
Molecular Dynamics Simulation of Association Processes in Aqueous Solutions of Maleate Salts of Drug-like Compounds: The Role of Counterion


Abstract
:1. Introduction
- (i)
- Develop a parametrization of classical force fields for the correct description of intramolecular H-bonds of various types and strengths. Check this on the example of aqueous solutions of two systems: [2AmPyr + Mle] and [2AmNic + Mle].
- (ii)
- Carry out MD simulations of the early stages of the co-crystallization process, that is, the associations of the [2AmPyr + Mle] and [2AmNic + Mle] systems in aqueous solutions and describe the association process at the atomic-molecular level.
- (iii)
- Localization of dimers, trimers, and their hydrates formed in aqueous solutions. Estimation of the average lifetime of localized structures and identification of spectral features of “long-lived” (~100 ps) associates.

2. Results and Discussion
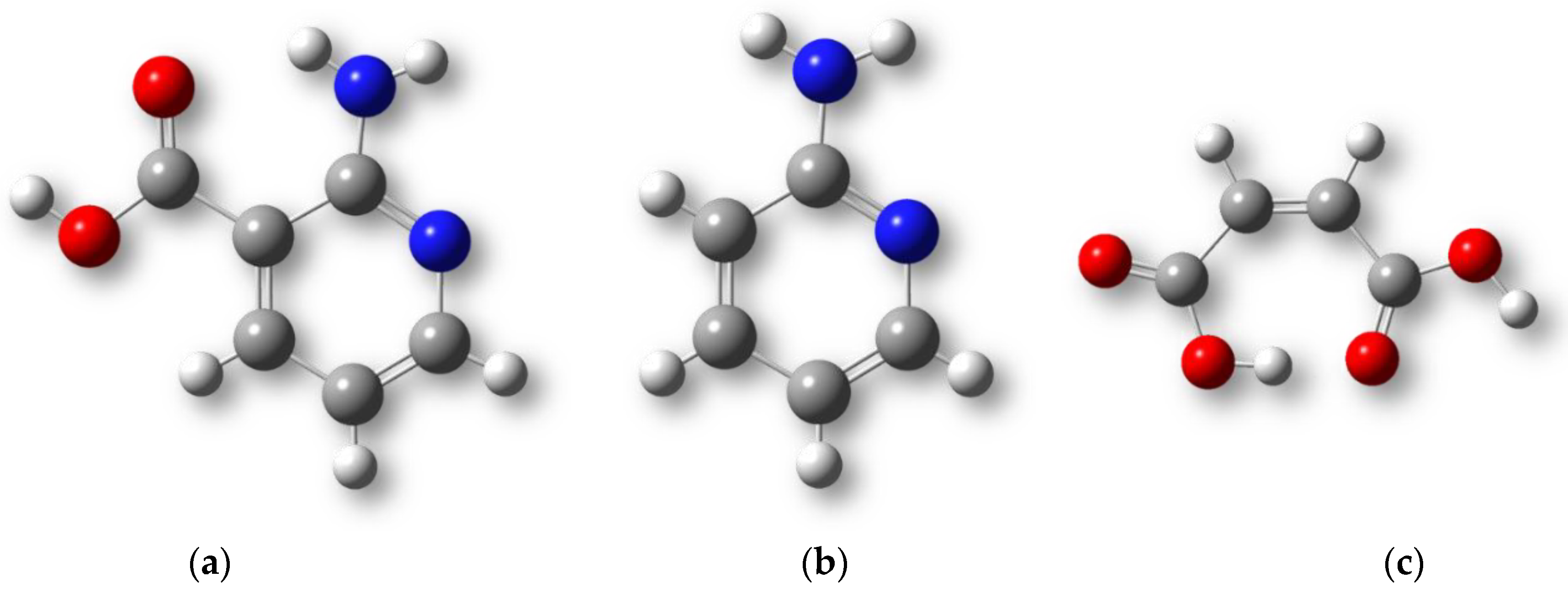
2.1. The Force Field Parameters of the Maleic Acid Monoanion
2.2. The 1 × 1 × 1 Cell
- (i)
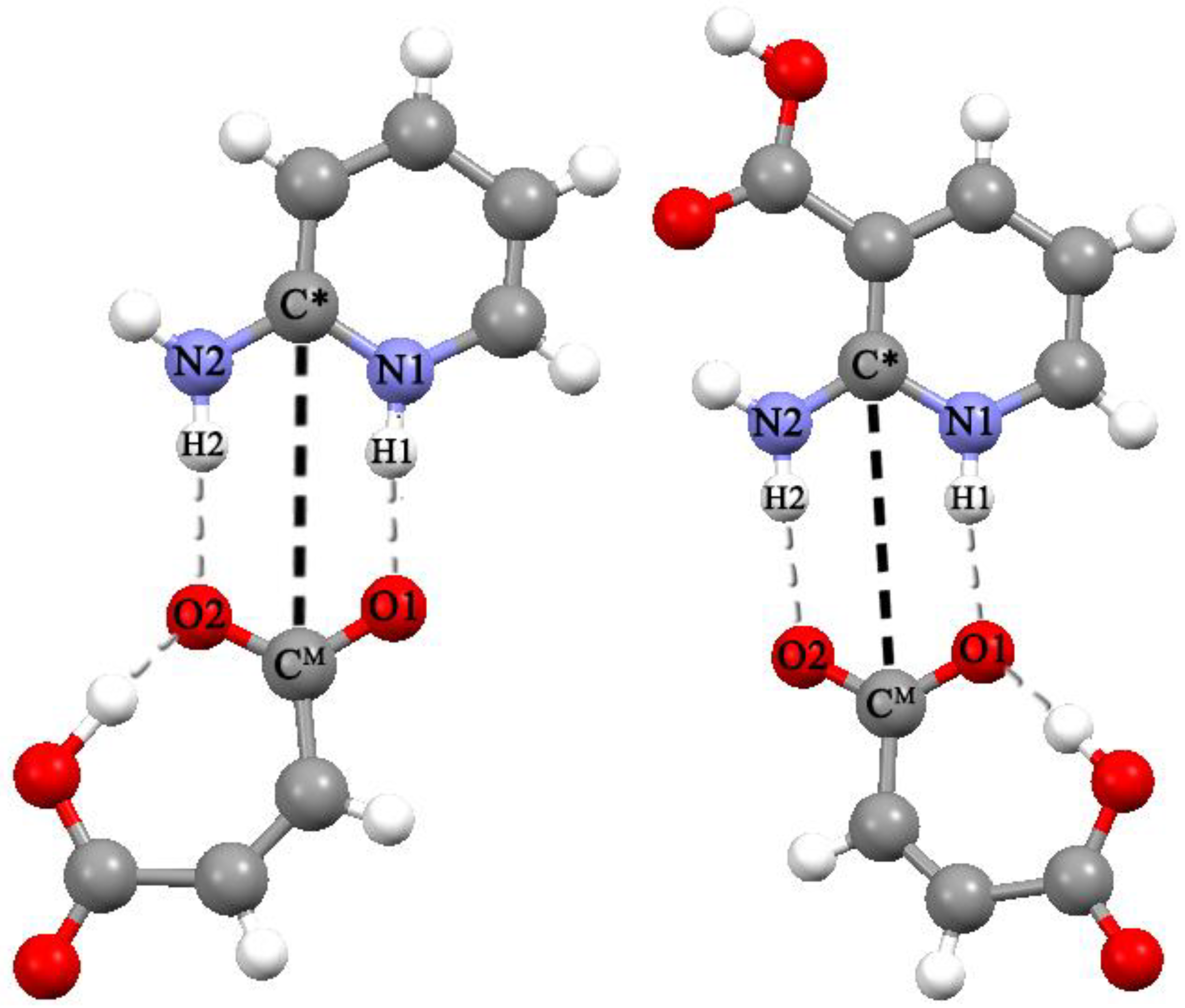
- The force field parameters have been found to satisfactorily describe the strong intramolecular H-bond in the maleic acid monoanion, as well as the formation of heterodimers [API + Mle] in an aqueous solution;
- (ii)
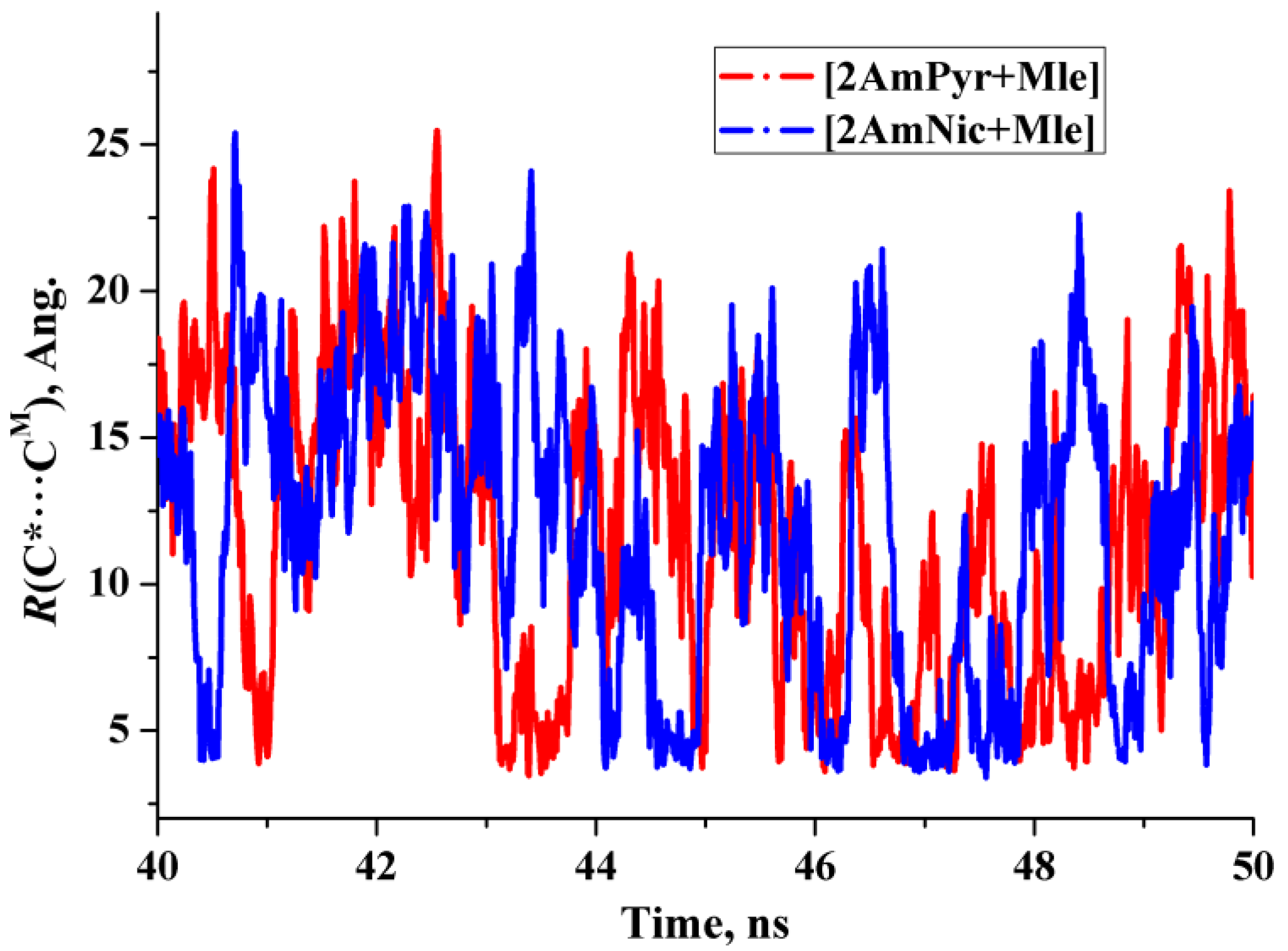
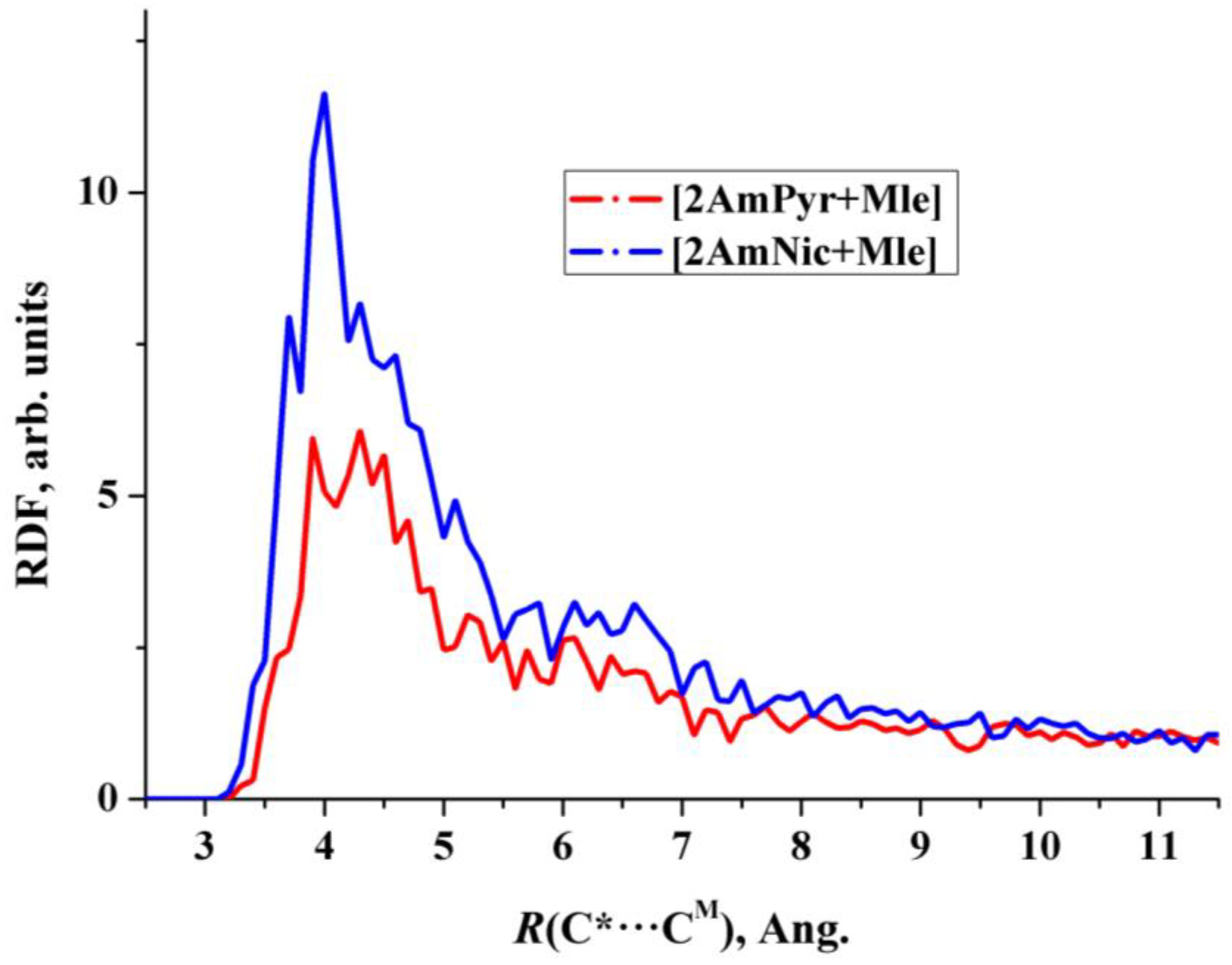
- Heterodimers [2AmPyr + Mle] and [2AmNic + Mle] exist for about 100 ps and can be treated as the long-lived species for which the IR spectrum can be evaluated [27].
