
The Dynamism of Transposon Methylation for Plant Development and Stress Adaptation


 ,
,  , ,
, , 
 ,
,
Abstract
:1. Introduction

2. TE Classification and Copy Number in Plants

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Subclass | Superfamily/Family | Plants | Autonomous Members | Non-Autonomous Members | Copy Number of the Entire Family | References |
|---|---|---|---|---|---|---|---|
| Class I | LTR Retrotransposons | copia-like | O. sativa | Tos17 | - | (2–5) 30 | [65] |
| copia-like | Hordeum sp. | BARE-1 | - | 5000–22,000 | [66] | ||
| copia-like | N. tabacum | Tto1 | - | 30 (300) | [67] | ||
| copia-like | N. tabacum | Tnt1A | - | >100 | [68] | ||
| copia-like | Z. mays | Hopscotch | - | 5–8 | [69] | ||
| copia-like | Z. mays | - | BS1 | 1–5 | [70] | ||
| copia-like | Z. mays | Opie-2 | - | 100,000 | [71] | ||
| gypsy-like | O. sativa | RIRE2 | Dasheng | 1200 | [72] | ||
| gypsy-like | Z. mays | Magellan | - | 4–8 | [73] | ||
| gypsy-like | Z. mays | Huck-2 | - | 200,000 | [71] | ||
| gypsy-like | Arabidopsis | Athila 4 | - | 22 | [74] | ||
| gypsy-like | Arabidopsis | Ta3 | - | 1 | [75] | ||
| gypsy-like | Arabidopsis | Athila 6 | - | 11 | [74] | ||
| gypsy-like | Arabidopsis | Tar17 | - | 2 | [67] | ||
| Non-LTR Retrotransposons | LINEs; L1-clade | Lilium speciosum | Del2 | - | 250,000 | [76] | |
| LINEs; L1-clade | Z. mays | Cin4 | - | 50–100 | [77] | ||
| LINEs; L1-clade | Arabidopsis | Tal1 | - | 1–6 | [78] | ||
| SINEs | N. tabacum | - | TS | 50,000 | [79] | ||
| SINEs | B. napus | - | S1 | 500 | [80] | ||
| Class II | DNA transposons | Mutator | Z. mays | MuDR | Mu1 | 10–100 | [81] |
| Mutator | Arabidopsis | AtMu1 | - | 1 (4) | [82] | ||
| CACTA | Z. mays | Spm | dSpm | 50–100 | [83] | ||
| CACTA | Arabidopsis | CAC1 | CAC2 | (4) 20 | [84] | ||
| hAT | Z. mays | Ac | Ds | 50–100 | [85] | ||
| PIF/Harbinger | Z. mays | PIFa | mPIF | 6000 | [86] | ||
| PIF/Harbinger | Angiosperms | PIF-like | Tourist-like | Variable | [86,87] | ||
| Tc1/Mariner | Angiosperms | MLEs | Stowaway-like | Variable | [88,89] |
3. Surprising Traits of TEs
4. Contribution of TEs in the Plant Genome
| Plant Genome | Total Genome Size (Mb) | Total TE Content (% of the Genome) | Total Class I or RNA (Retroelements) (% of the Genome) | Total Class II or DNA Transposons (% of the Genome) |
|---|---|---|---|---|
| Aegilops tauschii | 4.98 | 68.20 | 13.30 | 53.50 |
| Arabidopsis lyrata | 230.00 | 29.70 | 15.99 | 4.80 |
| Arabidopsis thaliana | 125.00 | 14.00–18.50 | 7.50 | 11.00 |
| Brachypodium distachyon | 355.00 | 28.10 | 23.33 | 4.77 |
| Brassica oleracea | 600.00 | 20.00 | 14.00 | 6.00 |
| Brassica rapa | 529.00 | 39.51 | 29.90 | 3.20 |
| Cajanus cajan | 833.00 | 51.67 | 19.18 | 4.53 |
| Carica papaya | 372.00 | 51.90 | 42.80 | 0.60 |
| Cicer arietinum | 738.00 | 49.41 | 45.64 | 9.32 |
| Citrus sinensis | 367.00 | 20.50 | 18.21 | 2.28 |
| Cucumis melo | 450.00 | 19.70 | 14.70 | 5.00 |
| Cucumis sativus | 367.00 | 24.01 | 12.16 | 1.24 |
| Fragaria vesca | 240.00 | 22.81 | 16.37 | 6.44 |
| Glycine max | 1115.00 | 58.74 | 42.24 | 16.50 |
| Gossypium herbaceum | 1660.00 | 52.10 | 52.00 | 0.10 |
| Gossypium raimondii | 880.00 | 56.95 | 48.99 | 4.54 |
| Gossypium raimondii | 880.00 | 61.30 | 54.90 | 1.50 |
| Hordeum vulgare | 5100.00 | 58.89 | 52.83 | 5.25 |
| Linum usitatissimum | 370.00 | 24.29 | 20.62 | 3.80 |
| Lotus japonicus | 472.00 | 30.80 | 10.4–19.23 | 0.97–8.10 |
| Malus domestica | 742.00 | 42.40 | 37.60 | 0.90 |
| Medicago truncatula | 475.00 | 38.00 | 9.60 | ND |
| Medicago truncatula | 550.00 | 30.50 | 26.50 | 3.40 |
| Musa acuminata | 523.00 | 32.63 | 31.17 | 1.42 |
| Oryza sativa | 389.00 | 34.79 | 19.35 | 12.96 |
| Phyllostachys edulis | 1908.00 | 45.45 | 38.20 | 7.25 |
| Populus trichocarpa | 485.00 | 42.00 | 10.30 | 2.50 |
| Populus trichocarpa | 550.00 | 34.90 | 7.02 | 2.10 |
| Pyrus bretschneideri | 527.00 | 53.10 | 45.97 | 12.12 |
| Ricinus communis | 320.00 | 50.33 | 18.16 | 0.91 |
| Secale cereale | 8090.00 | 69.30 | 64.30 | 5.00 |
| Setaria italica (Accession Zhang gu) | 510.00 | 46.30 | 31.60 | 9.40 |
| Setaria italica (Inbred Yugu1) | 510.00 | 40.00 | 25.00 | ND |
| Solanum lycopersicum | 900.00 | 63.20 | 62.30 | 0.90 |
| Solanum tuberosum | 844.00 | 62.20 | 32.29 | 3.94 |
| Sorghum bicolor | 730.00 | 62.00 | 54.52 | 7.46 |
| Theobroma cacao | 430.00 | 25.70 | 17.70 | 8.00 |
| Vitis vinifera | 475.00 | 41.40 | 17.04 | 0.43 |
| Zea mays | 2300.00 | 84.20 | 75.60 | 8.60 |
5. Distribution of TEs in the Plant Genome
6. TE-Induced Mutations
7. Association of RTEs with Genomes
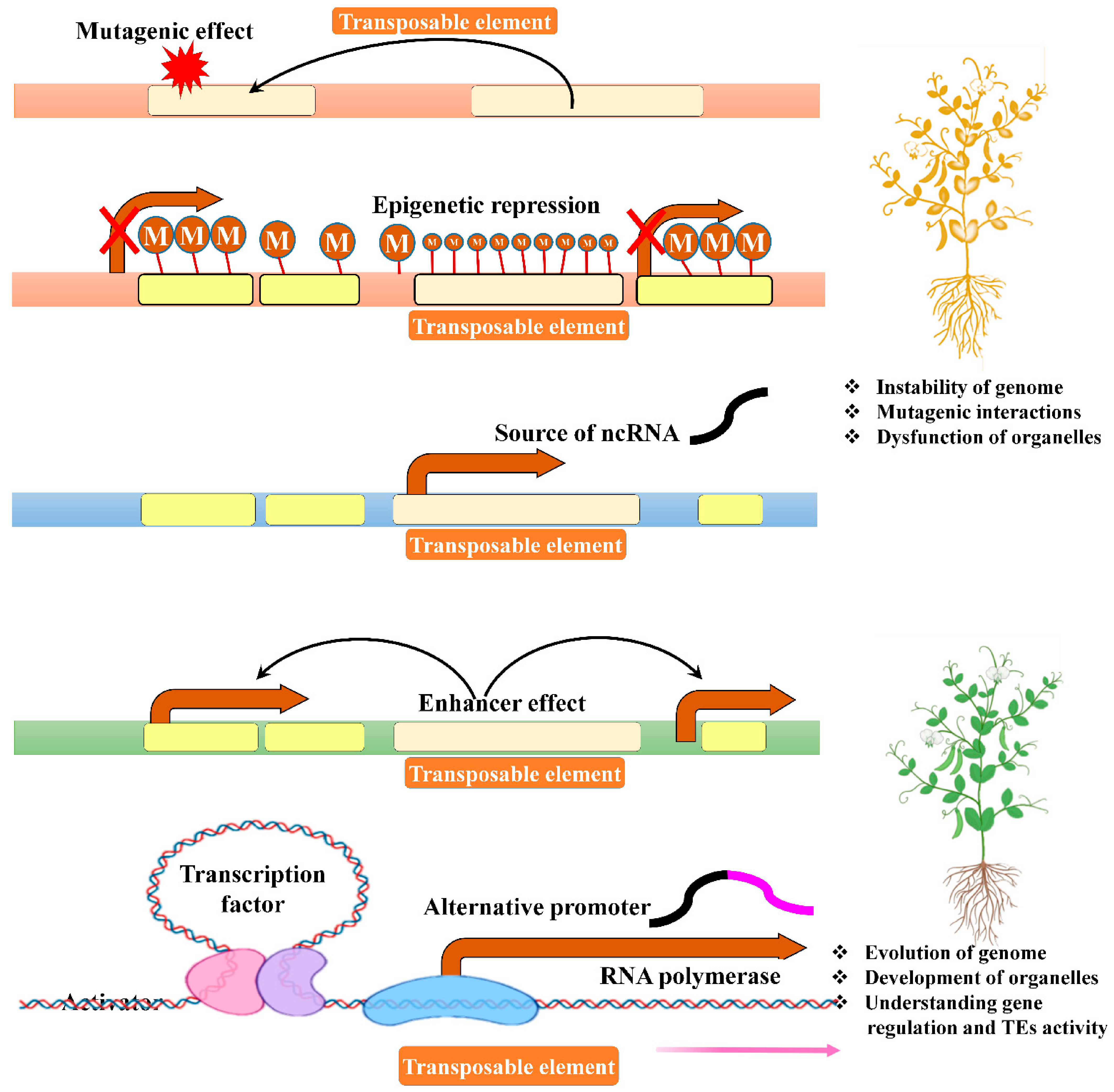
8. Balance between TE Expression and Repression
9. TE Transposition and Genome Stability
10. TE Is the Source of Non-Coding RNAs (ncRNAs)
11. Role of ncRNAs in Plant Response to Abiotic Stress
| Plant Species | siRNA | Mechanisms | Abiotic Stresses Induced/Suppressed | References |
|---|---|---|---|---|
| Arabidopsis | SRO5-P5CDH nat-siRNA | Regulation of proline metabolism | Salt stress ↓ | [180] |
| Arabidopsis | TAS1, TAS2, TAS3 ta-siRNA | Elevated expression | Hypoxia stress ↑ | [184,185,186] |
| Arabidopsis | HTT1, HTT2-TAS1 | NYE | Heat stress ↑ | [187,188] |
| Arabidopsis | TAS4 ta-siRNAs | Biosynthesis of anthocyanins | Phosphate deficiency ↑ | [189,190] |
| Arabidopsis | TAS4-siR81(-) | Accumulation of anthocyanin | Nitrogen deficiency ↑ | [190] |
| Arabidopsis | hcsiRNAs (ONSEN) | DNA methylation | Heat stress ↑ | [191,192,193] |
| Arabidopsis | hcsiRNAs (HD2C, HDA6) | DNA methylation | Drought and ABA stresses ↑ and ↓ | [194,195,196,197,198,199] |
| Arabidopsis | IPS1 * | miR399 target mimicry | Phosphate deficiency ↑ | [183,200,201] |
| Arabidopsis | lncRNAs * | Antisense transcription | Light stress ↑ | [202] |
| Arabidopsis | asHSFB2a * | Antisense transcription | Heat stress ↑ | [203] |
| Arabidopsis | COOLAIR * | Chromatin remodelling | Cold stress ↑ | [204] |
| Arabidopsis | lncRNAs * | Histone modification | Light stress ↑ | [202] |
| Arabidopsis | COLDAIR * | Histone modification | Cold stress ↑ | [205] |
| Arabidopsis | lncRNAs * | RdDM pathway | Heat stress ↑ | [206] |
| Arabidopsis | lncRNAs * | RdDM pathway | Salt stress ↓ | [207] |
| Brassica oleracea | nat-siRNAs | DNA methylation | Heat stress ↑ | [208,209] |
| Brassica rapa | nat-siRNAs | DNA methylation | Heat stress ↑ and ↓ | [209] |
| Brassica rapa | lincRNAs * | miRNA precursors | Cold and heat stresses ↑ and ↓ | [210] |
| Craterostigma plantagineum | CDT1-siRNA | NYE | Dehydration stress ↑ | [211] |
| Manihot esculenta | 2 nat-siRNA, 3 ta-siRNAs | NYE | Cold stress ↑ and ↓ | [212] |
| Oryza sativa | lncRNAs * | target mimicry | Phosphate deficiency ↑ and ↓ | [213] |
| Phaeodactylum tricornutum | pti-MIR5472 * | miR5472 precursors | Phosphate deficiency ↑ | [214] |
| Phaeodactylum tricornutum | pti-MIR5471 * | miR5471 precursors | Phosphate deficiency ↑ | [214] |
| Populus tomentosa | lincRNAs * | miRNA precursors | Nitrogen deficiency ↑ and ↓ | [215] |
| Populus tomentosa | lincRNAs * | Antisense transcription | Nitrogen deficiency ↑ and ↓ | [215] |
| Populus trichocarpa | lincRNA1128 * | ptc-miR482a.1 target mimicry | Drought stress ↓ | [216] |
| Populus trichocarpa | lincRNA1393 * | ptc-miR6459b target mimicry | Drought stress ↓ | [216] |
| Populus trichocarpa | lincRNA3018 * | ptc-miR399i target mimicry | Drought stress ↓ | [216] |
| Populus trichocarpa | lincRNA2752 * | ptc-miR169o target mimicry | Drought stress ↑ | [216] |
| Populus trichocarpa | lincRNA1795 * | ptc-miR476a target mimicry | Drought stress ↓ | [216] |
| Populus trichocarpa | lincRNA20 * | ptc-miR476a target mimicry | Drought stress ↑ | [216] |
| Populus trichocarpa | lincRNA2623 * | ptc-miR156k target mimicry | Drought stress ↓ | [216] |
| Populus trichocarpa | lincRNA2623 * | ptc-miR156c target mimicry | Drought stress ↓ | [216] |
| Populus trichocarpa | lincRNA967 * | ptc-miR6462e target mimicry | No response to drought stress | [216] |
| Populus trichocarpa | lincRNA2762 * | ptc-miR156k target mimicry | Drought stress ↓ | [216] |
| Populus trichocarpa | lincRNA1449 * | ptc-miR156k target mimicry | No response to drought stress | [216] |
| Populus trichocarpa | lincRNA179 * | ptc-miR156a target mimicry | No response to drought stress | [216] |
| Populus trichocarpa | lincRNA2198 *, lincRNA2131 *, lincRNA2085 *, lincRNA2962 * lincRNA1534 *, lincRNA1039 * lincRNA2962 * | NYE | Drought stress ↑ | [216] |
| Solanum lycopersicum | lncRNAs * | RdDM pathway | Salt and drought stresses ↓ | [217] |
| Triticum aestivum | 002061_0636_3054.1 siRNA | NYE | Heat, NaCl, and dehydration ↓ | [218] |
| Triticum aestivum | 005047_0654_1904.1 siRNA | NYE | Heat, NaCl, and dehydration ↓ | [218] |
| Triticum aestivum | 005047_0654_1904.1 siRNA | NYE | Cold stress ↑ | [218] |
| Triticum aestivum | 080621_1340_ 0098.1 siRNA | NYE | Cold stress ↑ and heat stress ↓ | [218] |
| Triticum aestivum | 007927_0100_2975.1 siRNA | NYE | Cold, NaCl, and dehydration ↓ | [218] |
| Triticum aestivum | ta-siRNA TAS3a-50D6 (+) | Auxin signalling pathway | Cold stress ↑ | [219] |
| Triticum aestivum | TalnRNA5 * | ta-miR2004 precursors | Heat stress ↑ | [218,220] |
| Triticum aestivum | TahlnRNA27 * | ta-miR2010 precursors | Heat stress ↑ | [218,220] |
| Triticum aestivum | TalnRNA21 *, TahlnRNA3 *, TahlnRNA14 *, TahlnRNA19 * TahlnRNA36 *, TahlnRNA41 * TahlnRNA42 *, TahlnRNA47 * TahlnRNA52 * | siRNA precursors | Heat stress ↑ | [218,220] |
| Zea mays | lncRNAs * | siRNA precursors and antisense transcription | Drought stress ↑ | [221] |
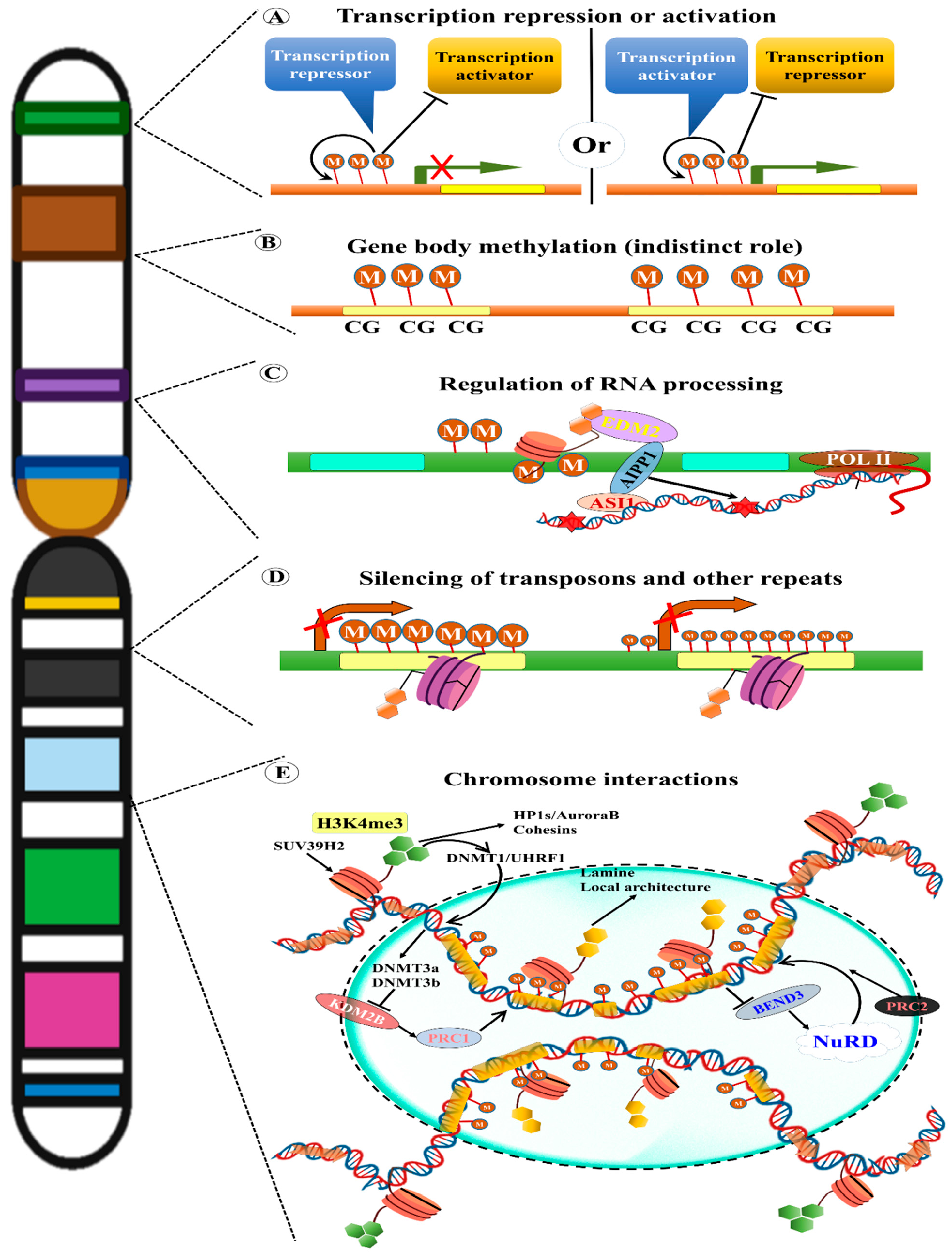
12. Epigenetic Effects of TEs
13. TE Methylation
14. TE Methylation in Plant Evolution
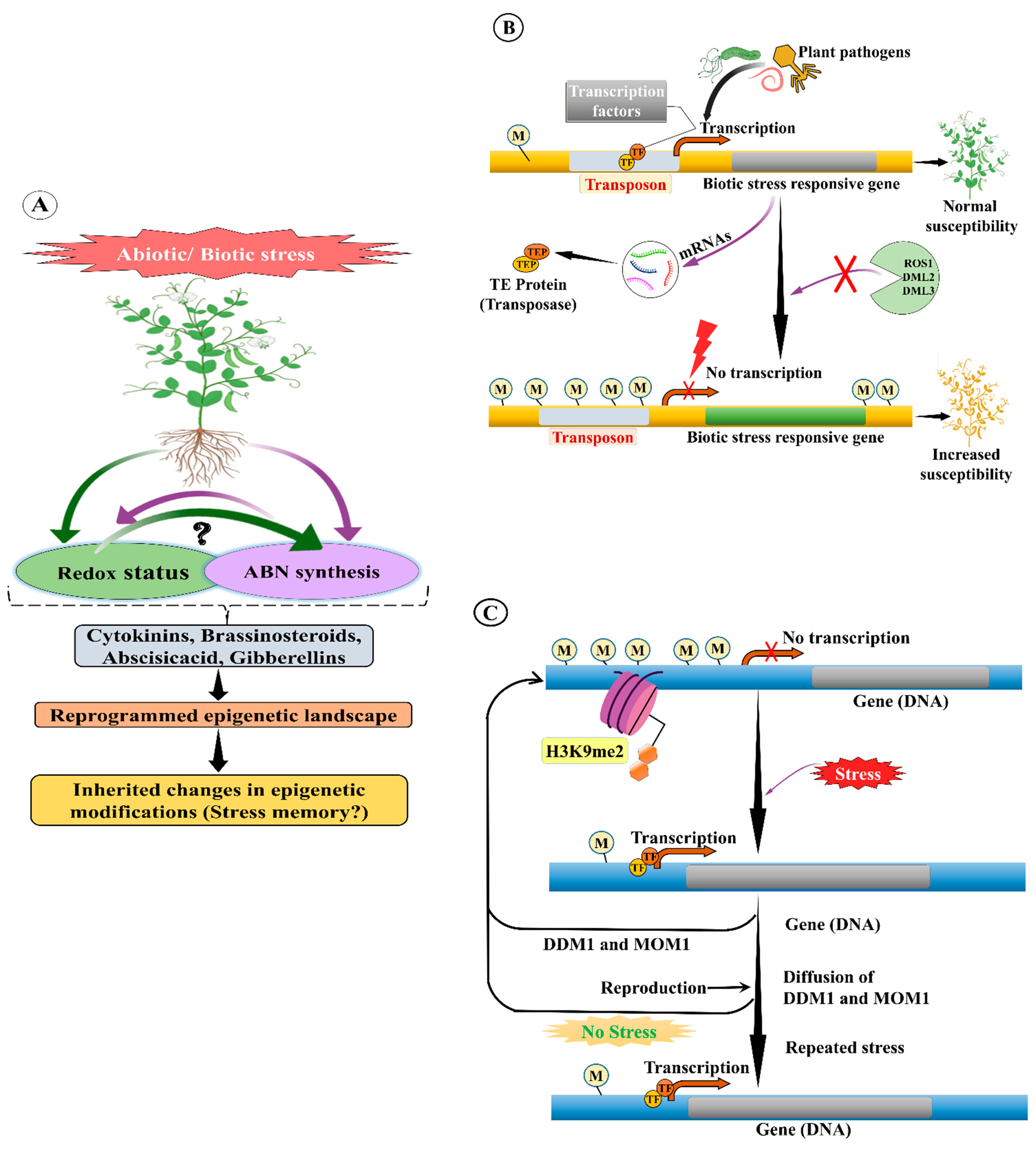
15. TE Methylation in Plant Stress Response
15.1. Abiotic Stress
| Abiotic Stress | Plants | Changes in DNA Methylation Levels | Major Effects | References |
