
Characterisation of LTR-Retrotransposons of Stevia rebaudiana and Their Use for the Analysis of Genetic Variability

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
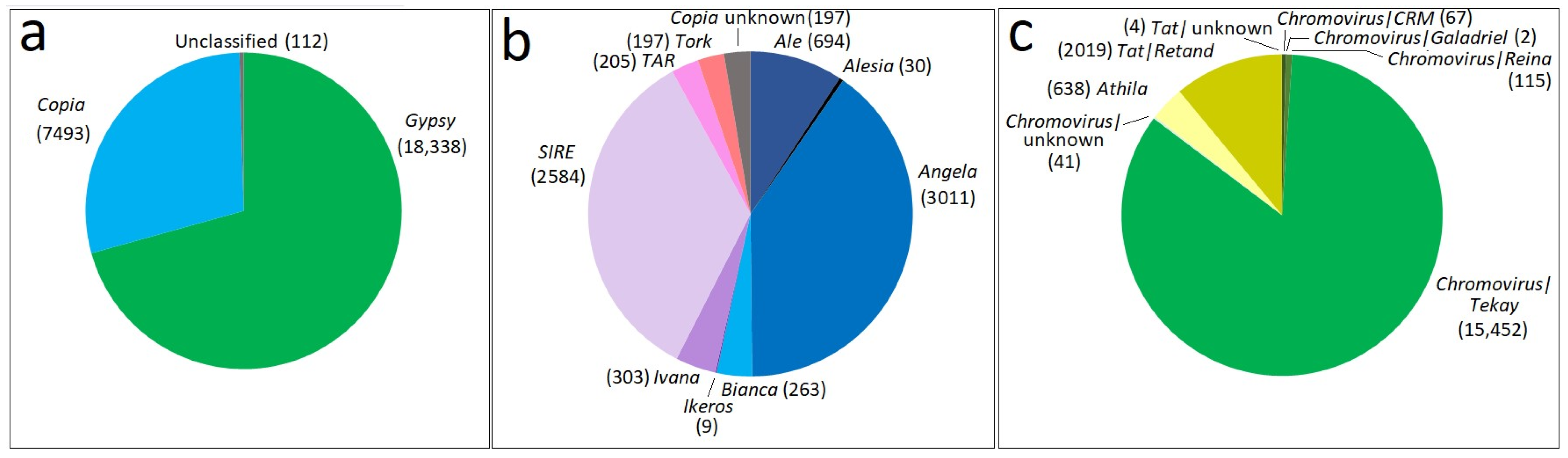
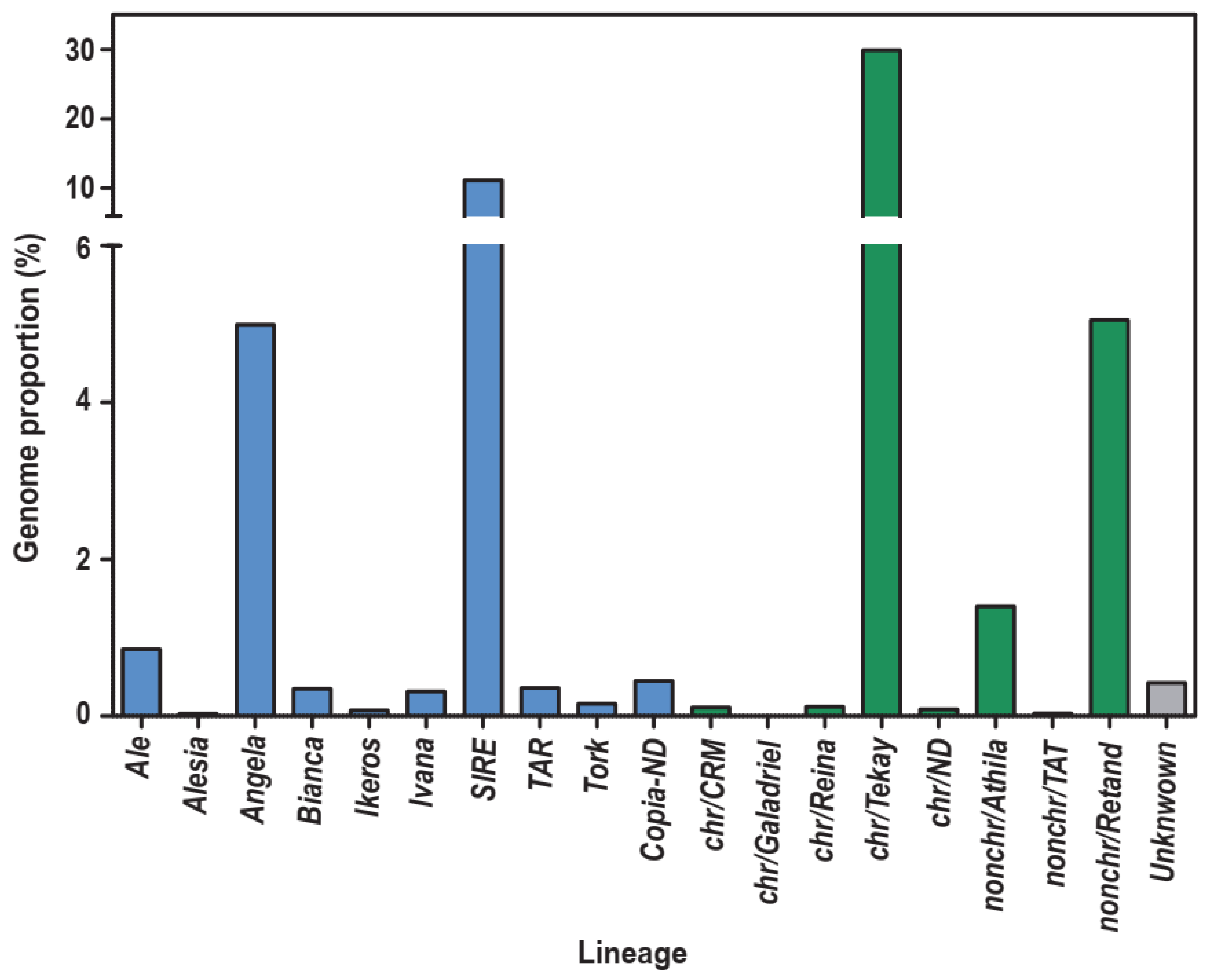
2.1. Identification and Characterisation of Full-Length LTR Retrotransposons in the S. rebaudiana Genome
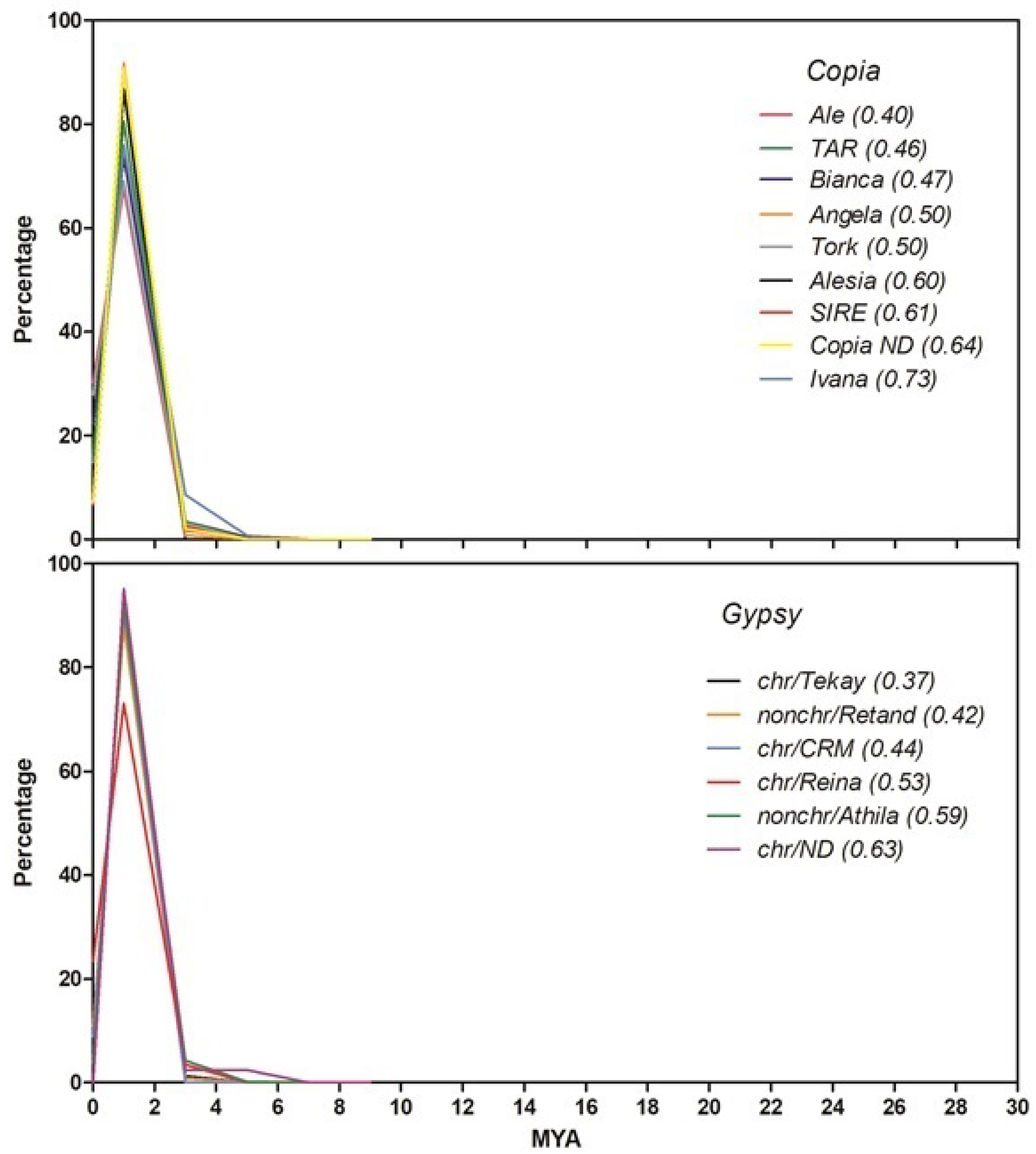
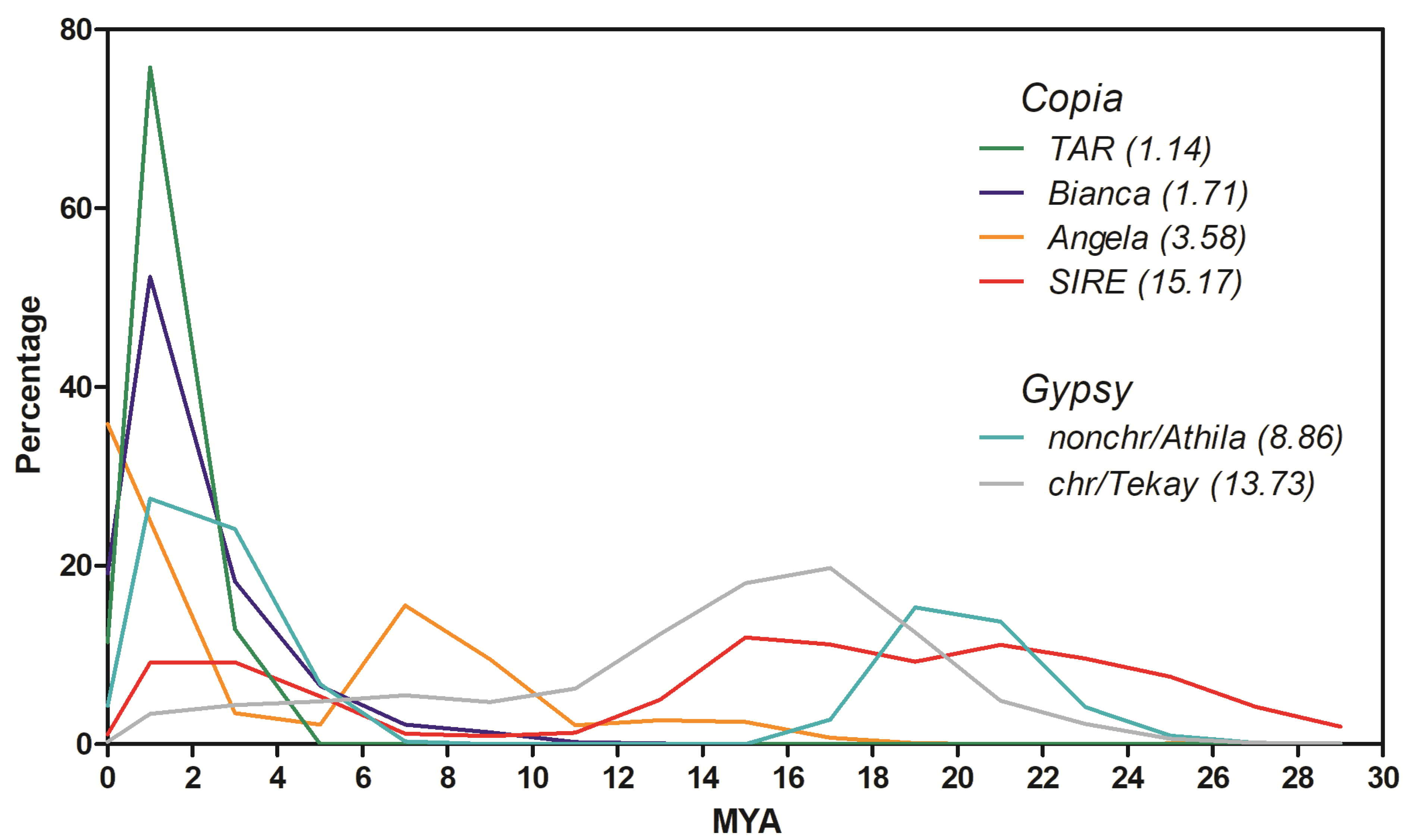
2.2. Insertion Time Profiles of Stevia LTR Retrotransposons
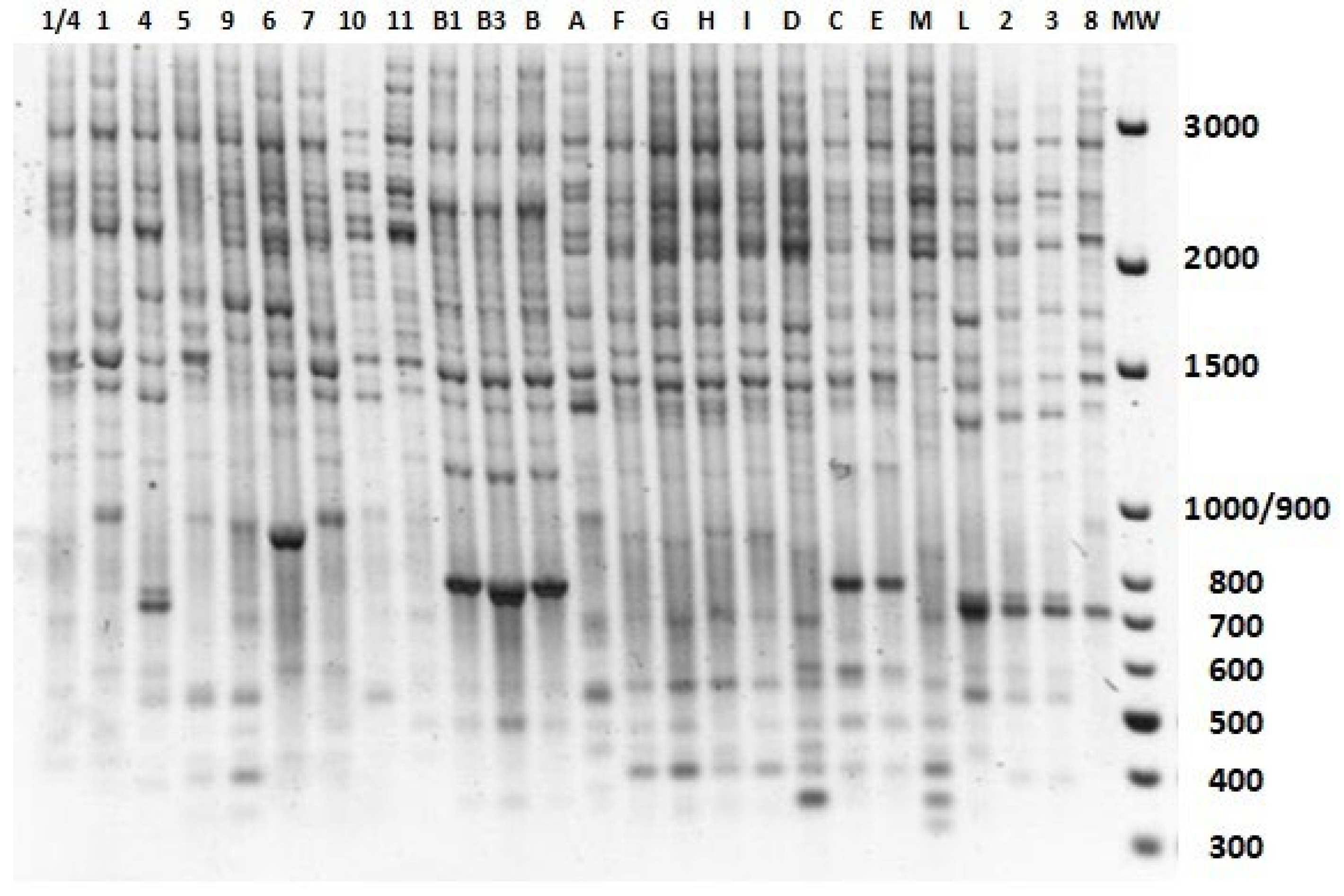
2.3. IRAP-Based Analysis of Genetic Variability among Stevia rebaudiana Accessions
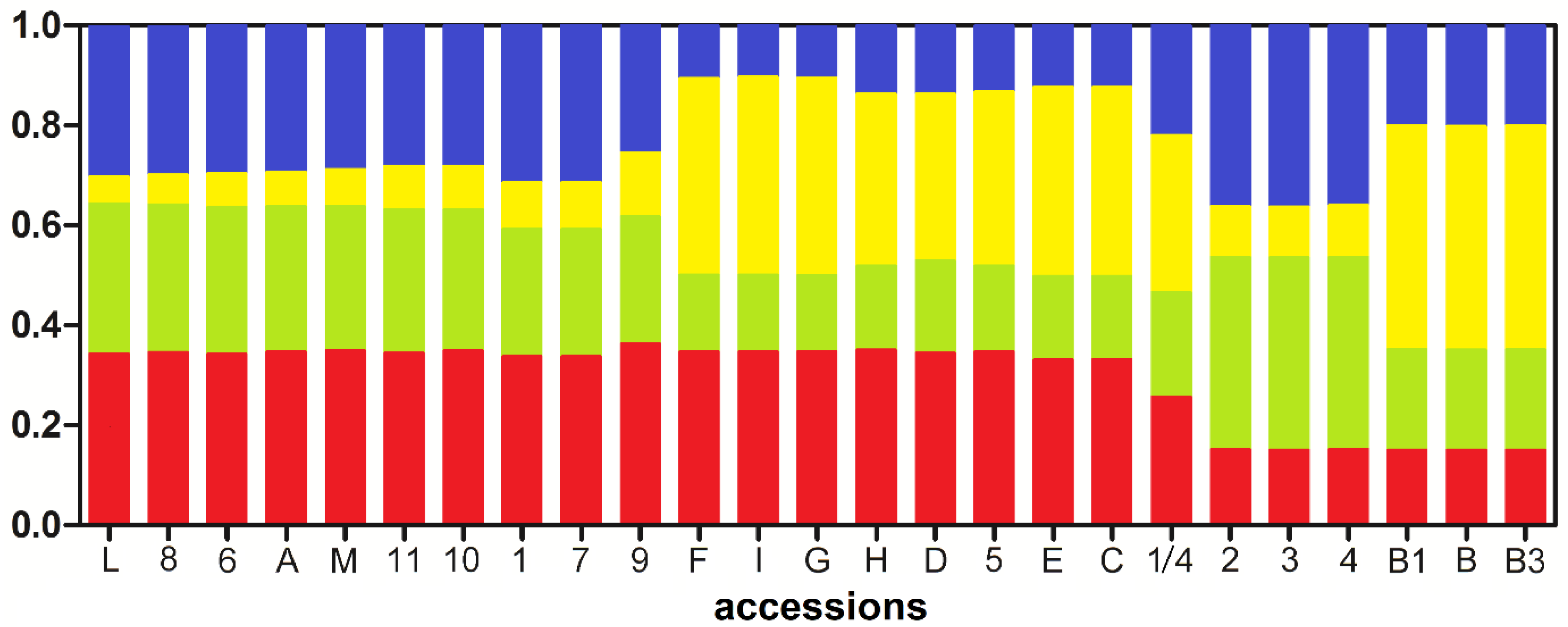
2.4. Analysis of Population Structure
3. Discussion
3.1. Repetitive Component of the S. rebaudiana Genome
3.2. Use of LTR-Retrotransposons for Genetic Variability Analyses
4. Materials and Methods
4.1. Full-Length LTR-Retrotransposons Collection and Characterisation
4.2. Plant Materials and DNA Isolation
4.3. IRAP Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Code | Sequence |
|---|---|
| ANG RNase+ (forward) | 5′-ATGGGACTTCGWTATTCTAGTG-3′ |
| ANG 1− (reverse) | 5′-TTTGAGAGCGGGTCAGTCCAA-3′ |
| ANG 3− (reverse) | 5′-CCATTCAATAACATCATCATCT-3′ |
| ANG 44− (reverse) | 5′-TTATTTACTTATGTTATTTACCA-3′ |
| ANG 44+ (forward) | 5′-ATTGGTAAATAACATAAGTAAAT-3′ |
| ANG 4− (reverse) | 5′-CACAAGCTTGTATACCCCAAG-3′ |
| ANG 5+ (forward) | 5′-TTCAAGAATCACACCCTCTA-3′ |
| ANG 55+ (forward) | 5′-TCATAACCTAGCCAAGACCT-3′ |
| ANG 55− (reverse) | 5′-AGGTCTTGGCTAGGTTATGA-3′ |
| ANG 6+ (forward) | 5′-AACAAACGCGACAAACTAAAAC-3′ |
| ANG gag− (reverse) | 5′-CAATTCTCAAGTTTCGATACCA-3′ |
4.4. Analysis of Population Structure
4.5. Analysis of Proximity between Retrotransposons and Genes Involved in Steviol Glycoside Accumulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| LTR | Long-terminal repeats |
| REs | Retrotransposons |
| TEs | Transposable elements |
| MYA | Million Years Ago |
| SVglys | Steviol glycosides |
References
- Woo, T.H.; Hong, T.H.; Kim, S.S.; Chung, W.H.; Kang, H.J.; Kim, C.B.; Seo, J.M. Repeatome: A database for repeat element comparative analysis in human and chimpanzee. Genom. Inform. 2007, 5, 179–187. [Google Scholar]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef]
- Galindo-González, L.; Mhiri, C.; Deyholos, M.K.; Grandbastien, M.A. LTR retrotransposons in plants: Engines of evolution. Gene 2017, 626, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Sabot, F.; Schulman, A.H. Parasitism and the retrotransposon life cycle in plants: A hitchhiker’s guide to the genome. Heredity 2006, 97, 381–388. [Google Scholar] [CrossRef] [Green Version]