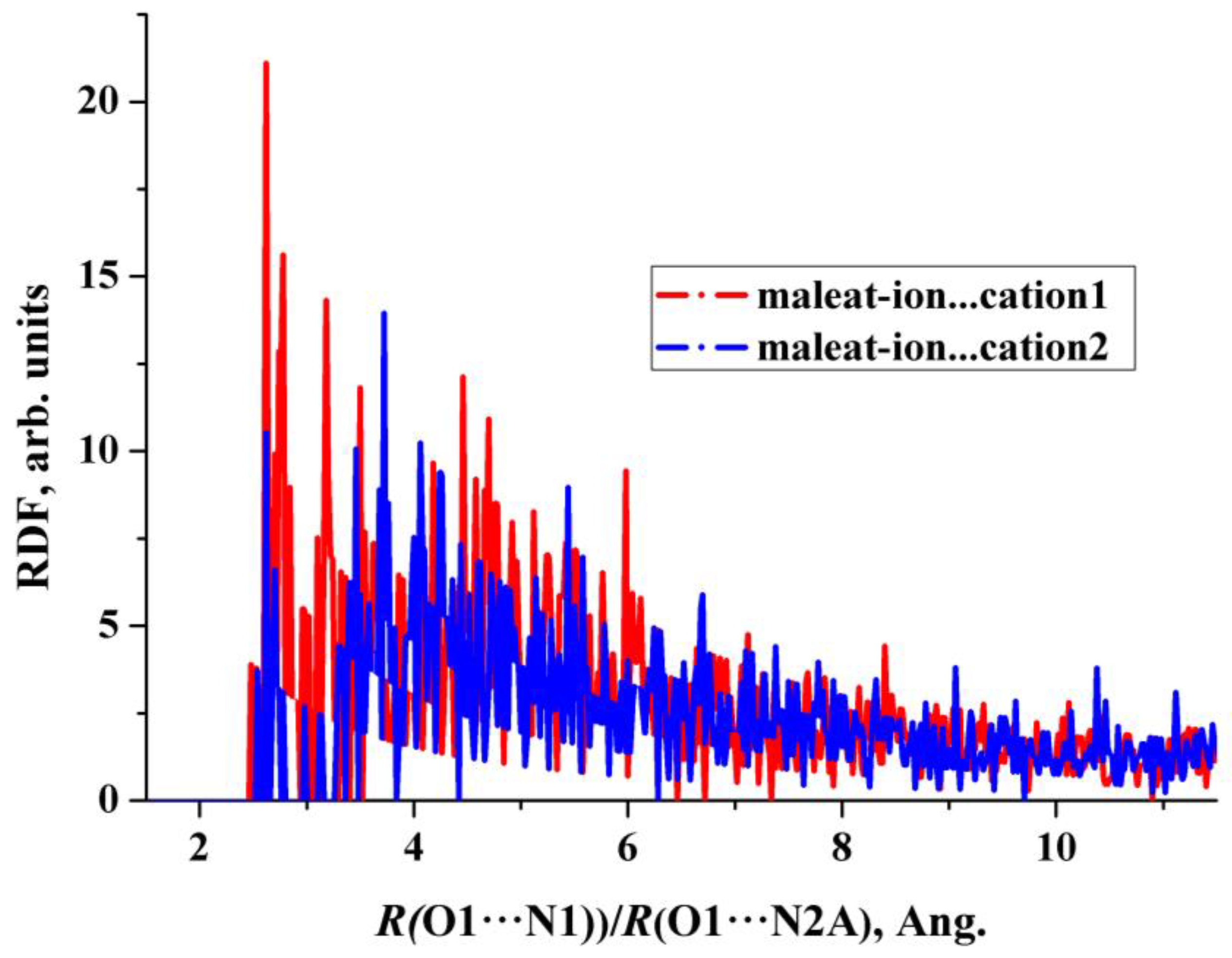
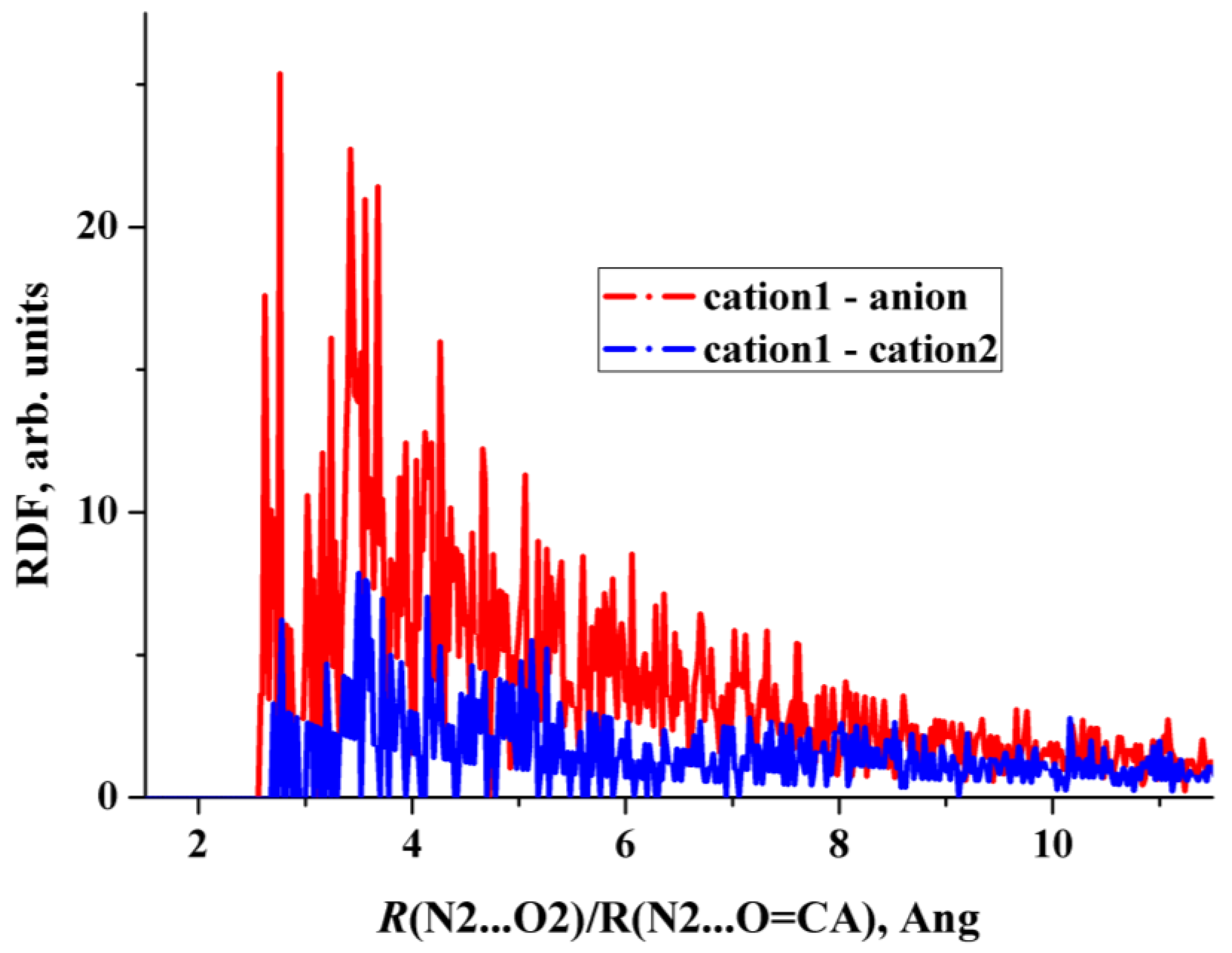
2.3. The 2 × 1 × 1 Cell
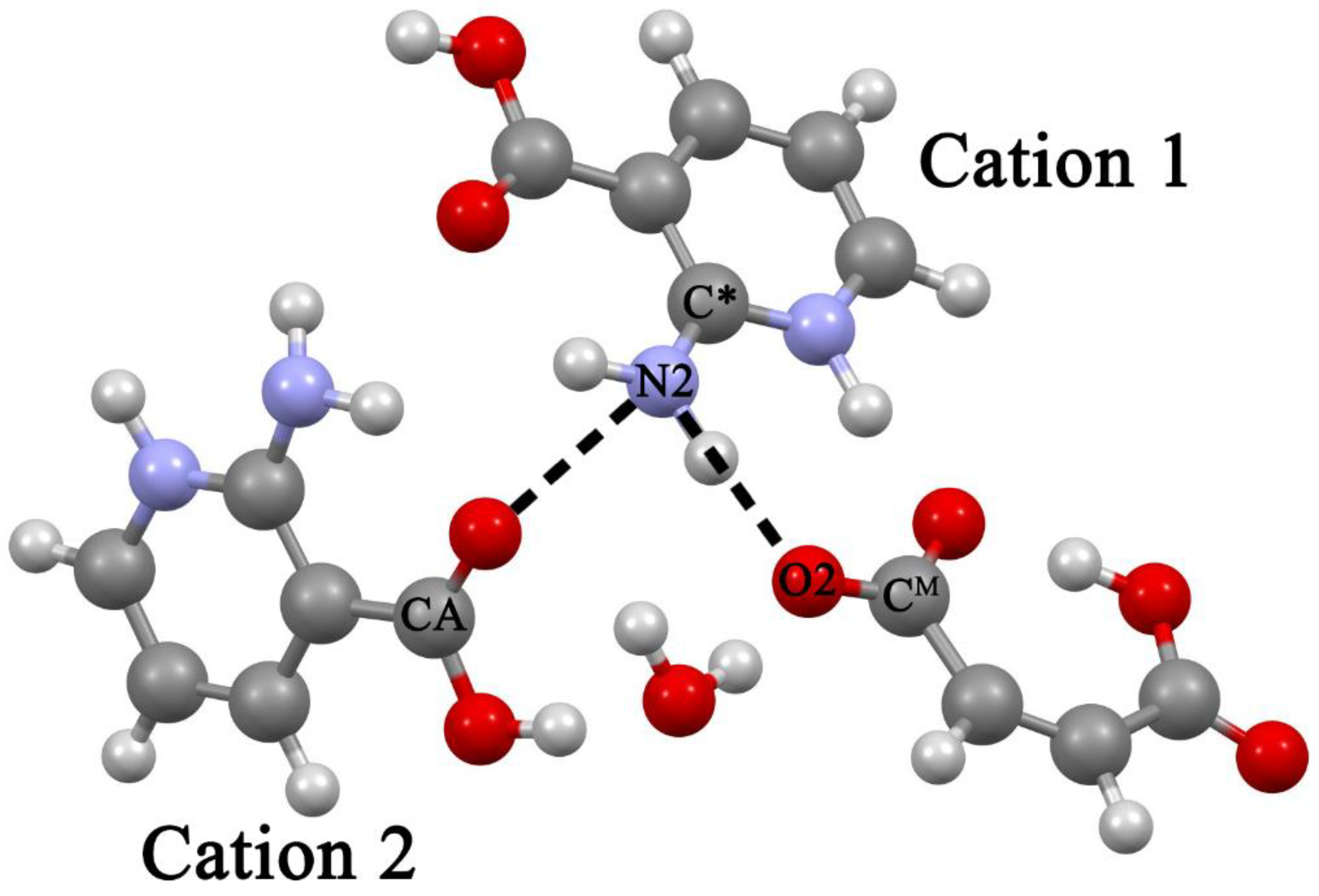
2.4. Structural Features of Heterodimers and Trimers
- (i)
- The calculated values of N···O distances in heterodimers differ significantly from the corresponding values in crystals. The experimental distances in the [2AmPyr + Mle] system differ by ~0.18 Å, while