|---|---|---|---|---|
| Cold stress | Arabidopsis | Enhanced methylation in the ALN promoter | Promotes seed dormancy | [276] |
| Cold stress | Arabidopsis | Variation in ICE1 methylation | Cold tolerance divergence in different accessions | [277,278] |
| Cold stress | B. rapa | Decreased DNA methylation levels in the BramMDH1 promoter | Increased heat tolerance and growth rate | [279] |
| Cold stress | B. rapa | Demethylation of BrCKA2 and BrCKB4 | Regulation of floral transition. Regulation of temperature-dependent sex determination | [280] |
| Cold stress | Cucumis sativus | Demethylation of CHH sites | Regulation of temperature-dependent sex determination | [281] |
| Cold stress | Rosa hybrida | Enhanced CHH methylation of the RhAG promoter | Regulation of floral organ development | [282] |
| Drought stress | Arabidopsis | Increased 5mC methylation partly depending on H1.3 | Adaptive response to water deficiency | [283] |
| Drought stress | Brachypodium distachyon | Decreased global 5mC while Bacillus subtilis strain B26 inoculation increases | Increased drought stress resilience | [284] |
| Drought stress | G. hirsutum | Global hypermethylation in all three contexts | Acclimation to drought stress | [285] |
| Drought stress | O. sativa | Differential 5mC methylation alterations | Constitutive drought tolerance | [286] |
| Drought stress | Populus trichocarpa | Increased methylation of upstream and downstream 2 kb and TEs | Regulation of drought responses | [287] |
| Drought stress | Z. mays | Suppression of ZmNAC111 by MITE through RdDM | Natural variation in maize drought tolerance | [288] |
| Heat stress | Arabidopsis | Altered methylation of transposon remnants | Regulation of basal thermotolerance | [206] |
| Heat stress | Arabidopsis | Changes in genome-wide CHH-methylation patterns | Natural adaptation to different temperatures | [289] |
| Heat stress | B. napus | DNA hypomethylation | Regulation of heat stress responses in cultured microspores | [290] |
| Heat stress | Brassica napus | Increased DNA methylation in heat-sensitive genotypes | Adaptation to heat stress | [291] |
| Heat stress | Glycine max | Hypomethylation in all contexts | Affects the expression of genes or TEs under heat stress | [292] |
| Heat stress | Gossypium hirsutum | Reduced DNA methylation level in a heat-sensitive line | Microspore sterility | [293,294] |
| Heat stress | O. sativa | Decreased DNA methylation levels of OsFIE1 | Regulation of seed size under heat stress | [295] |
| Heat, salt, cold stresses | O. sativa | Increased 6mA levels in heat and salt stress, decreased 6mA levels in cold stress | Regulation of plant responses to environmental stresses | [296] |
| Salt and drought stresses | S. melongena | Expression changes of C5-MTases and demethylases | Response to salt and drought stresses | [297] |
| Salt and drought stresses | Solanum lycopersicum | Activation of Rider retrotransposon | Modulation of salt and drought stress responses | [298] |
| Salt stress | B. napus | Decreased methylation in the salinity-tolerant cultivar but increased methylation in the salinity-sensitive cultivar | Acclimation to salt stress | [299] |
| Salt stress | O. sativa | Decreased 5mC levels in the OsMYB91promoter | Enhanced salt tolerance | [207] |
| Salt stress | O. sativa | Increased methylation level of the osa-miR393a promoter | Improved salt tolerance | [300] |
| Salt stress | T. aestivum | Increased 5mC levels in TaHKT2;1 and TaHKT2;3 | Improved salt tolerance | [301] |
| Salt stress | Triticum aestivum | Reduced methylation levels in the promoter of salinity-responsive genes | Contributes to superior salinity tolerance | [302] |
| Salt stress | Zea mays | Increased methylation of root ZmPP2C and demethylation of leaf ZmGST | Acclimation to salt stress | [303] |
| Salt, heat and drought stresses | O. sativa | Activation of an LTR retrotransposon, HUO | Modulation of stress responses | [304] |
15.2. Biotic Stress
16. Detection of TE Modifications and Measurement of TE Expression
17. Recent Machine Learning and Computational Tools for Analysing
| Approaches or Tools | Mapping or Pseudo-Mapping | Fate of Multimappers | Type of Quantification | Distinguishes Unit-Length Transcripts from other TE-Derived Transcripts | Includes Polymorphic TE Expression | Notes | References |
|---|---|---|---|---|---|---|---|
| Endogenous retrovirus (ERV) map | Reference genome | Discarded | Locus specific | - | - | Uses a curated full-length human ERV database | [328] |
| L1EM | Model transcriptome | EM algorithm | Locus specific | + | - | Proof-of-principle on human long interspersed element 1 (L1) could be generalized | [329] |
| Manual curation | Reference genome | Discarded | Locus specific | + | - | Difficult to generalize | [324] |
| Multi-omics 1 | Reference genome | NA | Locus specific | + | + | Combines targeted DNA sequencing, RNA-seq, and ChIP-seq (chromatin immunoprecipitation followed by sequencing) | [330] |
| Multi-omics 2 | Reference genome | NA | Locus specific | + | + | Combines whole-genome sequencing and RNA-seq | [331] |
| Random assignment of multimappers | Reference genome | Randomly assigned | Locus specific | - | - | Locus-specific transcription not reliable on youngest TEs | [332] |
| RE discover TE | Model transcriptome | EM algorithm F | Family specific | + | - | Uses Salmon TE algorithm | [333] |
| Rep Enrich | Reference genome | Remapped on TE pseudogenome | Family specific | - | - | - | [334] |
| Salmon TE | Consensus transcriptome | Expectation-maximization (EM) algorithm | Family specific | - | - | Rapid pseudo mapping | [335] |
| SQuIRE | Reference genome | EM algorithm | Locus specific | - | +/− | Polymorphic insertion can be added as extra chromosome if internal sequence known | [336] |
| TE tools | TE pseudo genome | Randomly assigned | Family specific | - | - | Applicable to unassembled genomes | [337] |
| TEcandidates | Reference genome | Remapped on partially masked reference genome | Locus specific | - | - | - | [338] |
| Telescope | Reference genome | EM algorithm | Locus specific | + | - | - | [339] |
| TEtranscripts | Reference genome | EM algorithm | Family specific | - | - | Commonly used tool, tested on a wide variety of organisms | [340] |
| TeXP | Reference genome | Randomly assigned | Family specific | +/- | - | Subtracts signal from pervasive transcription but not from other forms of chimeric transcripts | [341] |
18. Future Perspectives and Biotechnological Opportunities
19. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvák, Z.; Levin, H.L.; Macfarlan, T.S.; et al. Ten things you should know about transposable elements. Genome Biol. 2018, 19, 199. [Google Scholar] [CrossRef] [PubMed]
- Jangam, D.; Feschotte, C.; Betrán, E. Transposable Element Domestication as an Adaptation to Evolutionary Conflicts. Trends Genet. 2017, 33, 817–831. [Google Scholar] [CrossRef] [PubMed]
- Bennetzen, J.L.; Wang, H. The contributions of transposable elements to the structure, function, and evolution of plant genomes. Annu. Rev. Plant Biol. 2014, 65, 505–530. [Google Scholar] [CrossRef] [PubMed]
- Fedoroff, N. Transposons and genome evolution in plants. Proc. Natl. Acad. Sci. USA 2000, 97, 7002–7007. [Google Scholar] [CrossRef]
- Quesneville, H. Twenty years of transposable element analysis in the Arabidopsis thaliana genome. Mob. DNA 2020, 11, 28. [Google Scholar] [CrossRef]
- Kalendar, R.; Muterko, A.; Boronnikova, S. Retrotransposable Elements: DNA Fingerprinting and the Assessment of Genetic Diversity. Methods Mol. Biol. 2021, 2222, 263–286. [Google Scholar]
- Ariel, F.D.; Manavella, P.A. When junk DNA turns functional: Transposon-derived non-coding RNAs in plants. J. Exp. Bot. 2021, 72, 4132–4143. [Google Scholar] [CrossRef]
- Moelling, K.; Broecker, F.; Russo, G.; Sunagawa, S. RNase H As Gene Modifier, Driver of Evolution and Antiviral Defense. Front. Microbiol. 2017, 8, 1745. [Google Scholar] [CrossRef]
- Kumar, S. Epigenomics of Plant Responses to Environmental Stress. Epigenomes 2018, 2, 6. [Google Scholar] [CrossRef]
- Ayarpadikannan, S.; Kim, H.-S. The impact of transposable elements in genome evolution and genetic instability and their implications in various diseases. Genom. Inform. 2014, 12, 98–104. [Google Scholar] [CrossRef]
- Fattash, I.; Rooke, R.; Wong, A.; Hui, C.; Luu, T.; Bhardwaj, P.; Yang, G. Miniature inverted-repeat transposable elements: Discovery, distribution, and activity. Genome/Natl. Res. Counc. Can. = Genome/Cons. Natl. Rech. Can. 2013, 56, 475–486. [Google Scholar] [CrossRef]
- Sundaram, V.; Wysocka, J. Transposable elements as a potent source of diverse cis-regulatory sequences in mammalian genomes. Philos.Trans. R. Soc. Lond. B Biol. Sci. 2020, 375, 20190347. [Google Scholar] [CrossRef]
- Moschetti, R.; Palazzo, A.; Lorusso, P.; Viggiano, L.; Massimiliano Marsano, R. “What You Need, Baby, I Got It”: Transposable Elements as Suppliers of Cis-Operating Sequences in Drosophila. Biology 2020, 9, 25. [Google Scholar] [CrossRef]
- Sahebi, M.; Hanafi, M.M.; van Wijnen, A.J.; Rice, D.; Rafii, M.Y.; Azizi, P.; Osman, M.; Taheri, S.; Bakar, M.F.A.; Isa, M.N.M.; et al. Contribution of transposable elements in the plant’s genome. Gene 2018, 665, 155–166. [Google Scholar] [CrossRef]
- Payer, L.M.; Burns, K.H. Transposable elements in human genetic disease. Nat. Rev. Genet. 2019, 20, 760–772. [Google Scholar] [CrossRef]
- Hirsch, C.D.; Springer, N.M. Transposable element influences on gene expression in plants. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 157–165. [Google Scholar] [CrossRef]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory activities of transposable elements: From conflicts to benefits. Nat. Rev. Genet. 2017, 18, 71–86. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A.; Iordache, F.; Stanca, L.; Predoi, G.; Serban, A.I. Oxidative stress mitigation by antioxidants—An overview on their chemistry and influences on health status. Eur. J. Med. Chem. 2021, 209, 112891. [Google Scholar] [CrossRef]