- Orgel, L.; Crick, F. Selfish DNA: The ultimate parasite. Nature 1980, 284, 604–607. [Google Scholar] [CrossRef]
- Hawkins, J.S.; Kim, H.; Nason, J.D.; Wing, R.A.; Wendel, J.F. Differential lineage-specific amplification of transposable elements is responsible for genome size variation in Gossypium. Genome Res. 2006, 16, 1252–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubin, M.J.; Scheid, O.M.; Becker, C. Transposons: A blessing curse. Curr. Opin. Plant Biol. 2018, 42, 23–29. [Google Scholar] [CrossRef]
- Slotkin, R.K.; Martienssen, R. Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 2007, 8, 272. [Google Scholar] [CrossRef]
- Van Driel, R.; Fransz, P.F.; Verschure, P.J. The eukaryotic genome: A system regulated at different hierarchical levels. J. Cell Sci. 2003, 116, 4067–4075. [Google Scholar] [CrossRef] [Green Version]
- Song, J.T.; Lu, H.; McDowell, J.M.; Greenberg, J.T. A key role for ALD1 in activation of local and systemic defenses in Arabidopsis. Plant J. 2004, 40, 200–212. [Google Scholar] [CrossRef]
- Hollister, J.D.; Gaut, B.S. Epigenetic silencing of transposable elements: A tradeoff between reduced transposition and deleterious effects on neighboring gene expression. Genome Res. 2009, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollister, J.D.; Smith, L.M.; Guo, Y.L.; Ott, F.; Weigel, D.; Gaut, B.S. Transposable elements and small RNAs contribute to gene expression divergence between Arabidopsis thaliana and Arabidopsis lyrata. Proc. Natl. Acad. Sci. USA 2011, 108, 2322–2327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usai, G.; Vangelisti, A.; Simoni, S.; Giordani, T.; Natali, L.; Cavallini, A.; Mascagni, F. DNA modification patterns within the transposable elements of the fig (Ficus carica L.) genome. Plants 2021, 10, 451. [Google Scholar] [CrossRef] [PubMed]
- Wicker, T.; Keller, B. Genome-wide comparative analysis of copia retrotransposons in Triticeae, rice, and Arabidopsis reveals conserved ancient evolutionary lineages and distinct dynamics of individual copia families. Genome Res. 2007, 17, 1072–1081. [Google Scholar] [CrossRef] [Green Version]
- Llorens, C.; Futami, R.; Covelli, L.; Domínguez-Escribá, L.; Viu, J.M.; Tamarit, D.; Aguilar-Rodríguez, J.; Vicente-Ripolles, M.; Fuster, G.; Bernet, G.P.; et al. The Gypsy Database (GyDB) of mobile genetic elements: Release 2.0. Nucleic Acids Res. 2011, 39, 70–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barghini, E.; Mascagni, F.; Natali, L.; Giordani, T.; Cavallini, A. Analysis of the repetitive component and retrotransposon population in the genome of a marine angiosperm, Posidonia oceanica (L.) Delile. Mar. Genom. 2015, 24, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Usai, G.; Mascagni, F.; Natali, L.; Giordani, T.; Cavallini, A. Comparative genome-wide analysis of repetitive DNA in the genus Populus L. Tree Genet. Genomes 2017, 13, 96. [Google Scholar] [CrossRef] [Green Version]