the calculated values differ by ~0.06 Å. The reverse pattern is observed for the [2AmNic + Mle] system (Table 1).
- (ii)
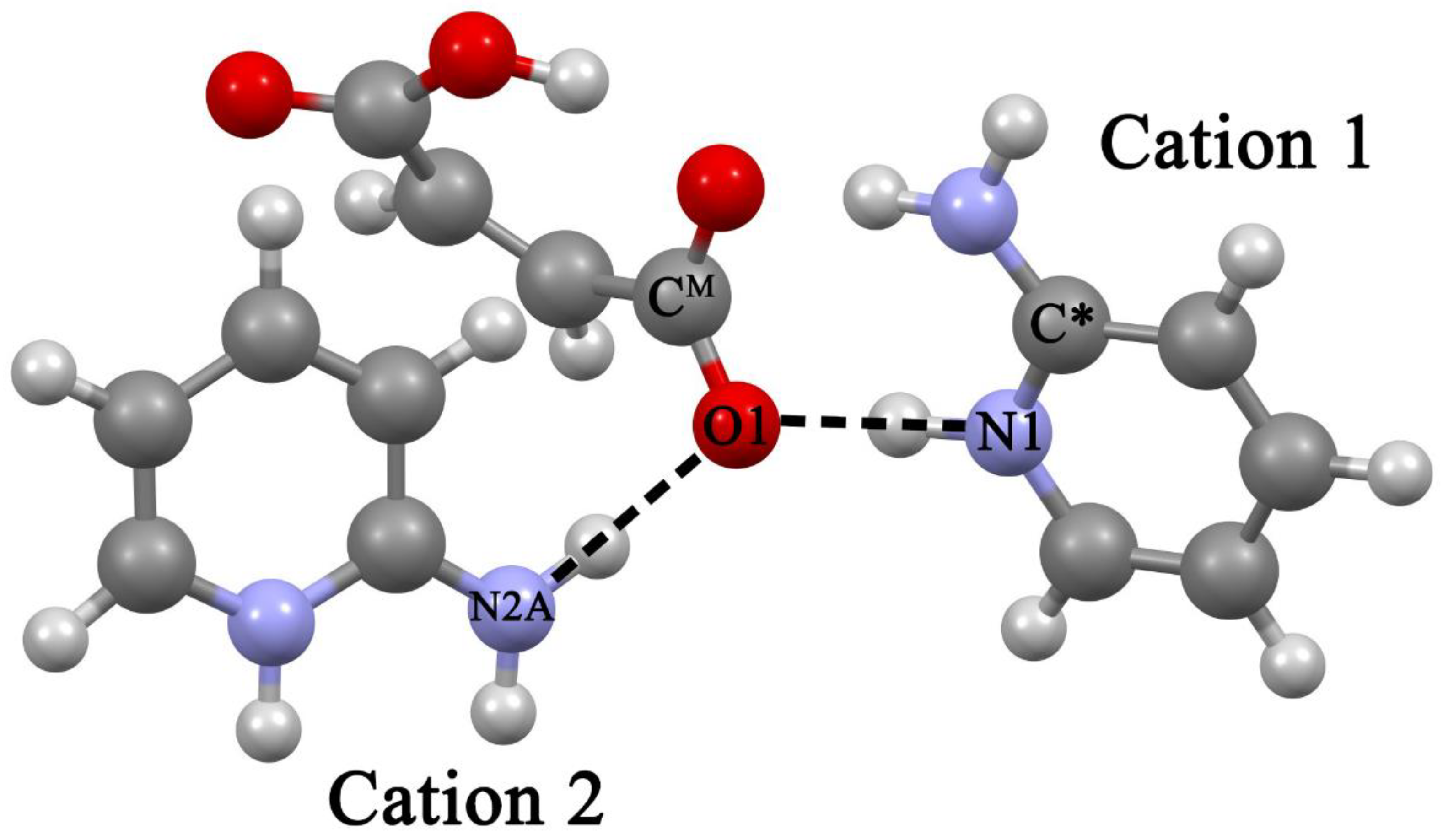
- The explicit allowance for the second cation in trimers leads to the fact that the calculated N···O distances of the cyclic eight-membered H-bonded ring of both systems agree with experiment much better than the corresponding values in heterodimers.