- Huang, S.; Tao, X.; Yuan, S.; Zhang, Y.; Li, P.; Beilinson, H.A.; Zhang, Y.; Yu, W.; Pontarotti, P.; Escriva, H.; et al. Discovery of an Active RAG Transposon Illuminates the Origins of V(D)J Recombination. Cell 2016, 166, 102–114. [Google Scholar] [CrossRef]
- Kapitonov, V.V.; Koonin, E.V. Evolution of the RAG1-RAG2 locus: Both proteins came from the same transposon. Biol. Direct 2015, 10, 20. [Google Scholar] [CrossRef]
- Sandoval-Villegas, N.; Nurieva, W.; Amberger, M.; Ivics, Z. Contemporary Transposon Tools: A Review and Guide through Mechanisms and Applications of Sleeping Beauty, piggyBac and Tol2 for Genome Engineering. Int. J. Mol. Sci. 2021, 22, 5084. [Google Scholar] [CrossRef]
- Tipanee, J.; VandenDriessche, T.; Chuah, M.K. Transposons: Moving Forward from Preclinical Studies to Clinical Trials. Hum. Gene Ther. 2017, 28, 1087–1104. [Google Scholar] [CrossRef]
- Palazzo, A.; Marsano, R.M. Transposable elements: A jump toward the future of expression vectors. Crit. Rev. Biotechnol. 2021, 41, 792–808. [Google Scholar] [CrossRef]
- Walker, J.A.; Jordan, V.E.; Steely, C.J.; Beckstrom, T.O.; McDaniel, C.L.; St. Romain, C.P.; Bennett, E.C.; Robichaux, A.; Clement, B.N.; Konkel, M.K.; et al. Papio Baboon Species Indicative Alu Elements. Genome Biol. Evol. 2017, 9, 1788–1796. [Google Scholar] [CrossRef]
- Steely, C.J.; Walker, J.A.; Jordan, V.E.; Beckstrom, T.O.; McDaniel, C.L.; St. Romain, C.P.; Bennett, E.C.; Robichaux, A.; Clement, B.N.; Raveendran, M.; et al. Alu Insertion Polymorphisms as Evidence for Population Structure in Baboons. Genome Biol. Evol. 2017, 9, 2418–2427. [Google Scholar] [CrossRef]
- Kalendar, R.; Amenov, A.; Daniyarov, A. Use of retrotransposon-derived genetic markers to analyse genomic variability in plants. Funct. Plant Biol. 2018, 46, 15–29. [Google Scholar] [CrossRef]
- Kalendar, R.; Raskina, O.; Belyayev, A.; Schulman, A.H. Long Tandem Arrays of Cassandra Retroelements and Their Role in Genome Dynamics in Plants. Int. J. Mol. Sci. 2020, 21, 2931. [Google Scholar] [CrossRef]
- Kalendar, R.; Tanskanen, J.; Chang, W.; Antonius, K.; Sela, H.; Peleg, O.; Schulman, A.H. Cassandra retrotransposons carry independently transcribed 5S RNA. Proc. Natl. Acad. Sci. USA 2008, 105, 5833–5838. [Google Scholar] [CrossRef]
- Kalendar, R.; Vicient, C.M.; Peleg, O.; Anamthawat-Jonsson, K.; Bolshoy, A.; Schulman, A.H. Large Retrotransposon Derivatives: Abundant, Conserved but Nonautonomous Retroelements of Barley and Related Genomes. Genetics 2004, 166, 1437–1450. [Google Scholar] [CrossRef]
- Orozco-Arias, S.; Isaza, G.; Guyot, R. Retrotransposons in Plant Genomes: Structure, Identification, and Classification through Bioinformatics and Machine Learning. Int. J. Mol. Sci. 2019, 20, 3837. [Google Scholar] [CrossRef]
- Hickman, A.B.; Dyda, F. DNA Transposition at Work. Chem. Rev. 2016, 116, 12758–12784. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Bano, A.; Ali, S.; Babar, M.A. Crosstalk amongst phytohormones from planta and PGPR under biotic and abiotic stresses. Plant Growth Regul. 2020, 90, 189–203. [Google Scholar] [CrossRef]
- Ashapkin, V.V.; Kutueva, L.I.; Aleksandrushkina, N.I.; Vanyushin, B.F. Epigenetic Mechanisms of Plant Adaptation to Biotic and Abiotic Stresses. Int. J. Mol. Sci. 2020, 21, 7457. [Google Scholar] [CrossRef] [PubMed]
- Deniz, Ö.; Frost, J.M.; Branco, M.R. Regulation of transposable elements by DNA modifications. Nat. Rev. Genet. 2019, 20, 417–431. [Google Scholar] [CrossRef]
- Mercé, C.; Bayer, P.E.; Tay Fernandez, C.; Batley, J.; Edwards, D. Induced Methylation in Plants as a Crop Improvement Tool: Progress and Perspectives. Agronomy 2020, 10, 1484. [Google Scholar] [CrossRef]
- Lindermayr, C.; Rudolf, E.E.; Durner, J.; Groth, M. Interactions between metabolism and chromatin in plant models. Mol. Metab. 2020, 38, 100951. [Google Scholar] [CrossRef]
- Ong-Abdullah, M.; Ordway, J.M.; Jiang, N.; Ooi, S.E.; Kok, S.Y.; Sarpan, N.; Azimi, N.; Hashim, A.T.; Ishak, Z.; Rosli, S.K.; et al. Loss of Karma transposon methylation underlies the mantled somaclonal variant of oil palm. Nature 2015, 525, 533–537. [Google Scholar] [CrossRef]
- Khan, T.; Relitti, N.; Brindisi, M.; Magnano, S.; Zisterer, D.; Gemma, S.; Butini, S.; Campiani, G. Autophagy modulators for the treatment of oral and esophageal squamous cell carcinomas. Med. Res. Rev. 2020, 40, 1002–1060. [Google Scholar] [CrossRef]
- Belyayev, A.; Kalendar, R.; Brodsky, L.; Nevo, E.; Schulman, A.H.; Raskina, O. Transposable elements in a marginal plant population: Temporal fluctuations provide new insights into genome evolution of wild diploid wheat. Mob. DNA 2010, 1, 6. [Google Scholar] [CrossRef]
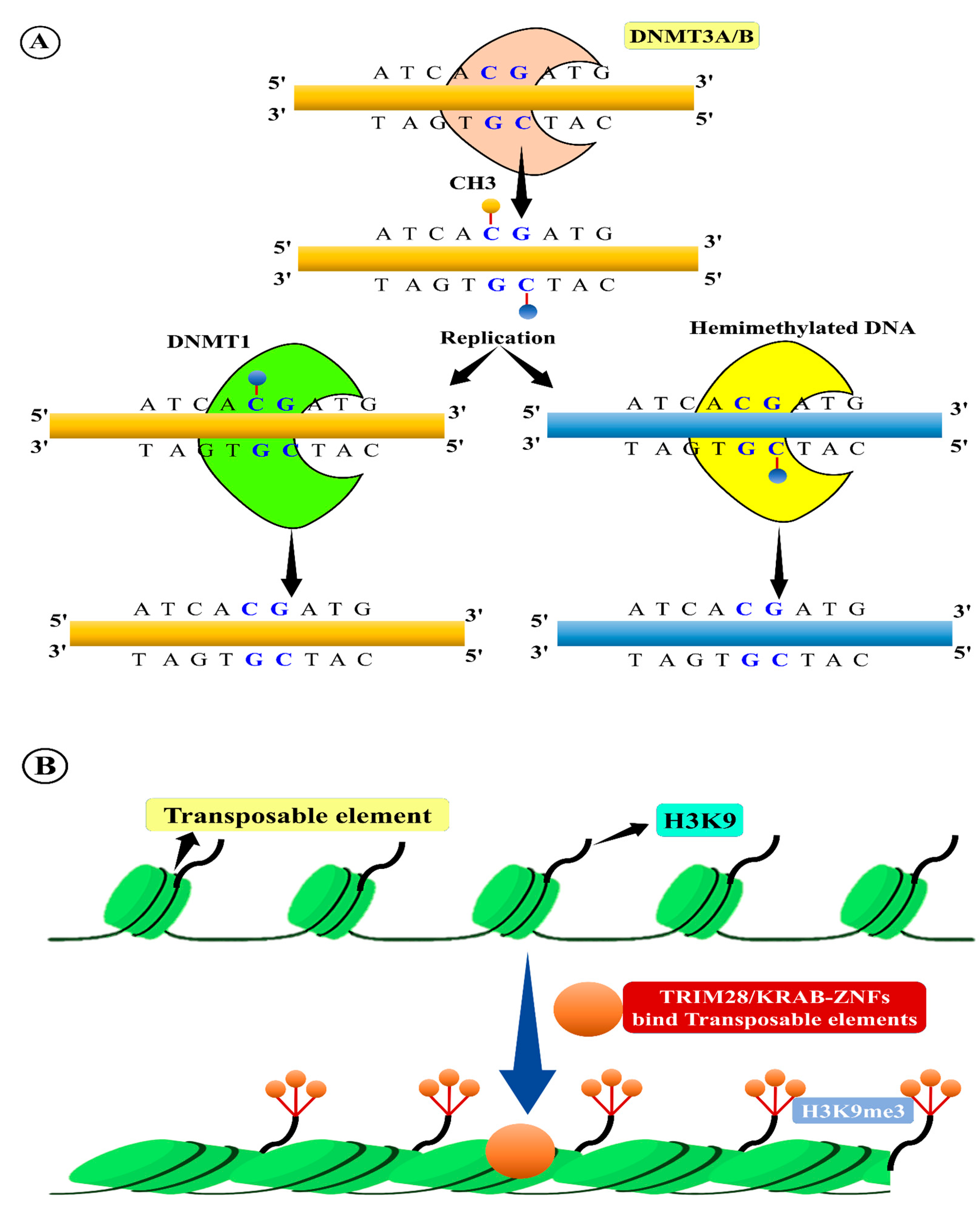
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef]
- Zhou, W.; Liang, G.; Molloy, P.L.; Jones, P.A. DNA methylation enables transposable element-driven genome expansion. Proc. Natl. Acad. Sci. USA 2020, 117, 19359–19366. [Google Scholar] [CrossRef]
- Zhang, H.; Lang, Z.; Zhu, J.K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 2018, 19, 489–506. [Google Scholar] [CrossRef]
- Jönsson, M.E.; Garza, R.; Johansson, P.A.; Jakobsson, J. Transposable Elements: A Common Feature of Neurodevelopmental and Neurodegenerative Disorders. Trends Genet. 2020, 36, 610–623. [Google Scholar] [CrossRef]
- Wambui Mbichi, R.; Wang, Q.-F.; Wan, T. RNA directed DNA methylation and seed plant genome evolution. Plant Cell Rep. 2020, 39, 983–996. [Google Scholar] [CrossRef]
- Slotkin, R.K.; Martienssen, R. Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 2007, 8, 272–285. [Google Scholar] [CrossRef]
- Kabelitz, T.; Brzezinka, K.; Friedrich, T.; Górka, M.; Graf, A.; Kappel, C.; Bäurle, I. A JUMONJI Protein with E3 Ligase and Histone H3 Binding Activities Affects Transposon Silencing in Arabidopsis. Plant Physiol. 2016, 171, 344–358. [Google Scholar] [CrossRef]
- Kalavacharla, V.; Subramani, M.; Ayyappan, V.; Dworkin, M.C.; Hayford, R.K. Plant Epigenomics. In Handbook of Epigenetics, 2nd ed.; Tollefsbol, T.O., Ed.; Academic Press: Cambridge, MA, USA, 2017; Chapter 16; pp. 245–258. [Google Scholar]
- Wang, X.; Weigel, D.; Smith, L.M. Transposon variants and their effects on gene expression in Arabidopsis. PLoS Genet. 2013, 9, e1003255. [Google Scholar] [CrossRef]
- Papareddy, R.K.; Páldi, K.; Paulraj, S.; Kao, P.; Lutzmayer, S.; Nodine, M.D. Chromatin regulates expression of small RNAs to help maintain transposon methylome homeostasis in Arabidopsis. Genome Biol. 2020, 21, 251. [Google Scholar] [CrossRef]
- Feng, J.X.; Riddle, N.C. Epigenetics and genome stability. Mamm. Genome 2020, 31, 181–195. [Google Scholar] [CrossRef]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef]
- Levin, H.L.; Moran, J.V. Dynamic interactions between transposable elements and their hosts. Nat. Rev. Genet. 2011, 12, 615–627. [Google Scholar] [CrossRef]
- Grabundzija, I.; Hickman, A.B.; Dyda, F. Helraiser intermediates provide insight into the mechanism of eukaryotic replicative transposition. Nat. Commun. 2018, 9, 1278. [Google Scholar] [CrossRef] [PubMed]
- Boeke, J.D.; Garfinkel, D.J.; Styles, C.A.; Fink, G.R. Ty elements transpose through an RNA intermediate. Cell 1985, 40, 491–500. [Google Scholar] [CrossRef]
- Griffiths, J.; Catoni, M.; Iwasaki, M.; Paszkowski, J. Sequence-Independent Identification of Active LTR Retrotransposons in Arabidopsis. Mol. Plant 2018, 11, 508–511. [Google Scholar] [CrossRef]
- Pastuzyn, E.D.; Day, C.E.; Kearns, R.B.; Kyrke-Smith, M.; Taibi, A.V.; McCormick, J.; Yoder, N.; Belnap, D.M.; Erlendsson, S.; Morado, D.R.; et al. The Neuronal Gene Arc Encodes a Repurposed Retrotransposon Gag Protein that Mediates Intercellular RNA Transfer. Cell 2018, 172, 275–288.e18. [Google Scholar] [CrossRef]
- Gao, D.; Jimenez-Lopez, J.C.; Iwata, A.; Gill, N.; Jackson, S.A. Functional and Structural Divergence of an Unusual LTR Retrotransposon Family in Plants. PLoS ONE 2012, 7, e48595. [Google Scholar] [CrossRef]
- Malik, H.S.; Eickbush, T.H. Phylogenetic analysis of ribonuclease H domains suggests a late, chimeric origin of LTR retrotransposable elements and retroviruses. Genome Res. 2001, 11, 1187–1197. [Google Scholar] [CrossRef]
- Feschotte, C.; Pritham, E.J. DNA transposons and the evolution of eukaryotic genomes. Annu. Rev. Genet. 2007, 41, 331–368. [Google Scholar] [CrossRef]
- Naorem, S.S.; Han, J.; Wang, S.; Lee, W.R.; Heng, X.; Miller, J.F.; Guo, H. DGR mutagenic transposition occurs via hypermutagenic reverse transcription primed by nicked template RNA. Proc. Natl. Acad. Sci. USA 2017, 114, E10187–E10195. [Google Scholar] [CrossRef]
- Piégu, B.; Bire, S.; Arensburger, P.; Bigot, Y. A survey of transposable element classification systems--a call for a fundamental update to meet the challenge of their diversity and complexity. Mol. Phylogenet. Evol. 2015, 86, 90–109. [Google Scholar] [CrossRef]
- Zhao, D.; Ferguson, A.A.; Jiang, N. What makes up plant genomes: The vanishing line between transposable elements and genes. Biochim. Biophys. Acta 2016, 1859, 366–380. [Google Scholar] [CrossRef] [PubMed]
- Berthelier, J.; Casse, N.; Daccord, N.; Jamilloux, V.; Saint-Jean, B.; Carrier, G. A transposable element annotation pipeline and expression analysis reveal potentially active elements in the microalga Tisochrysis lutea. BMC Genom. 2018, 19, 378. [Google Scholar] [CrossRef] [PubMed]
- Feschotte, C.; Jiang, N.; Wessler, S.R. Plant transposable elements: Where genetics meets genomics. Nat. Rev. Genet. 2002, 3, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Hirochika, H.; Sugimoto, K.; Otsuki, Y.; Tsugawa, H.; Kanda, M. Retrotransposons of rice involved in mutations induced by tissue culture. Proc. Natl. Acad. Sci. USA 1996, 93, 7783–7788. [Google Scholar] [CrossRef]