- Buti, M.; Moretto, M.; Barghini, E.; Mascagni, F.; Natali, L.; Brilli, M.; Lomsadze, A.; Sonego, P.; Giongo, L.; Alonge, M.; et al. The genome sequence and transcriptome of Potentilla micrantha and their comparison to Fragaria vesca (the woodland strawberry). GigaScience 2018, 7, giy010. [Google Scholar] [CrossRef] [Green Version]
- Neumann, P.; Novák, P.; Hoštáková, N.; Macas, J. Systematic survey of plant LTR retrotransposons elucidates phylogenetic relationships of their polyprotein domains and provides a reference for element classification. Mob. DNA 2019, 10, 1. [Google Scholar] [CrossRef]
- Gorinšek, B.; Gubenšek, F.; Kordiš, D. Evolutionary genomics of chromoviruses in eukaryotes. Mol. Biol. Evol. 2004, 21, 781–798. [Google Scholar] [CrossRef] [Green Version]
- Mascagni, F.; Vangelisti, A.; Usai, G.; Giordani, T.; Cavallini, A.; Natali, L. A computational genome-wide analysis of long terminal repeats retrotransposon expression in sunflower roots (Helianthus annuus L.). Genetica 2020, 148, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Lemus-Mondaca, R.; Vega-Gálvez, R.; Zura-Bravo, L.; Ah-Hen, K. Stevia rebaudiana Bertoni, source of a high-potency natural sweetener: A comprehensive review on the biochemical, nutritional and functional aspects. Food Chem. 2012, 132, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Angelini, L.G.; Martini, A.; Passera, B.; Tavarini, S. Cultivation of Stevia rebaudiana Bertoni and associated challenges. In Sweeteners; Mérillon, J.M., Ramawat, K., Eds.; Reference Series in Phytochemistry; Springer: Cham, Switzerland, 2018. [Google Scholar]
- Soejarto, D.D. Botany of Stevia and Stevia rebaudiana. In Stevia—The Genus Stevia; Kinghorn, A., Ed.; Taylor and Francis: Abingdon, UK; New York, NY, USA, 2002; pp. 18–39. [Google Scholar]
- Kumari, N.; Raina, R.; Sharma, P. Interpopulation variations in morphochemical characteristics of Stevia rebaudiana Bertoni. Turk. J. Bot. 2018, 42, 491–501. [Google Scholar] [CrossRef]
- Brandle, J.; Starratt, A.; Gijzen, M. Stevia rebaudiana: Its agricultural, biological, and chemical properties. Can. J. Plant Sci. 1998, 78, 527–536. [Google Scholar] [CrossRef] [Green Version]