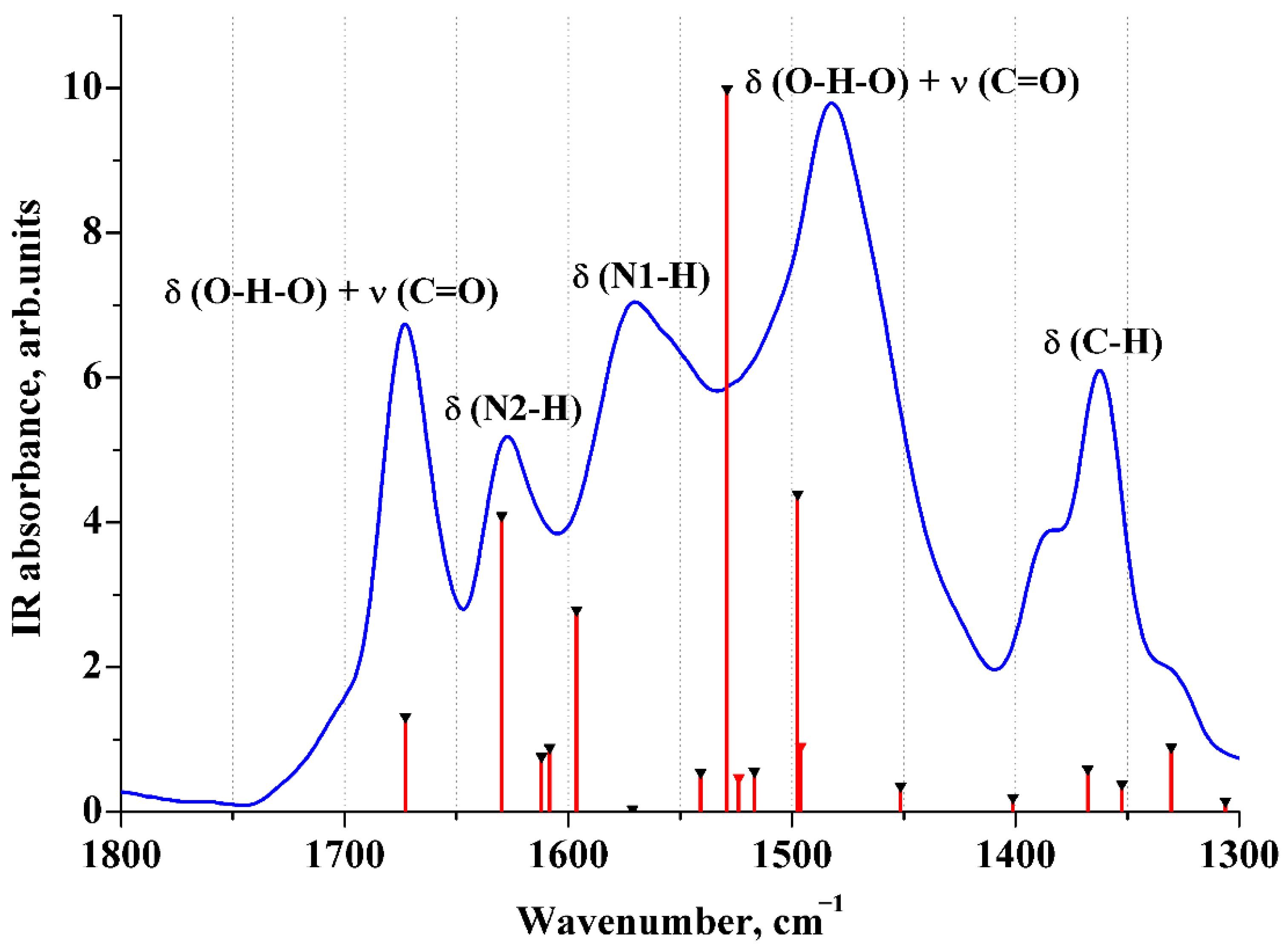
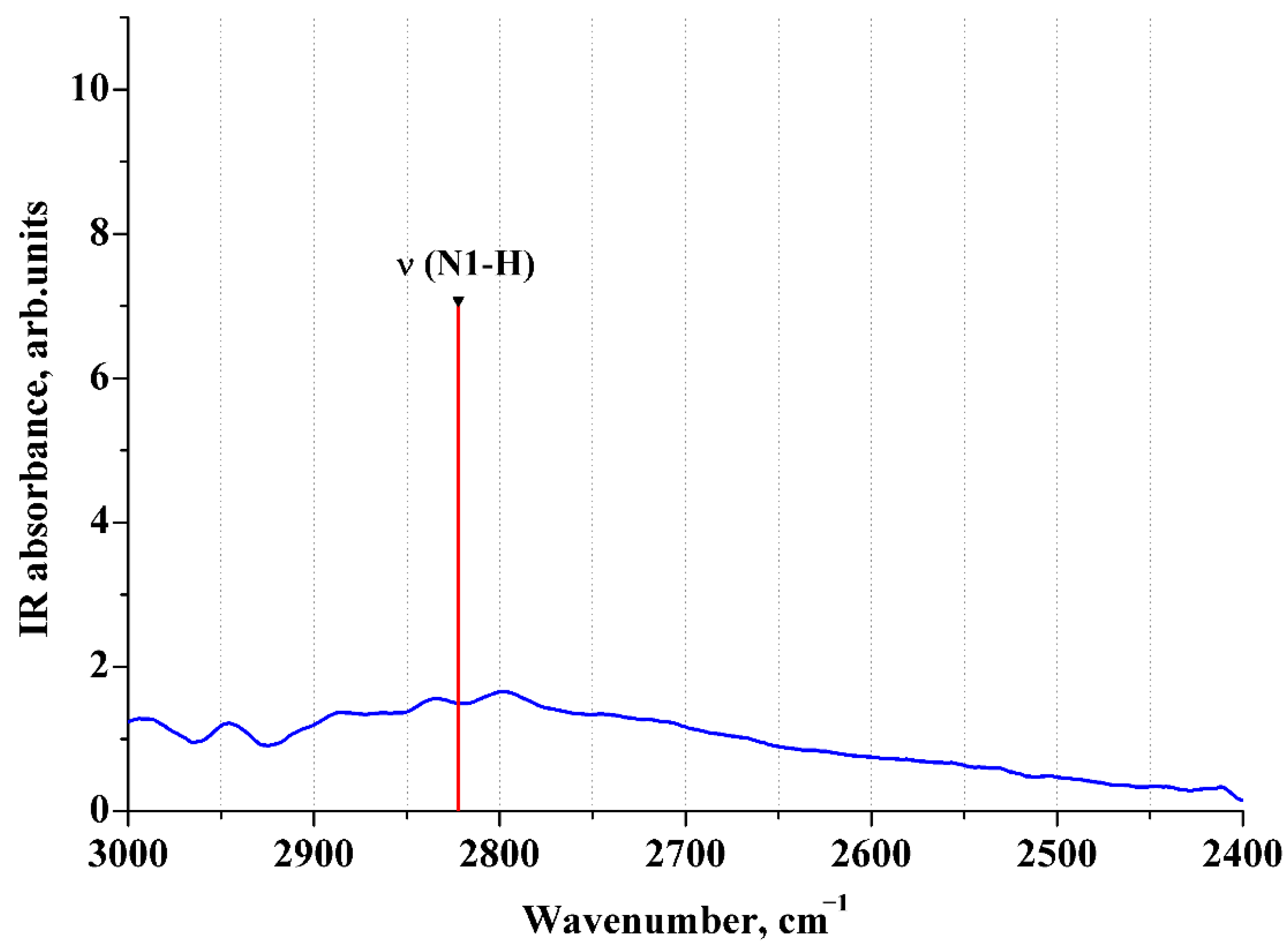
2.5. ATR IR Spectra of Aqueous Solutions of [2AmPyr + Mle]
3. Materials and Methods
3.1. Classical MD Simulations
3.2. DFT Computations
3.3. Enthalpy and Energy of Intermolecular H-Bonds
3.4. ATR-IR Spectroscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stahl, P.H.; Wermuth, C.G. Handbook of Pharmaceutical Salts Properties, Selection, and Use; Wiley: Hoboken, NJ, USA, 2008. [Google Scholar]
- Saal, C. The Relevance of Solid-State Forms for Solubility. In 9. Solubility in Pharmaceutical Chemistry; Christoph, S., Anita, N., Eds.; De Gruyter: Berlin, Germany, 2020; pp. 229–258. [Google Scholar]
- Paulekuhn, G.S.; Dressman, J.B.; Saal, C. Trends in Active Pharmaceutical Ingredient Salt Selection based on Analysis of the Orange Book Database. J. Med. Chem. 2007, 50, 6665–6672. [Google Scholar] [CrossRef]
- Prohotsky, D.L.; Zhao, F. A survey of Top 200 Drugs Inconsistent Practice of Drug Strength Expression for Drugs Containing Salt Forms. J. Pharm. Sci. 2012, 101, 1–6. [Google Scholar] [CrossRef]
- Bharate, S.S. Carboxylic Acid Counterions in FDA-Approved Pharmaceutical Salts. Pharm. Res. 2021, 38, 1307–1326. [Google Scholar] [CrossRef]
- Cadden, J.; Klooster, W.T.; Coles, S.J.; Aitipamula, S. Cocrystal Formulations: Evaluation of the Impact of Excipients on Dissolution by Molecular Simulation and Experimental Approaches. Cryst. Growth Des. 2021, 21, 1006–1018. [Google Scholar] [CrossRef]
- Good, D.J.; Rodriguez-Hornedo, N. Solubility Advantage of Pharmaceutical Cocrystals. Cryst. Growth Des. 2009, 9, 2252–2264. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Surov, A.O.; Vasilev, N.A.; Churakov, A.V.; Parashchuk, O.D.; Artobolevskii, S.V.; Alatortsev, O.A.; Makhrov, D.E.; Vener, M.V. Two faces of water in the formation and stabilization of multicomponent crystals of zwitterionic drug-like compounds. Symmetry 2021, 13, 425. [Google Scholar] [CrossRef]
- Voronin, A.P.; Surov, A.O.; Churakov, A.V.; Parashchuk, O.D.; Rykounov, A.A.; Vener, M.V. Combined X-ray Crystallographic, IR/Raman Spectroscopic, and Periodic DFT Investigations of New Multicomponent Crystalline Forms of Anthelmintic Drugs: A Case Study of Carbendazim Maleate. Molecules 2020, 25, 2386. [Google Scholar] [CrossRef] [PubMed]
- Babu, N.J.; Reddy, L.S.; Nangia, A. Amide N-Oxide Heterosynthon and Amide Dimer Homosynthon in Cocrystals of Carboxamide Drugs and Pyridine N-Oxides. Mol. Pharm. 2007, 4, 417–434. [Google Scholar] [CrossRef]
- Prikhodchenko, P.V.; Medvedev, A.G.; Tripol’skaya, T.A.; Churakov, A.V.; Wolanov, Y.; Howard, J.A.K.; Lev, O. Crystal Structures of Natural Amino Acid Perhydrates. CrystEngComm 2011, 13, 2399–2407. [Google Scholar] [CrossRef]
- Bolla, G.; Sanphui, P.; Nangia, A. Solubility Advantage of Tenoxicam Phenolic Cocrystals Compared to Salts. Cryst. Growth Des. 2013, 13, 1988–2003. [Google Scholar] [CrossRef]
- Bolla, G.; Nangia, A. Pharmaceutical Cocrystals: Walking the Talk. Chem. Commun. 2016, 52, 8342–8360. [Google Scholar] [CrossRef]
- Steiner, T. The Hydrogen Bond in the Solid State. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Surov, A.O.; Manin, A.N.; Voronin, A.P.; Churakov, A.V.; Perlovich, G.L.; Vener, M.V. Weak interactions cause packing polymorphism in pharmaceutical two-component crystals. The case study of the salicylamide cocrystal. Cryst. Growth Des. 2017, 17, 1425–1437. [Google Scholar] [CrossRef]
- Yadav, B.; Balasubramanian, S.; Chavan, R.B.; Thipparaboina, R.; Naidu, V.G.M.; Shastri, N.R. Hepatoprotective Cocrystals and Salts of Riluzole: Prediction, Synthesis, Solid State Characterization, and Evaluation. Cryst. Growth Des. 2018, 18, 1047–1061. [Google Scholar] [CrossRef]
- Surov, A.O.; Voronin, A.P.; Vener, M.V.; Churakov, A.V.; Perlovich, G.L. Specific features of supramolecular organisation and hydrogen bonding in proline cocrystals: A case study of fenamates and diclofenac. CrystEngComm 2018, 20, 6970–6981. [Google Scholar] [CrossRef]
- Mirzaei, M.; Sadeghi, F.; Molcanov, K.; Zare, J.K.; Gomila, R.M.; Frontera, A. Recurrent Supramolecular Motifs in a Series of Acid–Base Adducts Based on Pyridine-2,5-Dicarboxylic Acid N-Oxide and Organic Bases: Inter- and Intramolecular Hydrogen Bonding. Cryst. Growth Des. 2020, 20, 1738–1751. [Google Scholar] [CrossRef]
- Bolla, G.; Nangia, A. Novel pharmaceutical salts of albendazole. CrystEngComm 2018, 20, 6394–6405. [Google Scholar] [CrossRef] [Green Version]
- Voronin, A.P.; Vasilev, N.A.; Surov, A.O.; Churakov, A.V.; Perlovich, G.L. Exploring the solid form landscape of the antifungal drug isavuconazole: Crystal structure analysis, phase transformation behavior and dissolution performance. CrystEngComm 2021, 23, 8513–8526. [Google Scholar] [CrossRef]
