C(12) | 1.534(4) | C(13)-C(12)-C(15) | 104.3(2) |
| O(2)-C(11) | 1.202(3) | C(13)-C(12)-C(15) | 104.3(2) |
| N(3)-C(16) | 1.434(3) | C(14)-C(13)-C(12) | 106.9(2) |
| N(3)-C(15) | 1.342(3) | C(15)-N(3)-C(14) | 112.7(2) |
| N(3)-C(14) | 1.455(3) | N(3)-C(14)-C(13) | 106.3(3) |
| C(12)-C(13) | 1.518(4) | C(11)-N(2)-C(3)-C(10) | 134.3(2) |
| C(12)-C(15) | 1.519(3) | N(2)-C(11)-C(12)-C(13) | 131.6(2) |
| C(13)-C(14) | 1.478(4) | C(11)-N(2)-C(23)-C(28) | 160.1(3) |
| O(3)-C(15) | 1.224(3) | C(23)-N(2)-C(3)-C(2) | 70.4(3) |
| Compound | # | Ar1 | Ar2 | Mcf7 | VA13 | A549 | Hek293t | LogP(o/w) * |
|---|---|---|---|---|---|---|---|---|
 | 5a | 4-OMe-Ph | 4-OMe-Ph | 104.3 ± 20.0 | 103.1 ± 2.0 | 66.0 ± 8.7 | 27.0 ± 3.6 | 3.12 |
| 5b | 4-OMe-Ph | 4-Me-Ph | 27.1 ± 2.1 | 23. 5 ± 7.2 | 17.4 ± 1.7 | 14.0 ± 0.9 | 3.46 | |
| 5c | 4-OMe-Ph | 4-Cl-Ph | 5. 7 ± 1.1 | ~10 | 6.4 ± 0.4 | ~5 | 3.76 | |
| 5d | 3-Me-Ph | 3-Me-Ph | 17.9 ± 3.0 | 12.5 ± 0.4 | 8.0 ± 1.10 | 8.8 ± 0.8. | 3.88 | |
| 5e | 3-Me-Ph | 4-Cl-Ph | 7.6 ± 0.7 | 7.7 ± 1.0 | 7.10 ± 0.5 | 6.5 ± 0.4 | 4.14 | |
| 5f | 3-Me-Ph | 4-F,3-Cl-Ph | 5.6 ± 0.6 | 7.3 ± 1.1 | 6.9 ± 0.4 | 6.1 ± 0.4 | 4.32 | |
 | cis [27] | 4-Cl-Ph | 4-F,3-Cl-Ph | 14.4 ± 1.9 | 16.2 ± 2.2 | 8.6 ± 0.6 | 12.7 ± 0.7 | 4.85 |
| cis [38] | 4-Cl-Ph | 4-OMe-Ph | 41.9 ± 3.4 | 25.0 ± 1.9 | 28.4 ± 11.3 | 27.6 ± 4.1 | 4.30 | |
 | trans [27] | 4-Cl-Ph | 4-F,3-Cl-Ph | 18.4 ± 2.1 | 16.6 ± 2.9 | 9.6 ± 0.6 | 17.8 ± 4.1 | 4.85 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filatov, V.E.; Iuzabchuk, D.A.; Tafeenko, V.A.; Grishin, Y.K.; Roznyatovsky, V.A.; Lukianov, D.A.; Fedotova, Y.A.; Sukonnikov, M.A.; Skvortsov, D.A.; Zyk, N.V.; et al. Dispirooxindole-β-Lactams: Synthesis via Staudinger Ketene-Imine Cycloaddition and Biological Evaluation. Int. J. Mol. Sci. 2022, 23, 6666. https://doi.org/10.3390/ijms23126666
Filatov VE, Iuzabchuk DA, Tafeenko VA, Grishin YK, Roznyatovsky VA, Lukianov DA, Fedotova YA, Sukonnikov MA, Skvortsov DA, Zyk NV, et al. Dispirooxindole-β-Lactams: Synthesis via Staudinger Ketene-Imine Cycloaddition and Biological Evaluation. International Journal of Molecular Sciences. 2022; 23(12):6666. https://doi.org/10.3390/ijms23126666
Chicago/Turabian StyleFilatov, Vadim E., Dmitrii A. Iuzabchuk, Viktor A. Tafeenko, Yuri K. Grishin, Vitaly A. Roznyatovsky, Dmitrii A. Lukianov, Yulia A. Fedotova, Maxim A. Sukonnikov, Dmitry A. Skvortsov, Nikolai V. Zyk, and et al. 2022. "Dispirooxindole-β-Lactams: Synthesis via Staudinger Ketene-Imine Cycloaddition and Biological Evaluation" International Journal of Molecular Sciences 23, no. 12: 6666. https://doi.org/10.3390/ijms23126666