- Angelini, L.; Tavarini, S. Crop productivity, steviol glycoside yield, nutrient concentration and uptake of Stevia rebaudiana Bert. under Mediterranean field conditions. Commun. Soil Sci. Plant. Anal. 2014, 45, 2577–2592. [Google Scholar] [CrossRef]
- Barbet-Massin, C.; Giuliano, S.; Alletto, L.; Daydé, J.; Berger, M. Towards a semi-perennial culture of Stevia rebaudiana (Bertoni) under temperate climate: Effects of genotype, environment and plant age on steviol glycoside content and composition. Genet. Resour. Crop. Evol. 2016, 63, 685–694. [Google Scholar] [CrossRef]
- Hastoy, C.; Cossona, P.; Cavaignac, S.; Boutié, P.; Waffo-Teguo, P.; Rolin, D.; Schurdi-Levrauda, V. Deciphering performances of fifteen genotypes of Stevia rebaudiana in southwestern France through dry biomass and steviol glycoside evaluation. Ind. Crops Prod. 2019, 128, 607–619. [Google Scholar] [CrossRef]
- Tavarini, S.; Passera, B.; Angelini, L. Crop and steviol glycoside improvement in stevia by breeding. In Steviol Glycosides: Cultivation, Processing, Analysis and Applications in Food; The Royal Society of Chemistry: London, UK, 2018; pp. 1–31. [Google Scholar]
- Clemente, C.; Angelini, L.G.; Ascrizzi, R.; Tavarini, S. Stevia rebaudiana (Bertoni) as a multifunctional and sustainable crop for the mediterranean climate. Agriculture 2021, 11, 123. [Google Scholar] [CrossRef]
- Garcia, S.; Hidalg, O.; Jakovljević, I.; Siljak-Yakovlev, S.; Vigo, J.; Garnatje, T.; Vallès, J. New data on genome size in 128 Asteraceae species and subspecies, with first assessments for 40 genera, 3 tribes and 2 subfamilies. Plant Biosyst. 2013, 147, 1219–1227. [Google Scholar] [CrossRef]
- Xu, X.; Yuan, H.; Yu, X.; Huang, S.; Sun, Y.; Zhang, T.; Liu, Q.; Tong, H.; Zhang, Y.; Wang, Y.; et al. The chromosome-level Stevia genome provides insights into steviol glycoside biosynthesis. Hort. Res. 2021, 8, 129. [Google Scholar] [CrossRef]
- Schulman, A.H.; Flavell, A.J.; Ellis, T.H.N. The application of LTR retrotransposons as molecular markers in plants. Methods Mol. Biol. 2004, 260, 145–173. [Google Scholar] [PubMed]
- Kalendar, R.; Schulman, A.H. IRAP and REMAP for retrotransposon based genotyping and fingerprinting. Nat. Protoc. 2006, 1, 2478–2484. [Google Scholar] [CrossRef] [PubMed]
- Kalendar, R.; Grob, T.; Regina, M.; Suoniemi, A.; Schulman, A.H. IRAP and REMAP: Two new retrotransposon-based DNA fingerprinting techniques. Theor. Appl. Genet. 1999, 98, 704–711. [Google Scholar] [CrossRef]