- Vener, M.V.; Churakov, A.V.; Voronin, A.P.; Parashchuk, O.D.; Artobolevskii, S.V.; Alatortsev, O.A.; Makhrov, D.E.; Medvedev, A.G.; Filarowski, A. Comparison of Proton Acceptor and Proton Donor Properties of H2O and H2O2 in Organic Crystals of Drug-like Compounds: Peroxosolvates vs. Crystallohydrates. Molecules 2022, 27, 717. [Google Scholar] [CrossRef]
- Dunitz, J.D.; Gavezzotti, A. Supramolecular Synthons: Validation and Ranking of Intermolecular Interaction Energies. Cryst. Growth Des. 2012, 12, 5873–5877. [Google Scholar] [CrossRef]
- Anandha babu, G.; Ramasamy, P. Studies on the growth and characterization of 2-aminopyridinium maleate—A novel nonlinear optical crystal. J. Cryst. Growth 2009, 311, 1185–1189. [Google Scholar] [CrossRef]
- Choudhury, R.R.; Chitra, R. Investigation of hydrogen bond effects on the hyperpolarizability of 2-Aminopyridinium maleate (2APM) complex and determining the non-linear optical susceptibility of the molecular crystal of 2APM. Mol. Phys. 2011, 109, 1701–1708. [Google Scholar] [CrossRef]
- Chitra, R.; Roussel, P.; Capet, F.; Murli, C.; Choudhury, R.R. Molecular interactions in the anomalous salt: 2-Aminopyridinium maleate maleic acid. J. Mol. Struct. 2009, 923, 45–52. [Google Scholar] [CrossRef]
- Levina, E.O.; Lokshin, B.V.; Mai, B.D.; Vener, M.V. Spectral features of guanidinium-carboxylate salt bridges. The combined ATR IR and theoretical studies of aqueous solution of guanidinium acetate. Chem. Phys. Lett. 2016, 659, 117–120. [Google Scholar] [CrossRef]
- Singh, J.; Thornton, J.M.; Snarey, M.; Campbell, S.F. The geometries of interacting arginine-carboxyls in proteins. FEBS Lett. 1987, 224, 161–171. [Google Scholar] [PubMed] [Green Version]
- Yu, Z.; Jacobson, M.P.; Jasovitz, J.; Rapp, C.S.; Friesner, R.A. First-shell solvation of ion pairs: Correction of systematic errors in implicit solvent models. J. Phys. Chem. B 2004, 108, 6643–6654. [Google Scholar] [CrossRef]
- Rozanska, X.; Chipot, C. Modeling ion–ion interaction in proteins: A molecular dynamics free energy calculation of the guanidinium-acetate association. J. Chem. Phys. 2000, 112, 9691–9694. [Google Scholar] [CrossRef]
- Debiec, K.T.; Gronenborn, A.M.; Chong, L.T. Evaluating the Strength of Salt Bridges: A Comparison of Current Biomolecular Force Fields. J. Phys. Chem. B 2014, 118, 6561–6569. [Google Scholar] [CrossRef]
- Manin, A.N.; Voronin, A.P.; Shishkina, A.V.; Vener, M.V.; Churakov, A.V.; Perlovich, G.L. Influence of Secondary Interactions on the Structure, Sublimation Thermodynamics, and Solubility of Salicylate: 4-Hydroxybenzamide Cocrystals. Combined Experimental and Theoretical Study. J. Phys. Chem. B 2015, 119, 10466–10477. [Google Scholar] [CrossRef]
- Hetmanczyk, Ł.; Goremychkin, E.A.; Waliszewski, J.; Vener, M.V.; Lipkowski, P.; Tolstoy, P.M.; Filarowski, A. Spectroscopic Identification of Hydrogen Bond Vibrations and Quasi-Isostructural Polymorphism in N-Salicylideneaniline. Molecules 2021, 26, 5043. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Büyükgüngör, O.; Odabasoğlu, M. Bis(2-aminopyridinium) maleate. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2003, 59, 105–106. [Google Scholar] [CrossRef] [PubMed]
- Das, R.C.; Dash, U.N.; Panda, K.N. Thermodynamics of dissociation of DL-malic, maleic and fumaric acids in water and water+ dioxan mixtures. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1980, 76, 2152–2158. [Google Scholar] [CrossRef]
- Chitra, R.; Roussel, P.; Capet, F.; Murli, C.; Choudhury, R.R. 2-Aminopyridinium maleate: A structural study. J. Mol. Struct. 2008, 891, 103–109. [Google Scholar] [CrossRef]
- Vener, M.V.; Manaev, A.V.; Tsirelson, V.G. Proton Dynamics in Strong (Short) Intramolecular H-Bond. DFT Study of the KH Maleate Crystal. J. Phys. Chem. A 2008, 112, 13628–13632. [Google Scholar] [CrossRef]
- Di Muzio, S.; Ramondo, F.; Gontrani, L.; Ferella, F.; Nardone, M.; Benassi, P. Choline Hydrogen Dicarboxylate Ionic Liquids by X-ray Scattering, Vibrational Spectroscopy and Molecular Dynamics: H-Fumarate and H-Maleate and Their Conformations. Molecules 2020, 25, 4990. [Google Scholar] [CrossRef] [PubMed]
- Cole, D.J.; Vilseck, J.Z.; Tirado-Rives, J.; Payne, M.C.; Jorgensen, W.L. Biomolecular Force Field Parameterization via Atoms-in-Molecule Electron Density Partitioning. J. Chem. Theory Comput. 2016, 12, 2312–2323. [Google Scholar] [CrossRef]
- Cerezo, J.; Prampolini, G.; Cacelli, I. Developing accurate intramolecular force fields for conjugated systems through explicit coupling terms. Theor. Chem. Acc. 2018, 137, 80. [Google Scholar] [CrossRef]
- Landi, A.; Padula, D. Multiple charge separation pathways in new-generation non-fullerene acceptors: A computational study. J. Mater. Chem. A 2021, 9, 24849–24856. [Google Scholar] [CrossRef]