- Vicient, C.M.; Suoniemi, A.; Anamthawat-Jónsson, K.; Tanskanen, J.; Beharav, A.; Nevo, E.; Schulman, A.H. Retrotransposon BARE-1 and Its Role in Genome Evolution in the Genus Hordeum. Plant Cell 1999, 11, 1769–1784. [Google Scholar] [CrossRef]
- Hirochika, H. Activation of tobacco retrotransposons during tissue culture. EMBO J. 1993, 12, 2521–2528. [Google Scholar] [CrossRef] [PubMed]
- Grandbastien, M.A.; Spielmann, A.; Caboche, M. Tnt1, a mobile retroviral-like transposable element of tobacco isolated by plant cell genetics. Nature 1989, 337, 376–380. [Google Scholar] [CrossRef]
- White, S.E.; Habera, L.F.; Wessler, S.R. Retrotransposons in the flanking regions of normal plant genes: A role for copia-like elements in the evolution of gene structure and expression. Proc. Natl. Acad. Sci. USA 1994, 91, 11792–11796. [Google Scholar] [CrossRef]
- Jin, Y.K.; Bennetzen, J.L. Structure and coding properties of Bs1, a maize retrovirus-like transposon. Proc. Natl. Acad. Sci. USA 1989, 86, 6235–6239. [Google Scholar] [CrossRef]
- Meyers, B.C.; Tingey, S.V.; Morgante, M. Abundance, distribution, and transcriptional activity of repetitive elements in the maize genome. Genome Res. 2001, 11, 1660–1676. [Google Scholar] [CrossRef]
- Jiang, N.; Bao, Z.; Temnykh, S.; Cheng, Z.; Jiang, J.; Wing, R.A.; McCouch, S.R.; Wessler, S.R. Dasheng: A recently amplified nonautonomous long terminal repeat element that is a major component of pericentromeric regions in rice. Genetics 2002, 161, 1293–1305. [Google Scholar] [CrossRef] [PubMed]
- Purugganan, M.D.; Wessler, S.R. Molecular evolution of magellan, a maize Ty3/gypsy-like retrotransposon. Proc. Natl. Acad. Sci. USA 1994, 91, 11674–11678. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.A.; Voytas, D.F. Athila4 of Arabidopsis and Calypso of soybean define a lineage of endogenous plant retroviruses. Genome Res. 2002, 12, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Konieczny, A.; Voytas, D.F.; Cummings, M.P.; Ausubel, F.M. A superfamily of Arabidopsis thaliana retrotransposons. Genetics 1991, 127, 801–809. [Google Scholar] [CrossRef]
- Leeton, P.R.; Smyth, D.R. An abundant LINE-like element amplified in the genome of Lilium speciosum. Mol. Gen. Genet. 1993, 237, 97–104. [Google Scholar] [CrossRef]
- Schwarz-Sommer, Z.; Leclercq, L.; Göbel, E.; Saedler, H. Cin4, an insert altering the structure of the A1 gene in Zea mays, exhibits properties of nonviral retrotransposons. EMBO J. 1987, 6, 3873–3880. [Google Scholar] [CrossRef]
- Wright, D.A.; Ke, N.; Smalle, J.; Hauge, B.M.; Goodman, H.M.; Voytas, D.F. Multiple non-LTR retrotransposons in the genome of Arabidopsis thaliana. Genetics 1996, 142, 569–578. [Google Scholar] [CrossRef]
- Yoshioka, Y.; Matsumoto, S.; Kojima, S.; Ohshima, K.; Okada, N.; Machida, Y. Molecular characterization of a short interspersed repetitive element from tobacco that exhibits sequence homology to specific tRNAs. Proc. Natl. Acad. Sci. USA 1993, 90, 6562–6566. [Google Scholar] [CrossRef]
- Deragon, J.M.; Landry, B.S.; Pélissier, T.; Tutois, S.; Tourmente, S.; Picard, G. An analysis of retroposition in plants based on a family of SINEs from Brassica napus. J. Mol. Evol. 1994, 39, 378–386. [Google Scholar] [CrossRef]
- Chandler, V.; Rivin, C.; Walbot, V. Stable non-mutator stocks of maize have sequences homologous to the Mu1 transposable element. Genetics 1986, 114, 1007–1021. [Google Scholar] [CrossRef]
- Singer, T.; Yordan, C.; Martienssen, R.A. Robertson’s Mutator transposons in A. thaliana are regulated by the chromatin-remodeling gene Decrease in DNA Methylation (DDM1). Genes Dev. 2001, 15, 591–602. [Google Scholar] [CrossRef]
- Gierl, A. The En/Spm transposable element of maize. Curr. Top. Microbiol. Immunol. 1996, 204, 145–159. [Google Scholar]
- Miura, A.; Yonebayashi, S.; Watanabe, K.; Toyama, T.; Shimada, H.; Kakutani, T. Mobilization of transposons by a mutation abolishing full DNA methylation in Arabidopsis. Nature 2001, 411, 212–214. [Google Scholar] [CrossRef]
- Wessler, S.R. Phenotypic diversity mediated by the maize transposable elements Ac and Spm. Science 1988, 242, 399–405. [Google Scholar] [CrossRef]
- Zhang, X.; Feschotte, C.; Zhang, Q.; Jiang, N.; Eggleston, W.B.; Wessler, S.R. P instability factor: An active maize transposon system associated with the amplification of Tourist-like MITEs and a new superfamily of transposases. Proc. Natl. Acad. Sci. USA 2001, 98, 12572–12577. [Google Scholar] [CrossRef]
- Kapitonov, V.V.; Jurka, J. Molecular paleontology of transposable elements from Arabidopsis thaliana. Genetica 1999, 107, 27–37. [Google Scholar] [CrossRef]
- Turcotte, K.; Srinivasan, S.; Bureau, T. Survey of transposable elements from rice genomic sequences. Plant J. 2001, 25, 169–179. [Google Scholar] [CrossRef]
- Feschotte, C.; Wessler, S.R. Mariner-like transposases are widespread and diverse in flowering plants. Proc. Natl. Acad. Sci. USA 2002, 99, 280–285. [Google Scholar] [CrossRef]
- Svedberg, J.; Hosseini, S.; Chen, J.; Vogan, A.A.; Mozgova, I.; Hennig, L.; Manitchotpisit, P.; Abusharekh, A.; Hammond, T.M.; Lascoux, M.; et al. Convergent evolution of complex genomic rearrangements in two fungal meiotic drive elements. Nat. Commun. 2018, 9, 4242. [Google Scholar] [CrossRef]
- Cornejo, E.; Abreu, N.; Komeili, A. Compartmentalization and organelle formation in bacteria. Curr. Opin. Cell Biol. 2014, 26, 132–138. [Google Scholar] [CrossRef]
- Neumann, P.; Novák, P.; Hoštáková, N.; Macas, J. Systematic survey of plant LTR-retrotransposons elucidates phylogenetic relationships of their polyprotein domains and provides a reference for element classification. Mob. DNA 2019, 10, 1. [Google Scholar] [CrossRef]
- Bourgeois, Y.; Ruggiero, R.P.; Hariyani, I.; Boissinot, S. Disentangling the determinants of transposable elements dynamics in vertebrate genomes using empirical evidences and simulations. PLoS Genet. 2020, 16, e1009082. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H. Diverse transposable element landscapes in pathogenic and nonpathogenic yeast models: The value of a comparative perspective. Mob. DNA 2020, 11, 16. [Google Scholar] [CrossRef]
- Yang, N.; Yan, J. New genomic approaches for enhancing maize genetic improvement. Curr. Opin. Plant Biol. 2021, 60, 101977. [Google Scholar] [CrossRef] [PubMed]
- Schubert, I.; Vu, G.T.H. Genome Stability and Evolution: Attempting a Holistic View. Trends Plant Sci. 2016, 21, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Bennetzen, J.L.; Wang, X. Relationships between Gene Structure and Genome Instability in Flowering Plants. Mol. Plant 2018, 11, 407–413. [Google Scholar] [CrossRef]
- Soltis, P.S.; Marchant, D.B.; Van de Peer, Y.; Soltis, D.E. Polyploidy and genome evolution in plants. Curr. Opin. Genet. Dev. 2015, 35, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Ragupathy, R.; You, F.M.; Cloutier, S. Arguments for standardizing transposable element annotation in plant genomes. Trends Plant Sci. 2013, 18, 367–376. [Google Scholar] [CrossRef]
- Hu, T.T.; Pattyn, P.; Bakker, E.G.; Cao, J.; Cheng, J.-F.; Clark, R.M.; Fahlgren, N.; Fawcett, J.A.; Grimwood, J.; Gundlach, H.; et al. The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat. Genet. 2011, 43, 476–481. [Google Scholar] [CrossRef]
- Civáň, P.; Švec, M.; Hauptvogel, P. On the Coevolution of Transposable Elements and Plant Genomes. J. Bot. 2011, 2011, 893546. [Google Scholar] [CrossRef]
- Dodsworth, S.; Leitch, A.R.; Leitch, I.J. Genome size diversity in angiosperms and its influence on gene space. Curr. Opin. Genet. Dev. 2015, 35, 73–78. [Google Scholar] [CrossRef]
- Galindo-Gonzalez, L.; Mhiri, C.; Deyholos, M.K.; Grandbastien, M.A. LTR-retrotransposons in plants: Engines of evolution. Gene 2017, 626, 14–25. [Google Scholar] [CrossRef]
- Hollister, J.D.; Gaut, B.S. Epigenetic silencing of transposable elements: A trade-off between reduced transposition and deleterious effects on neighboring gene expression. Genome Res. 2009, 19, 1419–1428. [Google Scholar] [CrossRef]
- Haberer, G.; Kamal, N.; Bauer, E.; Gundlach, H.; Fischer, I.; Seidel, M.A.; Spannagl, M.; Marcon, C.; Ruban, A.; Urbany, C.; et al. European maize genomes highlight intraspecies variation in repeat and gene content. Nat. Genet. 2020, 52, 950–957. [Google Scholar] [CrossRef]
- Ma, B.; Xin, Y.; Kuang, L.; He, N. Distribution and Characteristics of Transposable Elements in the Mulberry Genome. Plant Genome 2019, 12, 180094. [Google Scholar] [CrossRef]
- Tenaillon, M.I.; Hollister, J.D.; Gaut, B.S. A triptych of the evolution of plant transposable elements. Trends Plant Sci. 2010, 15, 471–478. [Google Scholar] [CrossRef]
- The Arabidopsis Genome, I. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 2000, 408, 796–815. [Google Scholar] [CrossRef]
- Sasaki, T.; International Rice Genome Sequencing, P. The map-based sequence of the rice genome. Nature 2005, 436, 793–800. [Google Scholar] [CrossRef]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The B73 maize genome: Complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef]
- Estep, M.C.; DeBarry, J.D.; Bennetzen, J.L. The dynamics of LTR retrotransposon accumulation across 25 million years of panicoid grass evolution. Heredity 2013, 110, 194–204. [Google Scholar] [CrossRef]
- Piegu, B.; Guyot, R.; Picault, N.; Roulin, A.; Sanyal, A.; Kim, H.; Collura, K.; Brar, D.S.; Jackson, S.; Wing, R.A.; et al. Doubling genome size without polyploidization: Dynamics of retrotransposition-driven genomic expansions in Oryza australiensis, a wild relative of rice. Genome Res. 2006, 16, 1262–1269. [Google Scholar] [CrossRef]
- Bennett, M.D.; Leitch, I.J. “Plant DNA C-Values Database,” Release 5.0, December 2010. 2010. Available online: http://data.kew.org/cvalues/ (accessed on 29 June 2021).