- Ellinghaus, D.; Kurtz, S.; Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 2008, 9, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vangelisti, A.; Simoni, S.; Usai, G.; Ventimiglia, M.; Natali, L.; Cavallini, A.; Mascagni, F.; Giordani, T. LTR-retrotransposon dynamics in common fig (Ficus carica L.) genome. BMC Plant Biol. 2021, 21, 221. [Google Scholar] [CrossRef] [PubMed]
- SanMiguel, P.; Gaut, B.S.; Tikhonov, A.; Nakajima, Y.; Bennetzen, J.L. The paleontology of intergene retrotransposons of maize. Nat. Genet. 1998, 20, 43–45. [Google Scholar] [CrossRef]
- SanMiguel, P.; Tikhonov, A.; Jin, Y.K.; Motchoulskaia, N.; Zakharov, D.; Melake-Berhan, A.; Springer, P.S.; Edwards, K.J.; Lee, M.; Avramova, Z. Nested retrotransposons in the intergenic regions of the maize genome. Science 1996, 274, 765–768. [Google Scholar] [CrossRef] [Green Version]
- Mascagni, F.; Usai, G.; Natali, L.; Cavallini, A.; Giordani, T. A comparison of methods for LTR-retrotransposon insertion time profiling in the Populus trichocarpa genome. Caryologia 2018, 71, 85–92. [Google Scholar] [CrossRef]
- Mascagni, F.; Cavallini, A.; Giordani, T.; Natali, L. Different histories of two highly variable LTR retrotransposons in sunflower species. Gene 2017, 634, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Piegu, B.; Guyot, R.; Picault, N.; Roulin, A.; Saniyal, A.; Kim, H.; Collura, K.; Brar, D.S.; Jackson, S.; Wing, R.A.; et al. Doubling genome size without polyploidization: Dynamics of retrotransposition-driven genomic expansions in Oryza australiensis, a wild relative of rice. Genome Res. 2006, 16, 1262–1269. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Bennetzen, J.L. Plant retrotransposons. Ann. Rev. Genet. 1999, 33, 479–532. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Vigouroux, Y.; Glaubitz, J.C.; Matsuoka, Y.; Major, M.; Doebley, J. Population structure and genetic diversity of the new world maize races assessed by microsatellites. Am. J. Bot. 2008, 95, 1240–1253. [Google Scholar] [CrossRef] [PubMed]
- Castillo, A.; Dorado, G.; Feuillet, C.; Sourdille, P.; Hernandez, P. Genetic structure and ecogeographical adaptation in wild barley (Hordeum chilense Roemer et Schultes) as revealed by microsatellite markers. BMC Plant Biol. 2010, 10, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahid, S.; Slotkin, R.K. The current revolution in transposable element biology enabled by long reads. Curr. Opin. Plant Biol. 2020, 54, 49–56. [Google Scholar] [CrossRef]
- Buti, M.; Giordani, T.; Cattonaro, F.; Cossu, R.M.; Pistelli, L.; Vukich, M.; Morgante, M.; Cavallini, A.; Natali, L. Temporal dynamics in the evolution of the sunflower genome as revealed by sequencing and annotation of three large genomic regions. Theor. Appl. Genet. 2011, 123, 779–791. [Google Scholar] [CrossRef]