- Levina, E.O.; Penkov, N.V.; Rodionova, N.N.; Tarasov, S.A.; Barykina, D.V.; Vener, M.V. Hydration of the Carboxylate Group in Anti-Inflammatory Drugs: ATR-IR and Computational Studies of Aqueous Solution of Sodium Diclofenac. ACS Omega 2018, 3, 302–313. [Google Scholar] [CrossRef]
- Shishkina, A.V.; Ksenofontov, A.A.; Penkov, N.V.; Vener, M.V. Diclofenac Ion Hydration: Experimental and Theoretical Search for Anion Pairs. Molecules 2022, 27, 3350. [Google Scholar] [CrossRef] [PubMed]
- Clarke, H.D.; Arora, K.K.; Bass, H.; Kavuru, P.; Ong, T.T.; Pujari, T.; Wojtas, L.; Zaworotko, M.J. Structure−Stability Relationships in Cocrystal Hydrates: Does the Promiscuity of Water Make Crystalline Hydrates the Nemesis of Crystal Engineering? Cryst. Growth Des. 2010, 10, 2152–2167. [Google Scholar] [CrossRef]
- Hu, L.; Staples, R.J.; Shreeve, J.M. Energetic compounds based on a new fused triazolo[4,5-d]pyridazine ring: Nitroimino lights up energetic performance. Chem. Eng. J. 2021, 420, 129839. [Google Scholar] [CrossRef]
- Kalikmanov, V.I. Nucleation Theory; Lection Notes in Physics; Springer: Berlin/Heidelberg, Germany, 2012; Volume 860, ISBN 978-9048136421. [Google Scholar]
- Malarski, Z.; Rospenk, M.; Sobczyk, L.; Grech, E. Dielectric and spectroscopic studies of pentachlorophenol-amine complexes. J. Phys. Chem. 1982, 86, 401–406. [Google Scholar] [CrossRef]
- Asfin, R.E.; Denisov, G.S.; Mielke, Z.; Tokhadze, K.G. Particular Features of the n(OH) Absorption Band of Strongly Hydrogen-Bonded Complexes in the Gas Phase, Low-Temperature Matrices, and Crystalline Films at 12–600 K. Opt. Spectrosc. 2005, 99, 56–67. [Google Scholar] [CrossRef]
- Barone, V.; Biczysko, M.; Bloino, J. Fully anharmonic IR and Raman spectra of medium-size molecular systems: Accuracy and interpretation. Phys. Chem. Chem. Phys. 2014, 16, 1759–1787. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Jordan, M.J.T.; Gill, P.M.W.; Buckingham, A.D. An ab initio study of anharmonicity and matrix effects on the hydrogen-bonded BrH: NH3 complex. Mol. Phys. 1997, 92, 429–440. [Google Scholar] [CrossRef]
- Vener, M.V.; Sauer, J. Quantum anharmonic frequencies of the O⋯H⋯O fragment of the H5O2+ ion: A model three-dimensional study. Chem. Phys. Lett. 1999, 312, 591–597. [Google Scholar] [CrossRef]
- Rekik, N.; Ghalla, H.; Hanna, G. Explaining the Structure of the OH Stretching Band in the IR Spectra of Strongly Hydrogen-Bonded Dimers of Phosphinic Acid and Their Deuteraed Analogs in the Gas Phase: A Computational Study. J. Phys. Chem. A 2012, 116, 4495–4509. [Google Scholar] [CrossRef] [PubMed]
- Van Hoozen, B.L.; Petersen, P.B. Vibrational tug-of-war: The pKA dependence of the broad vibrational features of strongly hydrogen-bonded carboxylic acids. J. Chem. Phys. 2018, 148, 134309. [Google Scholar] [CrossRef] [PubMed]
- Vener, M.V.; Kühn, O.; Sauer, J. The infrared spectrum of the O⋯H⋯O fragment of H5O2+: Ab initio classical molecular dynamics and quantum 4D model calculations. J. Chem. Phys. 2001, 114, 240–249. [Google Scholar] [CrossRef]
- Brela, M.; Stare, J.; Pirc, G.; Sollner-Dolenc, M.; Boczar, M.; Wójcik, M.J.; Mavri, J. Car–Parrinello Simulation of the Vibrational Spectrum of a Medium Strong Hydrogen Bond by Two-Dimensional Quantization of the Nuclear Motion: Application to 2-Hydroxy-5-nitrobenzamide. J. Phys. Chem. B 2012, 116, 4510–4518. [Google Scholar] [CrossRef]
- Molčanov, K.; Stare, J.; Vener, M.V.; Kojić-Prodić, B.; Mali, G.; Grdadolnik, J.; Mohaček-Grošev, V. Nitranilic acid hexahydrate, a novel benchmark system of the Zundel cation in an intrinsically asymmetric environment: Spectroscopic features and hydrogen bond dynamics characterised by experimental and theoretical methods. Phys. Chem. Chem. Phys. 2014, 16, 998–1007. [Google Scholar] [CrossRef]
- Brela, M.Z.; Wójcik, M.J.; Witek, Ł.J.; Boczar, M.; Wrona, E.; Hashim, R.; Ozaki, Y. Born–Oppenheimer Molecular Dynamics Study on Proton Dynamics of Strong Hydrogen Bonds in Aspirin Crystals, with Emphasis on Differences between Two Crystal Forms. J. Phys. Chem. B 2016, 120, 3854–3862. [Google Scholar] [CrossRef] [PubMed]
- Gaigeot, M.-P.; Sprik, M. Ab Initio Molecular Dynamics Computation of the Infrared Spectrum of Aqueous Uracil. J. Phys. Chem. B 2003, 107, 10344–10358. [Google Scholar] [CrossRef]
- Sun, J.; Bousquet, D.; Forbert, H.; Marx, D. Glycine in Aqueous Solution: Solvation Shells, Interfacial Water, and Vibrational Spectroscopy from Ab Initio Molecular Dynamics. J. Chem. Phys. 2010, 133, 114508. [Google Scholar] [CrossRef] [PubMed]
- Vener, M.V.; Chernyshov, I.Y.; Rykounov, A.A.; Filarowski, A. Structural and Spectroscopic Features of Proton Hydrates in the Crystalline State. Solid-state DFT Study on HCl and Triflic Acid Hydrates. Mol. Phys. 2018, 116, 251–262. [Google Scholar] [CrossRef]
- Available online: https://ambermd.org/antechamber/ac.html#antechamber (accessed on 7 May 2022).