- Bartos, J.; Paux, E.; Kofler, R.; Havránková, M.; Kopecký, D.; Suchánková, P.; Safár, J.; Simková, H.; Town, C.D.; Lelley, T.; et al. A first survey of the rye (Secale cereale) genome composition through BAC end sequencing of the short arm of chromosome 1R. BMC Plant Biol. 2008, 8, 95. [Google Scholar] [CrossRef]
- Wang, H.; Liu, J.S. LTR retrotransposon landscape in Medicago truncatula: More rapid removal than in rice. BMC Genom. 2008, 9, 382. [Google Scholar] [CrossRef]
- Velasco, R.; Zharkikh, A.; Troggio, M.; Cartwright, D.A.; Cestaro, A.; Pruss, D.; Pindo, M.; Fitzgerald, L.M.; Vezzulli, S.; Reid, J.; et al. A high quality draft consensus sequence of the genome of a heterozygous grapevine variety. PLoS ONE 2007, 2, e1326. [Google Scholar] [CrossRef]
- Jaillon, O.; Aury, J.M.; Noel, B.; Policriti, A.; Clepet, C.; Casagrande, A.; Choisne, N.; Aubourg, S.; Vitulo, N.; Jubin, C.; et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 2007, 449, 463–467. [Google Scholar]
- Sato, S.; Nakamura, Y.; Kaneko, T.; Asamizu, E.; Kato, T.; Nakao, M.; Sasamoto, S.; Watanabe, A.; Ono, A.; Kawashima, K.; et al. Genome Structure of the Legume, Lotus japonicus. DNA Res. 2008, 15, 227–239. [Google Scholar] [CrossRef]
- Holligan, D.; Zhang, X.; Jiang, N.; Pritham, E.J.; Wessler, S.R. The transposable element landscape of the model legume Lotus japonicus. Genetics 2006, 174, 2215–2228. [Google Scholar] [CrossRef]
- Tuskan, G.A.; Difazio, S.; Jansson, S.; Bohlmann, J.; Grigoriev, I.; Hellsten, U.; Putnam, N.; Ralph, S.; Rombauts, S.; Salamov, A.; et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 2006, 313, 1596–1604. [Google Scholar]
- Wicker, T.; Zimmermann, W.; Perovic, D.; Paterson, A.H.; Ganal, M.; Graner, A.; Stein, N. A detailed look at 7 million years of genome evolution in a 439 kb contiguous sequence at the barley Hv-eIF4E locus: Recombination, rearrangements and repeats. Plant J. 2005, 41, 184–194. [Google Scholar] [CrossRef]
- Ming, R.; Hou, S.; Feng, Y.; Yu, Q.; Dionne-Laporte, A.; Saw, J.H.; Senin, P.; Wang, W.; Ly, B.V.; Lewis, K.L.; et al. The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature 2008, 452, 991–996. [Google Scholar] [CrossRef]
- Du, J.; Grant, D.; Tian, Z.; Nelson, R.T.; Zhu, L.; Shoemaker, R.C.; Ma, J. SoyTEdb: A comprehensive database of transposable elements in the soybean genome. BMC Genom. 2010, 11, 113. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Liu, J.; Geng, H.; Zhang, J.; Liu, Y.; Zhang, H.; Xing, S.; Du, J.; Ma, S.; Tian, Z. De novo assembly of a Chinese soybean genome. Sci. China Life Sci. 2018, 61, 871–884. [Google Scholar] [CrossRef] [PubMed]
- González, V.M.; Benjak, A.; Hénaff, E.M.; Mir, G.; Casacuberta, J.M.; Garcia-Mas, J.; Puigdomènech, P. Sequencing of 6.7 Mb of the melon genome using a BAC pooling strategy. BMC Plant Biol. 2010, 10, 246. [Google Scholar] [CrossRef] [PubMed]
- Ruggieri, V.; Alexiou, K.G.; Morata, J.; Argyris, J.; Pujol, M.; Yano, R.; Nonaka, S.; Ezura, H.; Latrasse, D.; Boualem, A.; et al. An improved assembly and annotation of the melon (Cucumis melo L.) reference genome. Sci. Rep. 2018, 8, 8088. [Google Scholar] [CrossRef]
- Hawkins, J.S.; Kim, H.; Nason, J.D.; Wing, R.A.; Wendel, J.F. Differential lineage-specific amplification of transposable elements is responsible for genome size variation in Gossypium. Genome Res. 2006, 16, 1252–1261. [Google Scholar] [CrossRef]
- Zhang, X.; Wessler, S.R. Genome-wide comparative analysis of the transposable elements in the related species Arabidopsis thaliana and Brassica oleracea. Proc. Natl. Acad. Sci. USA 2004, 101, 5589–5594. [Google Scholar] [CrossRef]
- Guo, Z.-H.; Ma, P.-F.; Yang, G.-Q.; Hu, J.-Y.; Liu, Y.-L.; Xia, E.-H.; Zhong, M.-C.; Zhao, L.; Sun, G.-L.; Xu, Y.-X.; et al. Genome Sequences Provide Insights into the Reticulate Origin and Unique Traits of Woody Bamboos. Mol. Plant 2019, 12, 1353–1365. [Google Scholar] [CrossRef]
- Jedlicka, P.; Lexa, M.; Vanat, I.; Hobza, R.; Kejnovsky, E. Nested plant LTR retrotransposons target specific regions of other elements, while all LTR retrotransposons often target palindromes and nucleosome-occupied regions: In silico study. Mob. DNA 2019, 10, 50. [Google Scholar] [CrossRef]
- Wei, L.; Xiao, M.; An, Z.; Ma, B.; Mason, A.S.; Qian, W.; Li, J.; Fu, D. New Insights into Nested Long Terminal Repeat Retrotransposons in Brassica Species. Mol. Plant 2013, 6, 470–482. [Google Scholar] [CrossRef]
- Pereira, H.S.; Barão, A.; Delgado, M.; Morais-Cecílio, L.; Viegas, W. Genomic analysis of Grapevine Retrotransposon 1 (Gret1) in Vitis vinifera. Theor. Appl. Genet. 2005, 111, 871–878. [Google Scholar] [CrossRef]
- Butelli, E.; Licciardello, C.; Zhang, Y.; Liu, J.; Mackay, S.; Bailey, P.; Reforgiato-Recupero, G.; Martin, C. Retrotransposons control fruit-specific, cold-dependent accumulation of anthocyanins in blood oranges. Plant Cell 2012, 24, 1242–1255. [Google Scholar] [CrossRef]
- Xiao, H.; Jiang, N.; Schaffner, E.; Stockinger, E.J.; van der Knaap, E. A Retrotransposon-Mediated Gene Duplication Underlies Morphological Variation of Tomato Fruit. Science 2008, 319, 1527–1530. [Google Scholar] [CrossRef]
- Michaels, S.D.; Ditta, G.; Gustafson-Brown, C.; Pelaz, S.; Yanofsky, M.; Amasino, R.M. AGL24 acts as a promoter of flowering in Arabidopsis and is positively regulated by vernalization. Plant J. 2003, 33, 867–874. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, F.; Peterson, T. Transposition of Reversed Ac Element Ends Generates Novel Chimeric Genes in Maize. PLoS Genet. 2006, 2, e164. [Google Scholar] [CrossRef]
- Yu, C.; Zhang, J.; Peterson, T. Genome rearrangements in maize induced by alternative transposition of reversed ac/ds termini. Genetics 2011, 188, 59–67. [Google Scholar] [CrossRef]
- Du, C.; Hoffman, A.; He, L.; Caronna, J.; Dooner, H.K. The complete Ac/Ds transposon family of maize. BMC Genom. 2011, 12, 588. [Google Scholar] [CrossRef]
- Xuan, Y.H.; Piao, H.L.; Je, B.I.; Park, S.J.; Park, S.H.; Huang, J.; Zhang, J.B.; Peterson, T.; Han, C.-D. Transposon Ac/Ds-induced chromosomal rearrangements at the rice OsRLG5 locus. Nucleic Acids Res. 2011, 39, e149. [Google Scholar] [CrossRef]
- Shen, D.; Song, C.; Miskey, C.; Chan, S.; Guan, Z.; Sang, Y.; Wang, Y.; Chen, C.; Wang, X.; Müller, F.; et al. A native, highly active Tc1/mariner transposon from zebrafish (ZB) offers an efficient genetic manipulation tool for vertebrates. Nucleic Acids Res. 2021, 49, 2126–2140. [Google Scholar] [CrossRef]
- Jiang, N.; Wessler, S.R. Insertion preference of maize and rice miniature inverted repeat transposable elements as revealed by the analysis of nested elements. Plant Cell 2001, 13, 2553–2564. [Google Scholar]
- Bureau, T.E.; Wessler, S.R. Stowaway: A new family of inverted repeat elements associated with the genes of both monocotyledonous and dicotyledonous plants. Plant Cell 1994, 6, 907–916. [Google Scholar]
- Panini, M.; Chiesa, O.; Troczka, B.J.; Mallott, M.; Manicardi, G.C.; Cassanelli, S.; Cominelli, F.; Hayward, A.; Mazzoni, E.; Bass, C. Transposon-mediated insertional mutagenesis unmasks recessive insecticide resistance in the aphid Myzus persicae. Proc. Natl. Acad. Sci. USA 2021, 118, e2100559118. [Google Scholar] [CrossRef]
- Quadrana, L.; Bortolini Silveira, A.; Mayhew, G.F.; LeBlanc, C.; Martienssen, R.A.; Jeddeloh, J.A.; Colot, V. The Arabidopsis thaliana mobilome and its impact at the species level. eLife 2016, 5, e15716. [Google Scholar] [CrossRef]
- Weng, M.L.; Becker, C.; Hildebrandt, J.; Neumann, M.; Rutter, M.T.; Shaw, R.G.; Weigel, D.; Fenster, C.B. Fine-Grained Analysis of Spontaneous Mutation Spectrum and Frequency in Arabidopsis thaliana. Genetics 2019, 211, 703–714. [Google Scholar] [CrossRef]
- Bourgeois, Y.; Boissinot, S. On the Population Dynamics of Junk: A Review on the Population Genomics of Transposable Elements. Genes 2019, 10, 419. [Google Scholar] [CrossRef]
- Wicker, T.; Gundlach, H.; Spannagl, M.; Uauy, C.; Borrill, P.; Ramírez-González, R.H.; De Oliveira, R.; Mayer, K.F.X.; Paux, E.; Choulet, F.; et al. Impact of transposable elements on genome structure and evolution in bread wheat. Genome Biol. 2018, 19, 103. [Google Scholar] [CrossRef]
- Middleton, C.P.; Senerchia, N.; Stein, N.; Akhunov, E.D.; Keller, B.; Wicker, T.; Kilian, B. Sequencing of chloroplast genomes from wheat, barley, rye and their relatives provides a detailed insight into the evolution of the Triticeae tribe. PLoS ONE 2014, 9, e85761. [Google Scholar] [CrossRef]
- Lisch, D. How important are transposons for plant evolution? Nat. Rev. Genet. 2013, 14, 49–61. [Google Scholar] [CrossRef]
- Negi, P.; Rai, A.N.; Suprasanna, P. Moving through the Stressed Genome: Emerging Regulatory Roles for Transposons in Plant Stress Response. Front. Plant Sci. 2016, 7, 1448. [Google Scholar] [CrossRef]
- Rejeb, I.B.; Pastor, V.; Mauch-Mani, B. Plant Responses to Simultaneous Biotic and Abiotic Stress: Molecular Mechanisms. Plants 2014, 3, 458–475. [Google Scholar] [CrossRef]
- Etchegaray, E.; Naville, M.; Volff, J.-N.; Haftek-Terreau, Z. Transposable element-derived sequences in vertebrate development. Mob. DNA 2021, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Pereira, V. Insertion bias and purifying selection of retrotransposons in the Arabidopsis thaliana genome. Genome Biol. 2004, 5, R79. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-I.; Kim, N.-S. Transposable elements and genome size variations in plants. Genom. Inform. 2014, 12, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Galindo-González, L.; Sarmiento, F.; Quimbaya, M.A. Shaping Plant Adaptability, Genome Structure and Gene Expression through Transposable Element Epigenetic Control: Focus on Methylation. Agronomy 2018, 8, 180. [Google Scholar] [CrossRef]
- Ramakrishnan, M.; Zhou, M.-B.; Pan, C.-F.; Hänninen, H.; Tang, D.-Q.; Vinod, K.K. Nuclear export signal (NES) of transposases affects the transposition activity of mariner-like elements Ppmar1 and Ppmar2 of moso bamboo. Mob. DNA 2019, 10, 35. [Google Scholar] [CrossRef]
- Ramakrishnan, M.; Zhou, M.; Pan, C.; Hanninen, H.; Yrjala, K.; Vinod, K.K.; Tang, D. Affinities of Terminal Inverted Repeats to DNA Binding Domain of Transposase Affect the Transposition Activity of Bamboo Ppmar2 Mariner-Like Element. Int. J. Mol. Sci. 2019, 20, 3692. [Google Scholar] [CrossRef]
- Lohe, A.R.; Hartl, D.L. Autoregulation of mariner transposase activity by overproduction and dominant-negative complementation. Mol. Biol. Evol. 1996, 13, 549–555. [Google Scholar] [CrossRef]
- Wang, X.; Duan, C.G.; Tang, K.; Wang, B.; Zhang, H.; Lei, M.; Lu, K.; Mangrauthia, S.K.; Wang, P.; Zhu, G.; et al. RNA-binding protein regulates plant DNA methylation by controlling mRNA processing at the intronic heterochromatin-containing gene IBM1. Proc. Natl. Acad. Sci. USA 2013, 110, 15467–15472. [Google Scholar] [CrossRef]
- Slotkin, R.K.; Vaughn, M.; Borges, F.; Tanurdzić, M.; Becker, J.D.; Feijó, J.A.; Martienssen, R.A. Epigenetic reprogramming and small RNA silencing of transposable elements in pollen. Cell 2009, 136, 461–472. [Google Scholar] [CrossRef]
- Klein, S.J.; O’Neill, R.J. Transposable elements: Genome innovation, chromosome diversity, and centromere conflict. Chromosome Res. 2018, 26, 5–23. [Google Scholar] [CrossRef]
- Martin, M.-L.; Jose, L.G.-P. DNA Transposons: Nature and Applications in Genomics. Curr. Genom. 2010, 11, 115–128. [Google Scholar]
- Sigman, M.J.; Slotkin, R.K. The First Rule of Plant Transposable Element Silencing: Location, Location, Location. Plant Cell 2016, 28, 304–313. [Google Scholar] [CrossRef]
- Joly-Lopez, Z.; Forczek, E.; Vello, E.; Hoen, D.R.; Tomita, A.; Bureau, T.E. Abiotic Stress Phenotypes Are Associated with Conserved Genes Derived from Transposable Elements. Front. Plant Sci. 2017, 8, 2027. [Google Scholar] [CrossRef]
- Ito, H.; Kim, J.-M.; Matsunaga, W.; Saze, H.; Matsui, A.; Endo, T.A.; Harukawa, Y.; Takagi, H.; Yaegashi, H.; Masuta, Y.; et al. A Stress-Activated Transposon in Arabidopsis Induces Transgenerational Abscisic Acid Insensitivity. Sci. Rep. 2016, 6, 23181. [Google Scholar] [CrossRef]
- Tikhonov, A.P.; SanMiguel, P.J.; Nakajima, Y.; Gorenstein, N.M.; Bennetzen, J.L.; Avramova, Z. Colinearity and its exceptions in orthologous adh regions of maize and sorghum. Proc. Natl. Acad. Sci. USA 1999, 96, 7409–7414. [Google Scholar] [CrossRef]
- Serrato-Capuchina, A.; Matute, D.R. The role of transposable elements in speciation. Genes 2018, 9, 254. [Google Scholar] [CrossRef]
- Li, S.; Ramakrishnan, M.; Vinod, K.K.; Kalendar, R.; Yrjälä, K.; Zhou, M. Development and Deployment of High-Throughput Retrotransposon-Based Markers Reveal Genetic Diversity and Population Structure of Asian Bamboo. Forests 2020, 11, 31. [Google Scholar] [CrossRef]
- Roy, S. Maintenance of genome stability in plants: Repairing DNA double strand breaks and chromatin structure stability. Front. Plant Sci. 2014, 5, 487. [Google Scholar] [CrossRef]
- Kovalchuk, I. Transgenerational Genome Instability in Plants. In Genome Stability; Kovalchuk, I., Kovalchuk, O., Eds.; Academic Press: Boston, MA, USA, 2016; Chpater 36; pp. 615–633. [Google Scholar]
- Manova, V.; Gruszka, D. DNA damage and repair in plants—From models to crops. Front. Plant Sci. 2015, 6, 885. [Google Scholar] [CrossRef]
- Waititu, J.K.; Zhang, C.; Liu, J.; Wang, H. Plant Non-Coding RNAs: Origin, Biogenesis, Mode of Action and Their Roles in Abiotic Stress. Int. J. Mol. Sci. 2020, 21, 8401. [Google Scholar] [CrossRef]
- Nosaka, M.; Itoh, J.; Nagato, Y.; Ono, A.; Ishiwata, A.; Sato, Y. Role of transposon-derived small RNAs in the interplay between genomes and parasitic DNA in rice. PLoS Genet. 2012, 8, e1002953. [Google Scholar] [CrossRef]