- Natali, L.; Cossu, R.M.; Barghini, E.; Giordani, T.; Buti, M.; Mascagni, F.; Morgante, M.; Gill, N.; Kane, N.C.; Rieseberg, L.; et al. The repetitive component of the sunflower genome as shown by different procedures for assembling next generation sequencing reads. BMC Genom. 2013, 14, 686. [Google Scholar] [CrossRef] [Green Version]
- Ventimiglia, M. ASTER-REP, a Database of Asteraceae Sequences for Studying Structure and Function of Transposable Elements. Ph.D. Thesis, University of Pisa, Pisa, Italy, 2022. [Google Scholar]
- Badouin, H.; Gouzy, J.; Grassa, C.J.; Murat, F.; Staton, S.E.; Cottret, L.; Lelandais-Brière, C.; Owens, G.L.; Carrère, S.; Mayjonade, B.; et al. The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature 2017, 546, 148–152. [Google Scholar] [CrossRef]
- Zagorski, D.; Hartmann, M.; Bertrand, Y.J.; Paštová, L.; Slavíková, R.; Josefiová, J.; Fehrer, J. Characterization and dynamics of repeatomes in closely related species of Hieracium (Asteraceae) and their synthetic and apomictic hybrids. Front. Plant Sci. 2020, 11, 591053. [Google Scholar] [CrossRef]
- Vitte, C.; Fustier, M.A.; Alix, K.; Tenaillon, M.I. The bright side of transposons in crop evolution. Brief. Funct. Genom. 2014, 13, 276–295. [Google Scholar] [CrossRef] [Green Version]
- Goubert, C.; Modolo, L.; Vieira, C.; Valiente Moro, C.; Mavingui, P.; Boulesteix, M. De novo assembly and annotation of the asian tiger mosquito (Aedes albopictus) repeatome with dnaPipeTE from raw genomic reads and comparative analysis with the yellow fever mosquito (Aedes aegypti). Genome Biol. Evol. 2015, 7, 1192–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staton, S.E.; Burke, J.M. Evolutionary transitions in the Asteraceae coincide with marked shifts in transposable element abundance. BMC Genom. 2015, 16, 623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mascagni, F.; Giordani, T.; Ceccarelli, M.; Cavallini, A.; Natali, L. Genome-wide analysis of LTR-retrotransposon diversity and its impact on the evolution of the genus Helianthus (L.). BMC Genom. 2017, 18, 634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mccann, J.; Jang, T.S.; Macas, J.; Schneeweiss, G.M.; Matzke, N.J.; Novák, P.; Stuessy, T.F.; Villaseñor, J.L.; Weiss-Schneeweiss, H. Dating the species network: Allopolyploidy and repetitive DNA evolution in American daisies (Melampodium sect. Melampodium, Asteraceae). Syst. Biol. 2018, 67, 1010–1024. [Google Scholar] [CrossRef]