- Available online: http://zarbi.chem.yale.edu/ligpargen (accessed on 7 May 2022).
- Available online: http://www.pi.iccom.cnr.it/joyce (accessed on 7 May 2022).
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Grigera, J.R.; Sraatsma, T.P. The missing term in effective pair potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 01410. [Google Scholar] [CrossRef] [Green Version]
- Hockney, R.W.; Goel, S.P.; Eastwood, J. Quiet high resolution computer models of a plasma. J. Comp. Phys. 1974, 14, 148–158. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N∙log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Essman, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Tuñón, I.; Rinaldi, D.; Ruiz-López, M.F.; Rivail, J.L. Hydroxide ion in liquid water: Structure, energetics, and proton transfer using a mixed discrete-continuum ab initio model. J. Phys. Chem. 1995, 99, 3798–3805. [Google Scholar] [CrossRef]
- Barone, V.; Carnimeo, I.; Scalmani, G. Computational Spectroscopy of Large Systems in Solution: The DFTB/PCM and TD-DFTB/PCM Approach. J. Chem. Theory Comput. 2013, 9, 2052–2071. [Google Scholar] [CrossRef]
- Vener, M.V.; Shenderovich, I.G.; Rykounov, A.A. A qualitative study of the effect of a counterion and polar environment on the structure and spectroscopic signatures of a hydrated hydroxyl anion. Theor. Chem. Acc. 2013, 132, 1361. [Google Scholar] [CrossRef]
- Merrick, J.P.; Moran, D.; Radom, L. An Evaluation of Harmonic Vibrational Frequency Scale Factors. J. Phys. Chem. A 2007, 111, 11683–11700. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, A.G.; Churakov, A.V.; Prikhodchenko, P.V.; Lev, O.; Vener, M.V. Crystalline Peroxosolvates: Nature of the Coformer, Hydrogen-Bonded Networks and Clusters, Intermolecular Interactions. Molecules 2021, 26, 26. [Google Scholar] [CrossRef]
- Rozenberg, M.; Loewenschuss, A.; Marcus, Y. An Empirical Correlation Between Stretching Vibration Redshift and Hydrogen Bond Length. Phys. Chem. Chem. Phys. 2000, 2, 2699–2702. [Google Scholar] [CrossRef]
- Mata, I.; Alkorta, I.; Espinosa, E.; Molins, E. Relationships Between Interaction Energy, Intermolecular Distance and Electron Density Properties in Hydrogen Bonded Complexes under External Electric Fields. Chem. Phys. Lett. 2011, 507, 185–189. [Google Scholar] [CrossRef]
- Tsirelson, V.G. The Quantum Theory of Atoms in Molecules: From Solid State to DNA and Drug Design; Matta, C.F., Boyd, R.J., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2007; Chapter 10. [Google Scholar]
- Keith, T.A. AIMAll (Version 19.10.12); TK Gristmill Software: Overland Park, KS, USA, 2019. [Google Scholar]
- Shih, O.; England, A.H.; Dallinger, G.C.; Smith, J.W.; Duffey, K.C.; Cohen, R.C.; Prendergast, D.; Saykally, R.J. Cation-cation contact pairing in water: Guanidinium. J. Chem. Phys. 2013, 139, 035104. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, R.; Nandi, S.; Halder, R.; Jana, B.; Bhattacharyya, K. Structural relaxation of acridine orange dimer in bulk water and inside a single live lung cell. J. Chem. Phys. 2016, 144, 065101. [Google Scholar] [CrossRef]
- Mak, C.H. Unraveling Base Stacking Driving Forces in DNA. J. Phys. Chem. B 2016, 120, 6010–6020. [Google Scholar] [CrossRef] [PubMed]
- Kruchinin, S.E.; Fedotova, M.V. Ion Pairing of the Neurotransmitters Acetylcholine and Glutamate in Aqueous Solutions. J. Phys. Chem. B 2021, 125, 11219–11231. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Distance/Angle a | X-ray | Heterodimer | Trimer |
|---|---|---|---|
| The [2AmPyr + Mle] System | |||
| N1···O1, Å | 2.727 | 2.823 | 2.781 |
| N2···O2, Å | 2.902 | 2.883 | 2.970 |
| O1···N2A, Å | 2.900 | - | 2.879 |
| Dihedral angle, deg | 102.1 | - | 88.2 |
| The [2AmNic + Mle] System | |||
| N1–H1···O1, Å | 2.816 | 2.765 | 2.810 |
| N2–H2···O2, Å | 2.816 | 2.901 | 2.880 |
| N2-H2···O=CA, Å | 3.01 | - | 3.31 |
| O2···H-Ow, Å | 2.717 | - | 2.702 |
| Ow···H-OA, Å | 2.567 | - | 2.559 |
| Dihedral angle, deg | 17.4 | - | 43.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vener, M.V.; Makhrov, D.E.; Voronin, A.P.; Shalafan, D.R. Molecular Dynamics Simulation of Association Processes in Aqueous Solutions of Maleate Salts of Drug-like Compounds: The Role of Counterion. Int. J. Mol. Sci. 2022, 23, 6302. https://doi.org/10.3390/ijms23116302
Vener MV, Makhrov DE, Voronin AP, Shalafan DR. Molecular Dynamics Simulation of Association Processes in Aqueous Solutions of Maleate Salts of Drug-like Compounds: The Role of Counterion. International Journal of Molecular Sciences. 2022; 23(11):6302. https://doi.org/10.3390/ijms23116302
Chicago/Turabian StyleVener, Mikhail V., Denis E. Makhrov, Alexander P. Voronin, and Daria R. Shalafan. 2022. "Molecular Dynamics Simulation of Association Processes in Aqueous Solutions of Maleate Salts of Drug-like Compounds: The Role of Counterion" International Journal of Molecular Sciences 23, no. 11: 6302. https://doi.org/10.3390/ijms23116302