- Ahmed, W.; Xia, Y.; Li, R.; Bai, G.; Siddique, K.H.M.; Guo, P. Non-coding RNAs: Functional roles in the regulation of stress response in Brassica crops. Genomics 2020, 112, 1419–1424. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; He, Q.; Chen, G.; Wang, L.; Jin, B. Regulation of Non-coding RNAs in Heat Stress Responses of Plants. Front. Plant Sci. 2016, 7, 1213. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Liu, Q.; Smith, N.A.; Liang, G.; Wang, M.-B. RNA Silencing in Plants: Mechanisms, Technologies and Applications in Horticultural Crops. Curr. Genom. 2016, 17, 476–489. [Google Scholar] [CrossRef] [PubMed]
- Asefpour Vakilian, K. Machine learning improves our knowledge about miRNA functions towards plant abiotic stresses. Sci. Rep. 2020, 10, 3041. [Google Scholar] [CrossRef]
- Huang, S.; Zhou, J.; Gao, L.; Tang, Y. Plant miR397 and its functions. Funct. Plant Biol. 2021, 48, 361–370. [Google Scholar] [CrossRef]
- Iglesias, M.J.; Terrile, M.C.; Windels, D.; Lombardo, M.C.; Bartoli, C.G.; Vazquez, F.; Estelle, M.; Casalongué, C.A. MiR393 Regulation of Auxin Signaling and Redox-Related Components during Acclimation to Salinity in Arabidopsis. PLoS ONE 2014, 9, e107678. [Google Scholar] [CrossRef]
- Borsani, O.; Zhu, J.; Verslues, P.E.; Sunkar, R.; Zhu, J.K. Endogenous siRNAs derived from a pair of natural cis-antisense transcripts regulate salt tolerance in Arabidopsis. Cell 2005, 123, 1279–1291. [Google Scholar] [CrossRef]
- Jha, U.C.; Nayyar, H.; Jha, R.; Khurshid, M.; Zhou, M.; Mantri, N.; Siddique, K.H.M. Long non-coding RNAs: Emerging players regulating plant abiotic stress response and adaptation. BMC Plant Biol. 2020, 20, 466. [Google Scholar] [CrossRef]
- Wang, L.; Cho, K.B.; Li, Y.; Tao, G.; Xie, Z.; Guo, B. Long Noncoding RNA (lncRNA)-Mediated Competing Endogenous RNA Networks Provide Novel Potential Biomarkers and Therapeutic Targets for Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 5758. [Google Scholar] [CrossRef]
- Wu, H.-J.; Wang, Z.-M.; Wang, M.; Wang, X.-J. Widespread Long Noncoding RNAs as Endogenous Target Mimics for MicroRNAs in Plants. Plant Physiol. 2013, 161, 1875–1884. [Google Scholar] [CrossRef]
- Allen, E.; Xie, Z.; Gustafson, A.M.; Carrington, J.C. microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 2005, 121, 207–221. [Google Scholar] [CrossRef]
- Rajagopalan, R.; Vaucheret, H.; Trejo, J.; Bartel, D.P. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev. 2006, 20, 3407–3425. [Google Scholar] [CrossRef]
- Lurin, C.; Andrés, C.; Aubourg, S.; Bellaoui, M.; Bitton, F.; Bruyère, C.; Caboche, M.; Debast, C.; Gualberto, J.; Hoffmann, B.; et al. Genome-wide analysis of Arabidopsis pentatricopeptide repeat proteins reveals their essential role in organelle biogenesis. Plant Cell 2004, 16, 2089–2103. [Google Scholar] [CrossRef]
- Kume, K.; Tsutsumi, K.; Saitoh, Y. TAS1 trans-acting siRNA targets are differentially regulated at low temperature, and TAS1 trans-acting siRNA mediates temperature-controlled At1g51670 expression. Biosci. Biotechnol. Biochem. 2010, 74, 1435–1440. [Google Scholar] [CrossRef]
- Li, S.; Liu, J.; Liu, Z.; Li, X.; Wu, F.; He, Y. HEAT-INDUCED TAS1 TARGET1 Mediates Thermotolerance via HEAT STRESS TRANSCRIPTION FACTOR A1a-Directed Pathways in Arabidopsis. Plant Cell 2014, 26, 1764–1780. [Google Scholar] [CrossRef]
- Hsieh, L.C.; Lin, S.I.; Shih, A.C.; Chen, J.W.; Lin, W.Y.; Tseng, C.Y.; Li, W.H.; Chiou, T.J. Uncovering small RNA-mediated responses to phosphate deficiency in Arabidopsis by deep sequencing. Plant Physiol. 2009, 151, 2120–2132. [Google Scholar] [CrossRef]
- Luo, Q.J.; Mittal, A.; Jia, F.; Rock, C.D. An autoregulatory feedback loop involving PAP1 and TAS4 in response to sugars in Arabidopsis. Plant Mol. Biol. 2012, 80, 117–129. [Google Scholar] [CrossRef]
- Ito, H.; Gaubert, H.; Bucher, E.; Mirouze, M.; Vaillant, I.; Paszkowski, J. An siRNA pathway prevents transgenerational retrotransposition in plants subjected to stress. Nature 2011, 472, 115–119. [Google Scholar] [CrossRef]
- Matsunaga, W.; Kobayashi, A.; Kato, A.; Ito, H. The effects of heat induction and the siRNA biogenesis pathway on the transgenerational transposition of ONSEN, a copia-like retrotransposon in Arabidopsis thaliana. Plant Cell Physiol. 2012, 53, 824–833. [Google Scholar] [CrossRef]
- Stief, A.; Brzezinka, K.; Lämke, J.; Bäurle, I. Epigenetic responses to heat stress at different time scales and the involvement of small RNAs. Plant Signal. Behav. 2014, 9, e970430. [Google Scholar] [CrossRef]
- Boyko, A.; Kovalchuk, I. Transgenerational response to stress in Arabidopsis thaliana. Plant Signal. Behav. 2010, 5, 995–998. [Google Scholar] [CrossRef]
- Chen, M.; Lv, S.; Meng, Y. Epigenetic performers in plants. Dev. Growth Differ. 2010, 52, 555–566. [Google Scholar] [CrossRef]
- Sridha, S.; Wu, K. Identification of AtHD2C as a novel regulator of abscisic acid responses in Arabidopsis. Plant J. 2006, 46, 124–133. [Google Scholar] [CrossRef]
- Aufsatz, W.; Mette, M.F.; Van der Winden, J.; Matzke, M.; Matzke, A.J. HDA6, a putative histone deacetylase needed to enhance DNA methylation induced by double-stranded RNA. EMBO J. 2002, 21, 6832–6841. [Google Scholar] [CrossRef]
- To, T.K.; Kim, J.-M.; Matsui, A.; Kurihara, Y.; Morosawa, T.; Ishida, J.; Tanaka, M.; Endo, T.; Kakutani, T.; Toyoda, T. Arabidopsis HDA6 regulates locus-directed heterochromatin silencing in cooperation with MET1. PLoS Genet. 2011, 7, e1002055. [Google Scholar] [CrossRef]
- Song, Y.; Wu, K.; Dhaubhadel, S.; An, L.; Tian, L. Arabidopsis DNA methyltransferase AtDNMT2 associates with histone deacetylase AtHD2s activity. Biochem. Biophys. Res. Commun. 2010, 396, 187–192. [Google Scholar] [CrossRef]
- Aung, K.; Lin, S.-I.; Wu, C.-C.; Huang, Y.-T.; Su, C.-l.; Chiou, T.J. pho2, a phosphate overaccumulator, is caused by a nonsense mutation in a microRNA399 target gene. Plant Physiol. 2006, 141, 1000–1011. [Google Scholar] [CrossRef]
- Franco-Zorrilla, J.M.; Valli, A.; Todesco, M.; Mateos, I.; Puga, M.I.; Rubio-Somoza, I.; Leyva, A.; Weigel, D.; García, J.A.; Paz-Ares, J. Target mimicry provides a new mechanism for regulation of microRNA activity. Nat. Genet. 2007, 39, 1033–1037. [Google Scholar] [CrossRef]
- Wang, H.; Chung, P.J.; Liu, J.; Jang, I.C.; Kean, M.J.; Xu, J.; Chua, N.H. Genome-wide identification of long noncoding natural antisense transcripts and their responses to light in Arabidopsis. Genome Res. 2014, 24, 444–453. [Google Scholar] [CrossRef]
- Wunderlich, M.; Gross-Hardt, R.; Schöffl, F. Heat shock factor HSFB2a involved in gametophyte development of Arabidopsis thaliana and its expression is controlled by a heat-inducible long non-coding antisense RNA. Plant Mol. Biol. 2014, 85, 541–550. [Google Scholar] [CrossRef]
- Csorba, T.; Questa, J.I.; Sun, Q.; Dean, C. Antisense COOLAIR mediates the coordinated switching of chromatin states at FLC during vernalization. Proc. Natl. Acad. Sci. USA 2014, 111, 16160–16165. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.B.; Sung, S. Vernalization-mediated epigenetic silencing by a long intronic noncoding RNA. Science 2011, 331, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Popova, O.V.; Dinh, H.Q.; Aufsatz, W.; Jonak, C. The RdDM pathway is required for basal heat tolerance in Arabidopsis. Mol. Plant. 2013, 6, 396–410. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Wang, Y.; Zheng, H.; Lu, W.; Wu, C.; Huang, J.; Yan, K.; Yang, G.; Zheng, C. Salt-induced transcription factor MYB74 is regulated by the RNA-directed DNA methylation pathway in Arabidopsis. J. Exp. Bot. 2015, 66, 5997–6008. [Google Scholar] [CrossRef]
- Wang, L.; Yu, X.; Wang, H.; Lu, Y.Z.; de Ruiter, M.; Prins, M.; He, Y.K. A novel class of heat-responsive small RNAs derived from the chloroplast genome of Chinese cabbage (Brassica rapa). BMC Genom. 2011, 12, 289. [Google Scholar] [CrossRef]
- Yu, X.; Yang, J.; Li, X.; Liu, X.; Sun, C.; Wu, F.; He, Y. Global analysis of cis-natural antisense transcripts and their heat-responsive nat-siRNAs in Brassica rapa. BMC Plant Biol. 2013, 13, 208. [Google Scholar] [CrossRef]
- Song, X.; Liu, G.; Huang, Z.; Duan, W.; Tan, H.; Li, Y.; Hou, X. Temperature expression patterns of genes and their coexpression with LncRNAs revealed by RNA-Seq in non-heading Chinese cabbage. BMC Genom. 2016, 17, 297. [Google Scholar] [CrossRef]
- Furini, A.; Koncz, C.; Salamini, F.; Bartels, D.J.T.E.J. High level transcription of a member of a repeated gene family confers dehydration tolerance to callus tissue of Craterostigma plantagineum. EMBO J. 1997, 16, 3599–3608. [Google Scholar] [CrossRef]
- Xia, J.; Zeng, C.; Chen, Z.; Zhang, K.; Chen, X.; Zhou, Y.; Song, S.; Lu, C.; Yang, R.; Yang, Z. Endogenous small-noncoding RNAs and their roles in chilling response and stress acclimation in Cassava. BMC Genom. 2014, 15, 634. [Google Scholar] [CrossRef]
- Xu, X.W.; Zhou, X.H.; Wang, R.R.; Peng, W.L.; An, Y.; Chen, L.L. Functional analysis of long intergenic non-coding RNAs in phosphate-starved rice using competing endogenous RNA network. Sci. Rep. 2016, 6, 20715. [Google Scholar] [CrossRef]
- Cruz de Carvalho, M.H.; Sun, H.X.; Bowler, C.; Chua, N.H. Noncoding and coding transcriptome responses of a marine diatom to phosphate fluctuations. New Phytol. 2016, 210, 497–510. [Google Scholar] [CrossRef]
- Chen, M.; Wang, C.; Bao, H.; Chen, H.; Wang, Y. Genome-wide identification and characterization of novel lncRNAs in Populus under nitrogen deficiency. Mol. Genet Genom. 2016, 291, 1663–1680. [Google Scholar] [CrossRef]
- Shuai, P.; Liang, D.; Tang, S.; Zhang, Z.; Ye, C.Y.; Su, Y.; Xia, X.; Yin, W. Genome-wide identification and functional prediction of novel and drought-responsive lincRNAs in Populus trichocarpa. J. Exp. Bot. 2014, 65, 4975–4983. [Google Scholar] [CrossRef]
- Huang, W.; Xian, Z.; Hu, G.; Li, Z. SlAGO4A, a core factor of RNA-directed DNA methylation (RdDM) pathway, plays an important role under salt and drought stress in tomato. Mol. Breed. 2016, 36, 28. [Google Scholar] [CrossRef]
- Yao, Y.; Ni, Z.; Peng, H.; Sun, F.; Xin, M.; Sunkar, R.; Zhu, J.K.; Sun, Q. Non-coding small RNAs responsive to abiotic stress in wheat (Triticum aestivum L.). Funct. Integr. Genom. 2010, 10, 187–190. [Google Scholar] [CrossRef]
- Tang, Z.; Zhang, L.; Xu, C.; Yuan, S.; Zhang, F.; Zheng, Y.; Zhao, C. Uncovering small RNA-mediated responses to cold stress in a wheat thermosensitive genic male-sterile line by deep sequencing. Plant Physiol. 2012, 159, 721–738. [Google Scholar] [CrossRef]
- Xin, M.; Wang, Y.; Yao, Y.; Song, N.; Hu, Z.; Qin, D.; Xie, C.; Peng, H.; Ni, Z.; Sun, Q. Identification and characterization of wheat long non-protein coding RNAs responsive to powdery mildew infection and heat stress by using microarray analysis and SBS sequencing. BMC Plant Biol. 2011, 11, 61. [Google Scholar] [CrossRef]
- Zhang, W.; Han, Z.; Guo, Q.; Liu, Y.; Zheng, Y.; Wu, F.; Jin, W. Identification of maize long non-coding RNAs responsive to drought stress. PLoS ONE 2014, 9, e98958. [Google Scholar] [CrossRef]
- Kalendar, R.; Sabot, F.; Rodriguez, F.; Karlov, G.I.; Natali, L.; Alix, K. Editorial: Mobile Elements and Plant Genome Evolution, Comparative Analyzes and Computational Tools. Front. Plant Sci. 2021, 12, 735134. [Google Scholar] [CrossRef]
- Lerat, E.; Casacuberta, J.; Chaparro, C.; Vieira, C. On the Importance to Acknowledge Transposable Elements in Epigenomic Analyses. Genes 2019, 10, 258. [Google Scholar] [CrossRef]
- Sanchez, D.H.; Paszkowski, J. Heat-Induced Release of Epigenetic Silencing Reveals the Concealed Role of an Imprinted Plant Gene. PLoS Genet. 2014, 10, e1004806. [Google Scholar] [CrossRef]
- Cavrak, V.V.; Lettner, N.; Jamge, S.; Kosarewicz, A.; Bayer, L.M.; Mittelsten Scheid, O. How a Retrotransposon Exploits the Plant’s Heat Stress Response for Its Activation. PLoS Genet. 2014, 10, e1004115. [Google Scholar] [CrossRef]
- Choi, J.Y.; Lee, Y.C.G. Double-edged sword: The evolutionary consequences of the epigenetic silencing of transposable elements. PLoS Genet. 2020, 16, e1008872. [Google Scholar] [CrossRef]
- Law, J.A.; Ausin, I.; Johnson, L.M.; Vashisht, A.A.; Zhu, J.K.; Wohlschlegel, J.A.; Jacobsen, S.E. A protein complex required for polymerase V transcripts and RNA- directed DNA methylation in Arabidopsis. Curr. Biol. 2010, 20, 951–956. [Google Scholar] [CrossRef]
- McCue, A.D.; Panda, K.; Nuthikattu, S.; Choudury, S.G.; Thomas, E.N.; Slotkin, R.K. ARGONAUTE 6 bridges transposable element mRNA-derived siRNAs to the establishment of DNA methylation. EMBO J. 2015, 34, 20–35. [Google Scholar] [CrossRef]
- Lang, Z.; Wang, Y.; Tang, K.; Tang, D.; Datsenka, T.; Cheng, J.; Zhang, Y.; Handa, A.K.; Zhu, J.K. Critical roles of DNA demethylation in the activation of ripening-induced genes and inhibition of ripening-repressed genes in tomato fruit. Proc. Natl. Acad. Sci. USA 2017, 114, E4511–E4519. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, J.K. Active DNA demethylation in plants and animals. Cold Spring Harb. Symp. Quant. Biol. 2012, 77, 161–173. [Google Scholar] [CrossRef]
- Zhang, X.; Yazaki, J.; Sundaresan, A.; Cokus, S.; Chan, S.W.; Chen, H.; Henderson, I.R.; Shinn, P.; Pellegrini, M.; Jacobsen, S.E.; et al. Genome-wide high-resolution mapping and functional analysis of DNA methylation in arabidopsis. Cell 2006, 126, 1189–1201. [Google Scholar] [CrossRef]
- Lei, M.; Zhang, H.; Julian, R.; Tang, K.; Xie, S.; Zhu, J.K. Regulatory link between DNA methylation and active demethylation in Arabidopsis. Proc. Natl. Acad. Sci. USA 2015, 112, 3553–3557. [Google Scholar] [CrossRef]