- Vitales, D.; Álvarez, I.; Garcia, S.; Hidalgo, O.; Nieto Feliner, G.; Pellicer, J.; Vallès, J.; Garnatje, T. Genome size variation at constant chromosome number is not correlated with repetitive DNA dynamism in Anacyclus (Asteraceae). Ann. Bot. 2019, 125, 611–623. [Google Scholar] [CrossRef]
- Arnaud, P.; Goubely, C.; Pelissier, T.; Deragon, J.M. SINE retroposons can be used in vivo as nucleation centers for de novo methylation. Mol. Cell Biol. 2000, 20, 3434–3441. [Google Scholar] [CrossRef]
- Studer, A.; Zhao, Q.; Ross-Ibarra, J.; Dobley, J. Identification of a functional transposon insertion in the maize domestication gene tb1. Nat. Genet. 2011, 43, 1160–1163. [Google Scholar] [CrossRef] [Green Version]
- Ou, S.; Jiang, N. LTR_FINDER_parallel: Parallelization of LTR_FINDER enabling rapid identification of long terminal repeat retrotransposons. Mob. DNA 2019, 10, 48. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Wang, H. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucl. Acids Res. 2007, 35, W265–W268. [Google Scholar] [CrossRef] [Green Version]
- Ou, S.; Jiang, N. LTR_retriever: A highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 2018, 176, 1410–1422. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European molecular biology open software suite. TIG 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Ammiraju, J.S.; Zuccolo, A.; Yu, Y.; Song, X.; Piegu, P.; Chevalier, F.; Walling, J.G.; Ma, J.; Talag, J.; Brar, D.S.; et al. Evolutionary dynamics of an ancient retrotransposon family provides insights into evolution of genome size in the genus Oryza. Plant J. 2007, 52, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 5 July 2021).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Doyle, J.J.; Doyle, J.L. Isolation of plant DNA from fresh tissue. Focus 1989, 12, 13–15. [Google Scholar]
- Rychlik, W.; Rhoads, R.E. A computer program for choosing optimal oligonudeotides for filter hybridization, sequencing and in vitro amplification of DNA. Nucleic Acids Res. 1989, 17, 8543–8551. [Google Scholar] [CrossRef]
- Vukich, M.; Schulman, A.H.; Giordani, T.; Natali, L.; Kalendar, R.; Cavallini, A. Genetic variability in sunflower (Helianthus annuus L.) and in the Helianthus genus as assessed by retrotransposon-based molecular markers. Theor. Appl. Genet. 2009, 119, 1027–1038. [Google Scholar] [CrossRef]
- Lynch, M.; Milligan, B.G. Analysis of population genetic structure with RAPD markers. Mol. Ecol. 1994, 3, 91–99. [Google Scholar] [CrossRef]