- Williams, B.P.; Pignatta, D.; Henikoff, S.; Gehring, M. Methylation-sensitive expression of a DNA demethylase gene serves as an epigenetic rheostat. PLoS Genet. 2015, 11, e1005142. [Google Scholar] [CrossRef]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Takuno, S.; Gaut, B.S. Gene body methylation is conserved between plant orthologs and is of evolutionary consequence. Proc. Natl. Acad. Sci. USA 2013, 110, 1797–1802. [Google Scholar] [CrossRef] [PubMed]
- Saze, H.; Kitayama, J.; Takashima, K.; Miura, S.; Harukawa, Y.; Ito, T.; Kakutani, T. Mechanism for full-length RNA processing of Arabidopsis genes containing intragenic heterochromatin. Nat. Commun. 2013, 4, 2301. [Google Scholar] [CrossRef]
- Lei, M.; La, H.; Lu, K.; Wang, P.; Miki, D.; Ren, Z.; Duan, C.G.; Wang, X.; Tang, K.; Zeng, L.; et al. Arabidopsis EDM2 promotes IBM1 distal polyadenylation and regulates genome DNA methylation patterns. Proc. Natl. Acad. Sci. USA 2014, 111, 527–532. [Google Scholar] [CrossRef]
- Duan, C.G.; Wang, X.; Zhang, L.; Xiong, X.; Zhang, Z.; Tang, K.; Pan, L.; Hsu, C.C.; Xu, H.; Tao, W.A.; et al. A protein complex regulates RNA processing of intronic heterochromatin-containing genes in Arabidopsis. Proc. Natl. Acad. Sci. USA 2017, 114, E7377–E7384. [Google Scholar] [CrossRef]
- Feng, S.; Cokus, S.J.; Schubert, V.; Zhai, J.; Pellegrini, M.; Jacobsen, S.E. Genome-wide Hi-C analyses in wild-type and mutants reveal high-resolution chromatin interactions in Arabidopsis. Mol. Cell 2014, 55, 694–707. [Google Scholar] [CrossRef]
- Grob, S.; Schmid, M.W.; Grossniklaus, U. Hi-C analysis in Arabidopsis identifies the KNOT, a structure with similarities to the flamenco locus of Drosophila. Mol. Cell 2014, 55, 678–693. [Google Scholar] [CrossRef]
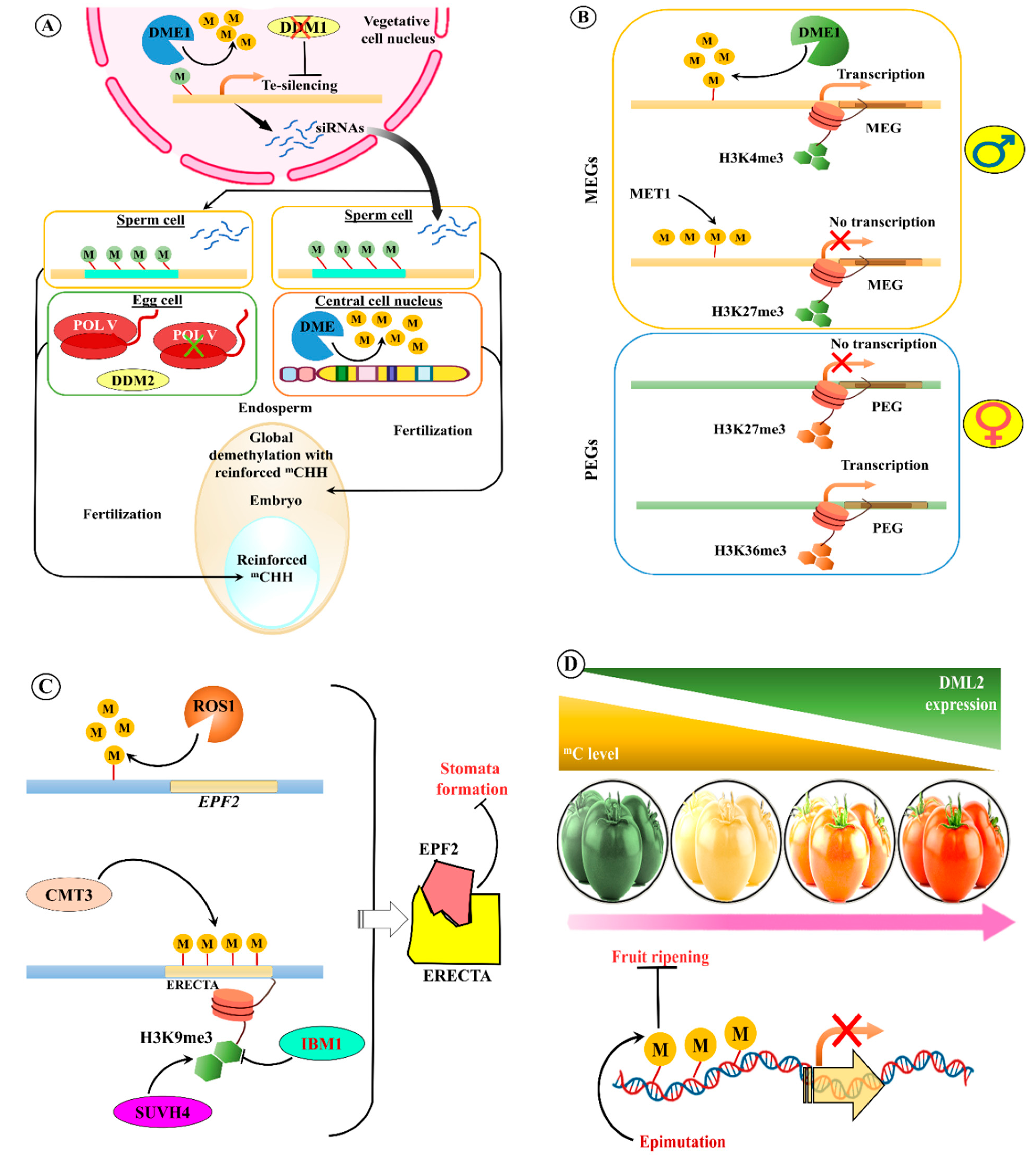
- Ibarra, C.A.; Feng, X.; Schoft, V.K.; Hsieh, T.F.; Uzawa, R.; Rodrigues, J.A.; Zemach, A.; Chumak, N.; Machlicova, A.; Nishimura, T.; et al. Active DNA demethylation in plant companion cells reinforces transposon methylation in gametes. Science 2012, 337, 1360–1364. [Google Scholar] [CrossRef]
- Martínez, G.; Panda, K.; Köhler, C.; Slotkin, R.K. Silencing in sperm cells is directed by RNA movement from the surrounding nurse cell. Nat. Plants 2016, 2, 16030. [Google Scholar] [CrossRef]
- Gehring, M.; Bubb, K.L.; Henikoff, S. Extensive demethylation of repetitive elements during seed development underlies gene imprinting. Science 2009, 324, 1447–1451. [Google Scholar] [CrossRef]
- Ingouff, M.; Selles, B.; Michaud, C.; Vu, T.M.; Berger, F.; Schorn, A.J.; Autran, D.; Van Durme, M.; Nowack, M.K.; Martienssen, R.A.; et al. Live-cell analysis of DNA methylation during sexual reproduction in Arabidopsis reveals context and sex-specific dynamics controlled by noncanonical RdDM. Genes Dev. 2017, 31, 72–83. [Google Scholar] [CrossRef]
- Gehring, M.; Huh, J.H.; Hsieh, T.F.; Penterman, J.; Choi, Y.; Harada, J.J.; Goldberg, R.B.; Fischer, R.L. DEMETER DNA glycosylase establishes MEDEA polycomb gene self-imprinting by allele-specific demethylation. Cell 2006, 124, 495–506. [Google Scholar] [CrossRef]
- Jullien, P.E.; Katz, A.; Oliva, M.; Ohad, N.; Berger, F. Polycomb group complexes self-regulate imprinting of the Polycomb group gene MEDEA in Arabidopsis. Curr. Biol. 2006, 16, 486–492. [Google Scholar] [CrossRef]
- Dong, X.; Zhang, M.; Chen, J.; Peng, L.; Zhang, N.; Wang, X.; Lai, J. Dynamic and Antagonistic Allele-Specific Epigenetic Modifications Controlling the Expression of Imprinted Genes in Maize Endosperm. Mol. Plant 2017, 10, 442–455. [Google Scholar] [CrossRef]
- Yamamuro, C.; Miki, D.; Zheng, Z.; Ma, J.; Wang, J.; Yang, Z.; Dong, J.; Zhu, J.K. Overproduction of stomatal lineage cells in Arabidopsis mutants defective in active DNA demethylation. Nat. Commun. 2014, 5, 4062. [Google Scholar] [CrossRef]
- Wang, Y.; Xue, X.; Zhu, J.K.; Dong, J. Demethylation of ERECTA receptor genes by IBM1 histone demethylase affects stomatal development. Development 2016, 143, 4452–4461. [Google Scholar] [CrossRef]
- Liu, R.; How-Kit, A.; Stammitti, L.; Teyssier, E.; Rolin, D.; Mortain-Bertrand, A.; Halle, S.; Liu, M.; Kong, J.; Wu, C.; et al. A DEMETER-like DNA demethylase governs tomato fruit ripening. Proc. Natl. Acad. Sci. USA 2015, 112, 10804–10809. [Google Scholar] [CrossRef]
- Torres, D.E.; Thomma, B.P.H.J.; Seidl, M.F. Transposable Elements Contribute to Genome Dynamics and Gene Expression Variation in the Fungal Plant Pathogen Verticillium dahliae. Genome Biol. Evol. 2021, 13, evab135. [Google Scholar] [CrossRef]
- Lisch, D. Epigenetic regulation of transposable elements in plants. Annu. Rev. Plant Biol. 2009, 60, 43–66. [Google Scholar] [CrossRef]
- Ahmed, I.; Sarazin, A.; Bowler, C.; Colot, V.; Quesneville, H. Genome-wide evidence for local DNA methylation spreading from small RNA-targeted sequences in Arabidopsis. Nucleic Acids Res. 2011, 39, 6919–6931. [Google Scholar] [CrossRef]
- Hollister, J.D.; Smith, L.M.; Guo, Y.L.; Ott, F.; Weigel, D.; Gaut, B.S. Transposable elements and small RNAs contribute to gene expression divergence between Arabidopsis thaliana and Arabidopsis lyrata. Proc. Natl. Acad. Sci. USA 2011, 108, 2322–2327. [Google Scholar] [CrossRef]
- Sotelo-Silveira, M.; Chávez Montes, R.A.; Sotelo-Silveira, J.R.; Marsch-Martínez, N.; de Folter, S. Entering the Next Dimension: Plant Genomes in 3D. Trends Plant Sci. 2018, 23, 598–612. [Google Scholar] [CrossRef]
- Kumar, A. Jump around: Transposons in and out of the laboratory. F1000Research 2020, 9, 135. [Google Scholar] [CrossRef]
- Rymen, B.; Ferrafiat, L.; Blevins, T. Non-coding RNA polymerases that silence transposable elements and reprogram gene expression in plants. Transcription 2020, 11, 172–191. [Google Scholar] [CrossRef]
- Xu, L.; Jiang, H. Writing and Reading Histone H3 Lysine 9 Methylation in Arabidopsis. Front. Plant Sci. 2020, 11, 452. [Google Scholar] [CrossRef]
- Scheid, R.; Chen, J.; Zhong, X. Biological role and mechanism of chromatin readers in plants. Curr. Opin. Plant Biol. 2021, 61, 102008. [Google Scholar] [CrossRef]
- Pease, N.A.; Nguyen, P.H.B.; Woodworth, M.A.; Ng, K.K.H.; Irwin, B.; Vaughan, J.C.; Kueh, H.Y. Tunable, division-independent control of gene activation timing by a polycomb switch. Cell Rep. 2021, 34, 108888. [Google Scholar] [CrossRef]
- Alix, K.; Gérard, P.R.; Schwarzacher, T.; Heslop-Harrison, J.S. Polyploidy and interspecific hybridization: Partners for adaptation, speciation and evolution in plants. Ann. Bot. 2017, 120, 183–194. [Google Scholar] [CrossRef]
- Kyriakidou, M.; Tai, H.H.; Anglin, N.L.; Ellis, D.; Strömvik, M.V. Current Strategies of Polyploid Plant Genome Sequence Assembly. Front. Plant Sci. 2018, 9, 1660. [Google Scholar] [CrossRef]
- Ritter, E.J.; Niederhuth, C.E. Intertwined evolution of plant epigenomes and genomes. Curr. Opin. Plant Biol. 2021, 61, 101990. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, S.L.; Scheben, A.; Edwards, D.; Spillane, C.; Ortiz, R. Assessing and Exploiting Functional Diversity in Germplasm Pools to Enhance Abiotic Stress Adaptation and Yield in Cereals and Food Legumes. Front. Plant Sci. 2017, 8, 1461. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H. Chromatin Remodeling and Epigenetic Regulation in Plant DNA Damage Repair. Int. J. Mol. Sci. 2019, 20, 4093. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Fromm, M.; Avramova, Z. H3K27me3 and H3K4me3 chromatin environment at super-induced dehydration stress memory genes of Arabidopsis thaliana. Mol. Plant 2014, 7, 502–513. [Google Scholar] [CrossRef]
- Mehraj, H.; Takahashi, S.; Miyaji, N.; Akter, A.; Suzuki, Y.; Seki, M.; Dennis, E.S.; Fujimoto, R. Characterization of Histone H3 Lysine 4 and 36 Tri-methylation in Brassica rapa L. Front. Plant Sci. 2021, 12, 785. [Google Scholar] [CrossRef]
- Gan, E.-S.; Xu, Y.; Ito, T. Dynamics of H3K27me3 methylation and demethylation in plant development. Plant Signal. Behav. 2015, 10, e1027851. [Google Scholar] [CrossRef]
- Bhadouriya, S.L.; Mehrotra, S.; Basantani, M.K.; Loake, G.J.; Mehrotra, R. Role of Chromatin Architecture in Plant Stress Responses: An Update. Front. Plant Sci. 2021, 11, 603380. [Google Scholar] [CrossRef]
- Tricker, P.J.; Gibbings, J.G.; Rodríguez López, C.M.; Hadley, P.; Wilkinson, M.J. Low relative humidity triggers RNA-directed de novo DNA methylation and suppression of genes controlling stomatal development. J. Exp. Bot. 2012, 63, 3799–3813. [Google Scholar] [CrossRef]
- Miryeganeh, M. Plants’ Epigenetic Mechanisms and Abiotic Stress. Genes 2021, 12, 1106. [Google Scholar] [CrossRef]
- Kong, L.; Liu, Y.; Wang, X.; Chang, C. Insight into the Role of Epigenetic Processes in Abiotic and Biotic Stress Response in Wheat and Barley. Int. J. Mol. Sci. 2020, 21, 1480. [Google Scholar] [CrossRef]
- Secco, D.; Wang, C.; Shou, H.; Schultz, M.D.; Chiarenza, S.; Nussaume, L.; Ecker, J.R.; Whelan, J.; Lister, R. Stress induced gene expression drives transient DNA methylation changes at adjacent repetitive elements. eLife 2015, 4, e09343. [Google Scholar] [CrossRef]
- Liu, J.; He, Z. Small DNA Methylation, Big Player in Plant Abiotic Stress Responses and Memory. Front. Plant Sci. 2020, 11, 1977. [Google Scholar] [CrossRef]
- Iwasaki, M.; Hyvärinen, L.; Piskurewicz, U.; Lopez-Molina, L. Non-canonical RNA-directed DNA methylation participates in maternal and environmental control of seed dormancy. eLife 2019, 8, e37434. [Google Scholar] [CrossRef]
- Xie, H.J.; Li, H.; Liu, D.; Dai, W.M.; He, J.Y.; Lin, S.; Duan, H.; Liu, L.L.; Chen, S.G.; Song, X.L.; et al. ICE1 demethylation drives the range expansion of a plant invader through cold tolerance divergence. Mol. Ecol. 2015, 24, 835–850. [Google Scholar] [CrossRef]
- Xie, H.; Sun, Y.; Cheng, B.; Xue, S.; Cheng, D.; Liu, L.; Meng, L.; Qiang, S. Variation in ICE1 Methylation Primarily Determines Phenotypic Variation in Freezing Tolerance in Arabidopsis thaliana. Plant Cell Physiol. 2019, 60, 152–165. [Google Scholar] [CrossRef]
- Liu, T.; Li, Y.; Duan, W.; Huang, F.; Hou, X. Cold acclimation alters DNA methylation patterns and confers tolerance to heat and increases growth rate in Brassica rapa. J. Exp. Bot. 2017, 68, 1213–1224. [Google Scholar] [CrossRef]
- Duan, W.; Zhang, H.; Zhang, B.; Wu, X.; Shao, S.; Li, Y.; Hou, X.; Liu, T. Role of vernalization-mediated demethylation in the floral transition of Brassica rapa. Planta 2017, 245, 227–233. [Google Scholar] [CrossRef]
- Lai, Y.S.; Zhang, X.; Zhang, W.; Shen, D.; Wang, H.; Xia, Y.; Qiu, Y.; Song, J.; Wang, C.; Li, X. The association of changes in DNA methylation with temperature-dependent sex determination in cucumber. J. Exp. Bot. 2017, 68, 2899–2912. [Google Scholar] [CrossRef]
- Ma, N.; Chen, W.; Fan, T.; Tian, Y.; Zhang, S.; Zeng, D.; Li, Y. Low temperature-induced DNA hypermethylation attenuates expression of RhAG, an AGAMOUS homolog, and increases petal number in rose (Rosa hybrida). BMC Plant Biol. 2015, 15, 237. [Google Scholar] [CrossRef]
- Rutowicz, K.; Puzio, M.; Halibart-Puzio, J.; Lirski, M.; Kotliński, M.; Kroteń, M.A.; Knizewski, L.; Lange, B.; Muszewska, A.; Śniegowska-Świerk, K.; et al. A Specialized Histone H1 Variant Is Required for Adaptive Responses to Complex Abiotic Stress and Related DNA Methylation in Arabidopsis. Plant Physiol. 2015, 169, 2080–2101. [Google Scholar] [CrossRef]
- Gagné-Bourque, F.; Mayer, B.F.; Charron, J.B.; Vali, H.; Bertrand, A.; Jabaji, S. Accelerated Growth Rate and Increased Drought Stress Resilience of the Model Grass Brachypodium distachyon Colonized by Bacillus subtilis B26. PLoS ONE 2015, 10, e0130456. [Google Scholar] [CrossRef]
- Lu, X.; Wang, X.; Chen, X.; Shu, N.; Wang, J.; Wang, D.; Wang, S.; Fan, W.; Guo, L.; Guo, X.; et al. Single-base resolution methylomes of upland cotton (Gossypium hirsutum L.) reveal epigenome modifications in response to drought stress. BMC Genom. 2017, 18, 297. [Google Scholar] [CrossRef]
- Wang, W.; Qin, Q.; Sun, F.; Wang, Y.; Xu, D.; Li, Z.; Fu, B. Genome-Wide Differences in DNA Methylation Changes in Two Contrasting Rice Genotypes in Response to Drought Conditions. Front. Plant Sci. 2016, 7, 1675. [Google Scholar] [CrossRef]
- Liang, D.; Zhang, Z.; Wu, H.; Huang, C.; Shuai, P.; Ye, C.Y.; Tang, S.; Wang, Y.; Yang, L.; Wang, J.; et al. Single-base-resolution methylomes of Populus trichocarpa reveal the association between DNA methylation and drought stress. BMC Genet. 2014, 15 (Suppl. 1), S9. [Google Scholar] [CrossRef]
- Mao, H.; Wang, H.; Liu, S.; Li, Z.; Yang, X.; Yan, J.; Li, J.; Tran, L.S.; Qin, F. A transposable element in a NAC gene is associated with drought tolerance in maize seedlings. Nat. Commun. 2015, 6, 8326. [Google Scholar] [CrossRef]
- Shen, X.; De Jonge, J.; Forsberg, S.K.; Pettersson, M.E.; Sheng, Z.; Hennig, L.; Carlborg, Ö. Natural CMT2 variation is associated with genome-wide methylation changes and temperature seasonality. PLoS Genet. 2014, 10, e1004842. [Google Scholar] [CrossRef]
- Li, J.; Huang, Q.; Sun, M.; Zhang, T.; Li, H.; Chen, B.; Xu, K.; Gao, G.; Li, F.; Yan, G.; et al. Global DNA methylation variations after short-term heat shock treatment in cultured microspores of Brassica napus cv. Topas. Sci. Rep. 2016, 6, 38401. [Google Scholar] [CrossRef]
- Gao, G.; Li, J.; Li, H.; Li, F.; Xu, K.; Yan, G.; Chen, B.; Qiao, J.; Wu, X. Comparison of the heat stress induced variations in DNA methylation between heat-tolerant and heat-sensitive rapeseed seedlings. Breed. Sci. 2014, 64, 125–133. [Google Scholar] [CrossRef]
- Hossain, M.S.; Kawakatsu, T.; Kim, K.D.; Zhang, N.; Nguyen, C.T.; Khan, S.M.; Batek, J.M.; Joshi, T.; Schmutz, J.; Grimwood, J.; et al. Divergent cytosine DNA methylation patterns in single-cell, soybean root hairs. New Phytol. 2017, 214, 808–819. [Google Scholar] [CrossRef]
- Ma, Y.; Min, L.; Wang, M.; Wang, C.; Zhao, Y.; Li, Y.; Fang, Q.; Wu, Y.; Xie, S.; Ding, Y.; et al. Disrupted Genome Methylation in Response to High Temperature Has Distinct Affects on Microspore Abortion and Anther Indehiscence. Plant Cell 2018, 30, 1387–1403. [Google Scholar] [CrossRef]
- Min, L.; Li, Y.; Hu, Q.; Zhu, L.; Gao, W.; Wu, Y.; Ding, Y.; Liu, S.; Yang, X.; Zhang, X. Sugar and auxin signaling pathways respond to high-temperature stress during anther development as revealed by transcript profiling analysis in cotton. Plant Physiol. 2014, 164, 1293–1308. [Google Scholar] [CrossRef] [PubMed]
- Folsom, J.J.; Begcy, K.; Hao, X.; Wang, D.; Walia, H. Rice fertilization-Independent Endosperm1 regulates seed size under heat stress by controlling early endosperm development. Plant Physiol. 2014, 165, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liang, Z.; Cui, X.; Ji, C.; Li, Y.; Zhang, P.; Liu, J.; Riaz, A.; Yao, P.; Liu, M.; et al. N(6)-Methyladenine DNA Methylation in Japonica and Indica Rice Genomes and Its Association with Gene Expression, Plant Development, and Stress Responses. Mol. Plant 2018, 11, 1492–1508. [Google Scholar] [CrossRef] [PubMed]
- Moglia, A.; Gianoglio, S.; Acquadro, A.; Valentino, D.; Milani, A.M.; Lanteri, S.; Comino, C. Identification of DNA methyltransferases and demethylases in Solanum melongena L. and their transcription dynamics during fruit development and after salt and drought stresses. PLoS ONE 2019, 14, e0223581. [Google Scholar] [CrossRef]
- Benoit, M.; Drost, H.G.; Catoni, M.; Gouil, Q.; Lopez-Gomollon, S.; Baulcombe, D.; Paszkowski, J. Environmental and epigenetic regulation of Rider retrotransposons in tomato. PLoS Genet. 2019, 15, e1008370. [Google Scholar] [CrossRef]
- Marconi, G.; Pace, R.; Traini, A.; Raggi, L.; Lutts, S.; Chiusano, M.; Guiducci, M.; Falcinelli, M.; Benincasa, P.; Albertini, E. Use of MSAP markers to analyse the effects of salt stress on DNA methylation in rapeseed (Brassica napus var. oleifera). PLoS ONE 2013, 8, e75597. [Google Scholar] [CrossRef]
- Ganie, S.A.; Dey, N.; Mondal, T.K. Promoter methylation regulates the abundance of osa-miR393a in contrasting rice genotypes under salinity stress. Funct. Integr. Genom. 2016, 16, 1–11. [Google Scholar] [CrossRef]
- Kumar, S.; Beena, A.S.; Awana, M.; Singh, A. Salt-Induced Tissue-Specific Cytosine Methylation Downregulates Expression of HKT Genes in Contrasting Wheat (Triticum aestivum L.) Genotypes. DNA Cell Biol. 2017, 36, 283–294. [Google Scholar] [CrossRef]
- Wang, M.; Qin, L.; Xie, C.; Li, W.; Yuan, J.; Kong, L.; Yu, W.; Xia, G.; Liu, S. Induced and constitutive DNA methylation in a salinity-tolerant wheat introgression line. Plant Cell Physiol. 2014, 55, 1354–1365. [Google Scholar] [CrossRef]
- Tan, M.P. Analysis of DNA methylation of maize in response to osmotic and salt stress based on methylation-sensitive amplified polymorphism. Plant Physiol. Biochem. 2010, 48, 21–26. [Google Scholar] [CrossRef]
- Peng, Y.; Zhang, Y.; Gui, Y.; An, D.; Liu, J.; Xu, X.; Li, Q.; Wang, J.; Wang, W.; Shi, C.; et al. Elimination of a Retrotransposon for Quenching Genome Instability in Modern Rice. Mol. Plant 2019, 12, 1395–1407. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, N.; Liu, J.; Liu, W.; Wang, G.-L. The role of effectors and host immunity in plant-necrotrophic fungal interactions. Virulence 2014, 5, 722–732. [Google Scholar] [CrossRef]
- Zou, B.; Sun, Q.; Zhang, W.; Ding, Y.; Yang, D.L.; Shi, Z.; Hua, J. The Arabidopsis Chromatin-Remodeling Factor CHR5 Regulates Plant Immune Responses and Nucleosome Occupancy. Plant Cell Physiol. 2017, 58, 2202–2216. [Google Scholar] [CrossRef]
- Choi, S.-M.; Song, H.-R.; Han, S.-K.; Han, M.; Kim, C.-Y.; Park, J.; Lee, Y.-H.; Jeon, J.-S.; Noh, Y.-S.; Noh, B. HDA19 is required for the repression of salicylic acid biosynthesis and salicylic acid-mediated defense responses in Arabidopsis. Plant J. 2012, 71, 135–146. [Google Scholar] [CrossRef]
- Li, T.; Chen, X.; Zhong, X.; Zhao, Y.; Liu, X.; Zhou, S.; Cheng, S.; Zhou, D.-X. Jumonji C domain protein JMJ705-mediated removal of histone H3 lysine 27 trimethylation is involved in defense-related gene activation in rice. Plant Cell 2013, 25, 4725–4736. [Google Scholar] [CrossRef]
- Cui, X.; Jin, P.; Cui, X.; Gu, L.; Lu, Z.; Xue, Y.; Wei, L.; Qi, J.; Song, X.; Luo, M.; et al. Control of transposon activity by a histone H3K4 demethylase in rice. Proc. Natl. Acad. Sci. USA 2013, 110, 1953–1958. [Google Scholar] [CrossRef]
- Soyer, J.L.; El Ghalid, M.; Glaser, N.; Ollivier, B.; Linglin, J.; Grandaubert, J.; Balesdent, M.-H.; Connolly, L.R.; Freitag, M.; Rouxel, T.; et al. Epigenetic control of effector gene expression in the plant pathogenic fungus Leptosphaeria maculans. PLoS Genet. 2014, 10, e1004227. [Google Scholar] [CrossRef]
- Zhang, X. Dynamic differential methylation facilitates pathogen stress response in Arabidopsis. Proc. Natl. Acad. Sci. USA 2012, 109, 12842–12843. [Google Scholar] [CrossRef]
- Schumann, U.; Lee, J.; Kazan, K.; Ayliffe, M.; Wang, M.-B. DNA-Demethylase Regulated Genes Show Methylation-Independent Spatiotemporal Expression Patterns. Front. Plant Sci. 2017, 8, 1449. [Google Scholar] [CrossRef]
- Annacondia, M.L.; Magerøy, M.H.; Martinez, G. Stress response regulation by epigenetic mechanisms: Changing of the guards. Physiol. Plant. 2018, 162, 239–250. [Google Scholar] [CrossRef]
- Liu, S.; de Jonge, J.; Trejo-Arellano, M.S.; Santos-González, J.; Köhler, C.; Hennig, L. Role of H1 and DNA methylation in selective regulation of transposable elements during heat stress. New Phytol. 2021, 229, 2238–2250. [Google Scholar] [CrossRef] [PubMed]
- Le, T.-N.; Schumann, U.; Smith, N.A.; Tiwari, S.; Au, P.C.K.; Zhu, Q.-H.; Taylor, J.M.; Kazan, K.; Llewellyn, D.J.; Zhang, R.; et al. DNA demethylases target promoter transposable elements to positively regulate stress responsive genes in Arabidopsis. Genome Biol. 2014, 15, 458. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, M.; Paszkowski, J. Identification of genes preventing transgenerational transmission of stress-induced epigenetic states. Proc. Natl. Acad. Sci. USA 2014, 111, 8547–8552. [Google Scholar] [CrossRef] [PubMed]
- Lanciano, S.; Cristofari, G. Measuring and interpreting transposable element expression. Nat. Rev. Genet. 2020, 21, 721–736. [Google Scholar] [CrossRef]
- Kulski, J.K. Next-generation sequencing—an overview of the history, tools, and “Omic” applications. In Next Generation Sequencing—Advances, Applications and Challenges; Kulski, J.K., Ed.; IntechOpen: London, UK, 2016; Volume 3, p. 60. [Google Scholar]
- Boo, S.H.; Kim, Y.K. The emerging role of RNA modifications in the regulation of mRNA stability. Exp. Mol. Med. 2020, 52, 400–408. [Google Scholar] [CrossRef]
- Satheesh, V.; Fan, W.; Chu, J.; Cho, J. Recent advancement of NGS technologies to detect active transposable elements in plants. Genes Genom. 2021, 43, 289–294. [Google Scholar] [CrossRef]
- Cho, J.; Benoit, M.; Catoni, M.; Drost, H.G.; Brestovitsky, A.; Oosterbeek, M.; Paszkowski, J. Sensitive detection of pre-integration intermediates of long terminal repeat retrotransposons in crop plants. Nat. Plants 2019, 5, 26–33. [Google Scholar] [CrossRef]
- Wang, B.; Tseng, E.; Regulski, M.; Clark, T.A.; Hon, T.; Jiao, Y.; Lu, Z.; Olson, A.; Stein, J.C.; Ware, D. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 2016, 7, 11708. [Google Scholar] [CrossRef]
- Panda, K.; Slotkin, R.K. Long-Read cDNA Sequencing Enables a “Gene-Like” Transcript Annotation of Transposable Elements. Plant Cell 2020, 32, 2687–2698. [Google Scholar] [CrossRef]
- Deininger, P.; Morales, M.E.; White, T.B.; Baddoo, M.; Hedges, D.J.; Servant, G.; Srivastav, S.; Smither, M.E.; Concha, M.; DeHaro, D.L.; et al. A comprehensive approach to expression of L1 loci. Nucleic Acids Res. 2017, 45, e31. [Google Scholar] [CrossRef]
- Belancio, V.P.; Roy-Engel, A.M.; Pochampally, R.R.; Deininger, P. Somatic expression of LINE-1 elements in human tissues. Nucleic Acids Res. 2010, 38, 3909–3922. [Google Scholar] [CrossRef]
- Morillon, A.; Bénard, L.; Springer, M.; Lesage, P. Differential effects of chromatin and Gcn4 on the 50-fold range of expression among individual yeast Ty1 retrotransposons. Mol. Cell Biol. 2002, 22, 2078–2088. [Google Scholar] [CrossRef]
- Orozco-Arias, S.; Isaza, G.; Guyot, R.; Tabares-Soto, R. A systematic review of the application of machine learning in the detection and classification of transposable elements. PeerJ 2019, 7, e8311. [Google Scholar] [CrossRef]
- Tokuyama, M.; Kong, Y.; Song, E.; Jayewickreme, T.; Kang, I.; Iwasaki, A. ERVmap analysis reveals genome-wide transcription of human endogenous retroviruses. Proc. Natl. Acad. Sci. USA 2018, 115, 12565–12572. [Google Scholar] [CrossRef]
- McKerrow, W.; Fenyö, D. L1EM: A tool for accurate locus specific LINE-1 RNA quantification. Bioinformatics 2020, 36, 1167–1173. [Google Scholar] [CrossRef]
- Philippe, C.; Vargas-Landin, D.B.; Doucet, A.J.; van Essen, D.; Vera-Otarola, J.; Kuciak, M.; Corbin, A.; Nigumann, P.; Cristofari, G. Activation of individual L1 retrotransposon instances is restricted to cell-type dependent permissive loci. eLife 2016, 5, e13926. [Google Scholar] [CrossRef]
- Scott, E.C.; Gardner, E.J.; Masood, A.; Chuang, N.T.; Vertino, P.M.; Devine, S.E. A hot L1 retrotransposon evades somatic repression and initiates human colorectal cancer. Genome Res. 2016, 26, 745–755. [Google Scholar] [CrossRef]
- Teissandier, A.; Servant, N.; Barillot, E.; Bourc’his, D. Tools and best practices for retrotransposon analysis using high-throughput sequencing data. Mob. DNA 2019, 10, 52. [Google Scholar] [CrossRef]
- Kong, Y.; Rose, C.M.; Cass, A.A.; Williams, A.G.; Darwish, M.; Lianoglou, S.; Haverty, P.M.; Tong, A.J.; Blanchette, C.; Albert, M.L.; et al. Transposable element expression in tumors is associated with immune infiltration and increased antigenicity. Nat. Commun. 2019, 10, 5228. [Google Scholar] [CrossRef]
- Criscione, S.W.; Zhang, Y.; Thompson, W.; Sedivy, J.M.; Neretti, N. Transcriptional landscape of repetitive elements in normal and cancer human cells. BMC Genom. 2014, 15, 583. [Google Scholar] [CrossRef]
- Jeong, H.H.; Yalamanchili, H.K.; Guo, C.; Shulman, J.M.; Liu, Z. An ultra-fast and scalable quantification pipeline for transposable elements from next generation sequencing data. Pac. Symp. Biocomput. 2018, 23, 168–179. [Google Scholar] [PubMed]
- Yang, W.R.; Ardeljan, D.; Pacyna, C.N.; Payer, L.M.; Burns, K.H. SQuIRE reveals locus-specific regulation of interspersed repeat expression. Nucleic Acids Res. 2019, 47, e27. [Google Scholar] [CrossRef] [PubMed]
- Lerat, E.; Fablet, M.; Modolo, L.; Lopez-Maestre, H.; Vieira, C. TEtools facilitates big data expression analysis of transposable elements and reveals an antagonism between their activity and that of piRNA genes. Nucleic Acids Res. 2017, 45, e17. [Google Scholar] [CrossRef] [PubMed]
- Valdebenito-Maturana, B.; Riadi, G. TEcandidates: Prediction of genomic origin of expressed transposable elements using RNA-seq data. Bioinformatics 2018, 34, 3915–3916. [Google Scholar] [CrossRef]
- Bendall, M.L.; de Mulder, M.; Iñiguez, L.P.; Lecanda-Sánchez, A.; Pérez-Losada, M.; Ostrowski, M.A.; Jones, R.B.; Mulder, L.C.F.; Reyes-Terán, G.; Crandall, K.A.; et al. Telescope: Characterization of the retrotranscriptome by accurate estimation of transposable element expression. PLoS Comput. Biol. 2019, 15, e1006453. [Google Scholar] [CrossRef]
- Jin, Y.; Tam, O.H.; Paniagua, E.; Hammell, M. TEtranscripts: A package for including transposable elements in differential expression analysis of RNA-seq datasets. Bioinformatics 2015, 31, 3593–3599. [Google Scholar] [CrossRef]
- Navarro, F.C.; Hoops, J.; Bellfy, L.; Cerveira, E.; Zhu, Q.; Zhang, C.; Lee, C.; Gerstein, M.B. TeXP: Deconvolving the effects of pervasive and autonomous transcription of transposable elements. PLoS Comput. Biol. 2019, 15, e1007293. [Google Scholar] [CrossRef]
- Lacal, I.; Ventura, R. Epigenetic Inheritance: Concepts, Mechanisms and Perspectives. Front. Mol. Neurosci. 2018, 11, 292. [Google Scholar] [CrossRef]
- Shahid, S.; Slotkin, R.K. The current revolution in transposable element biology enabled by long reads. Curr. Opin. Plant Biol. 2020, 54, 49–56. [Google Scholar] [CrossRef]
- Hu, J.; Manduzio, S.; Kang, H. Epitranscriptomic RNA Methylation in Plant Development and Abiotic Stress Responses. Front. Plant Sci. 2019, 10, 500. [Google Scholar] [CrossRef]
- Valihrach, L.; Androvic, P.; Kubista, M. Platforms for Single-Cell Collection and Analysis. Int. J. Mol. Sci. 2018, 19, 807. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramakrishnan, M.; Satish, L.; Kalendar, R.; Narayanan, M.; Kandasamy, S.; Sharma, A.; Emamverdian, A.; Wei, Q.; Zhou, M. The Dynamism of Transposon Methylation for Plant Development and Stress Adaptation. Int. J. Mol. Sci. 2021, 22, 11387. https://doi.org/10.3390/ijms222111387
Ramakrishnan M, Satish L, Kalendar R, Narayanan M, Kandasamy S, Sharma A, Emamverdian A, Wei Q, Zhou M. The Dynamism of Transposon Methylation for Plant Development and Stress Adaptation. International Journal of Molecular Sciences. 2021; 22(21):11387. https://doi.org/10.3390/ijms222111387
Chicago/Turabian StyleRamakrishnan, Muthusamy, Lakkakula Satish, Ruslan Kalendar, Mathiyazhagan Narayanan, Sabariswaran Kandasamy, Anket Sharma, Abolghassem Emamverdian, Qiang Wei, and Mingbing Zhou. 2021. "The Dynamism of Transposon Methylation for Plant Development and Stress Adaptation" International Journal of Molecular Sciences 22, no. 21: 11387. https://doi.org/10.3390/ijms222111387