- Earl, D.A.; Vonholdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Res. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Rosenberg, N.A.; Pritchard, J.K.; Weber, J.L.; Cann, H.M.; Kidd, K.K.; Zhivotovsky, L.A.; Feldman, M.W. The genetic structure of human populations. Science 2002, 298, 2381–2385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]






| Gene Family | Number of Genes |
|---|---|
| UDP-glycosyltransferase | 18 |
| Ent-kaurene oxidase | 5 |
| Geranylgeranyl pyrophosphate synthase | 3 |
| Methyl-erythritol-phosphate cytidylyltransferase | 3 |
| Hydroxy-methylbutenyl diphosphate synthase (ferredoxin) | 2 |
| Deoxy-xylulose-phosphate reductoisomerase | 1 |
| Deoxy-xylulose-phosphate synthase | 1 |
| Isopentenyl-diphosphate Delta-isomerase | 1 |
| Methyl-erythritol cyclodiphosphate synthase | 1 |
| ID Code | Accession Name | Origin | Cultivated in |
|---|---|---|---|
| 1/4 | MA1/4 | Brasil | pot |
| 1 | MApt1 | Brasil | pot |
| 2 | MA13/1 | Brasil | pot |
| 3 | MApt2 | Brasil | pot |
| 4 | MA10/1 | Brasil | pot |
| 5 | MApt5 | Brasil | pot |
| 6 | MA7/3 | Brasil | pot |
| 7 | MApt3 | Brasil | pot |
| 8 | MApt4 | Brasil | pot |
| 9 | SV1 | Brasil | pot |
| 10 | Criolla | Paraguay | pot |
| 11 | Sweet Herb | Paraguay | pot |
| A | PL | Israel | field |
| B | BR16 | Brasil | pot |
| C | RGm | Italy | field |
| D | SL | Israel | field |
| E | Num | Italy | pot |
| F | CO | Israel | field |
| G | BR5 | Brasil | field |
| H | SW30 | Italy | field |
| I | BR1 | Brasil | field |
| L | MASV4-2/2 | Brasil | pot |
| M | Eirete | Paraguay | pot |
| B1 | BR16 | Brasil | pot |
| B3 | BR16 | Brasil | pot |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simoni, S.; Clemente, C.; Usai, G.; Vangelisti, A.; Natali, L.; Tavarini, S.; Angelini, L.G.; Cavallini, A.; Mascagni, F.; Giordani, T. Characterisation of LTR-Retrotransposons of Stevia rebaudiana and Their Use for the Analysis of Genetic Variability. Int. J. Mol. Sci. 2022, 23, 6220. https://doi.org/10.3390/ijms23116220
Simoni S, Clemente C, Usai G, Vangelisti A, Natali L, Tavarini S, Angelini LG, Cavallini A, Mascagni F, Giordani T. Characterisation of LTR-Retrotransposons of Stevia rebaudiana and Their Use for the Analysis of Genetic Variability. International Journal of Molecular Sciences. 2022; 23(11):6220. https://doi.org/10.3390/ijms23116220
Chicago/Turabian StyleSimoni, Samuel, Clarissa Clemente, Gabriele Usai, Alberto Vangelisti, Lucia Natali, Silvia Tavarini, Luciana G. Angelini, Andrea Cavallini, Flavia Mascagni, and Tommaso Giordani. 2022. "Characterisation of LTR-Retrotransposons of Stevia rebaudiana and Their Use for the Analysis of Genetic Variability" International Journal of Molecular Sciences 23, no. 11: 6220. https://doi.org/10.3390/ijms23116220